
Combinational Inhibition of P-Glycoprotein-Mediated Etoposide Transport by Zosuquidar and Polysorbate 20
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Determination of Zosuquidar’s pKa Values
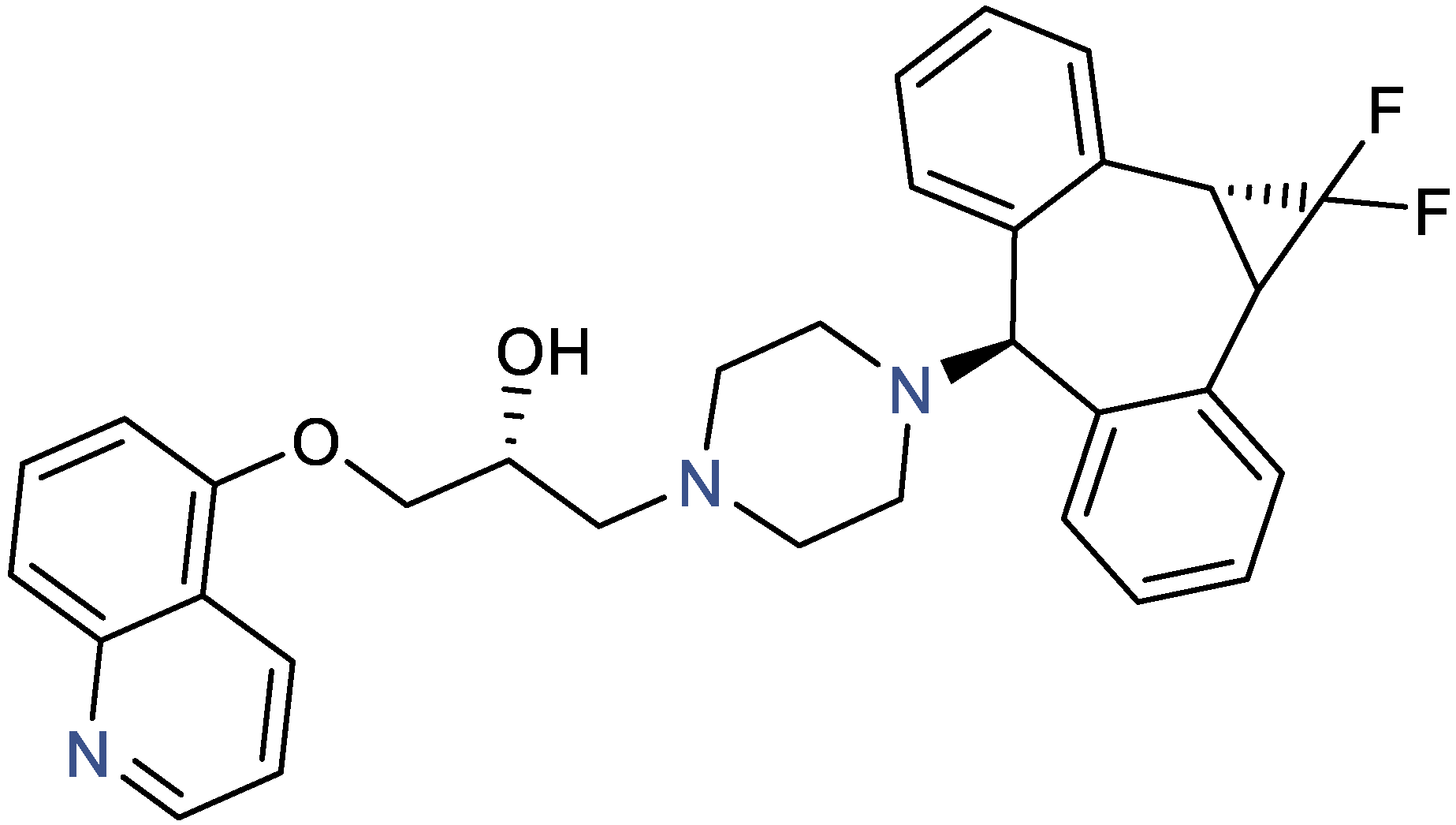
2.2. Zosuquidar Solubility
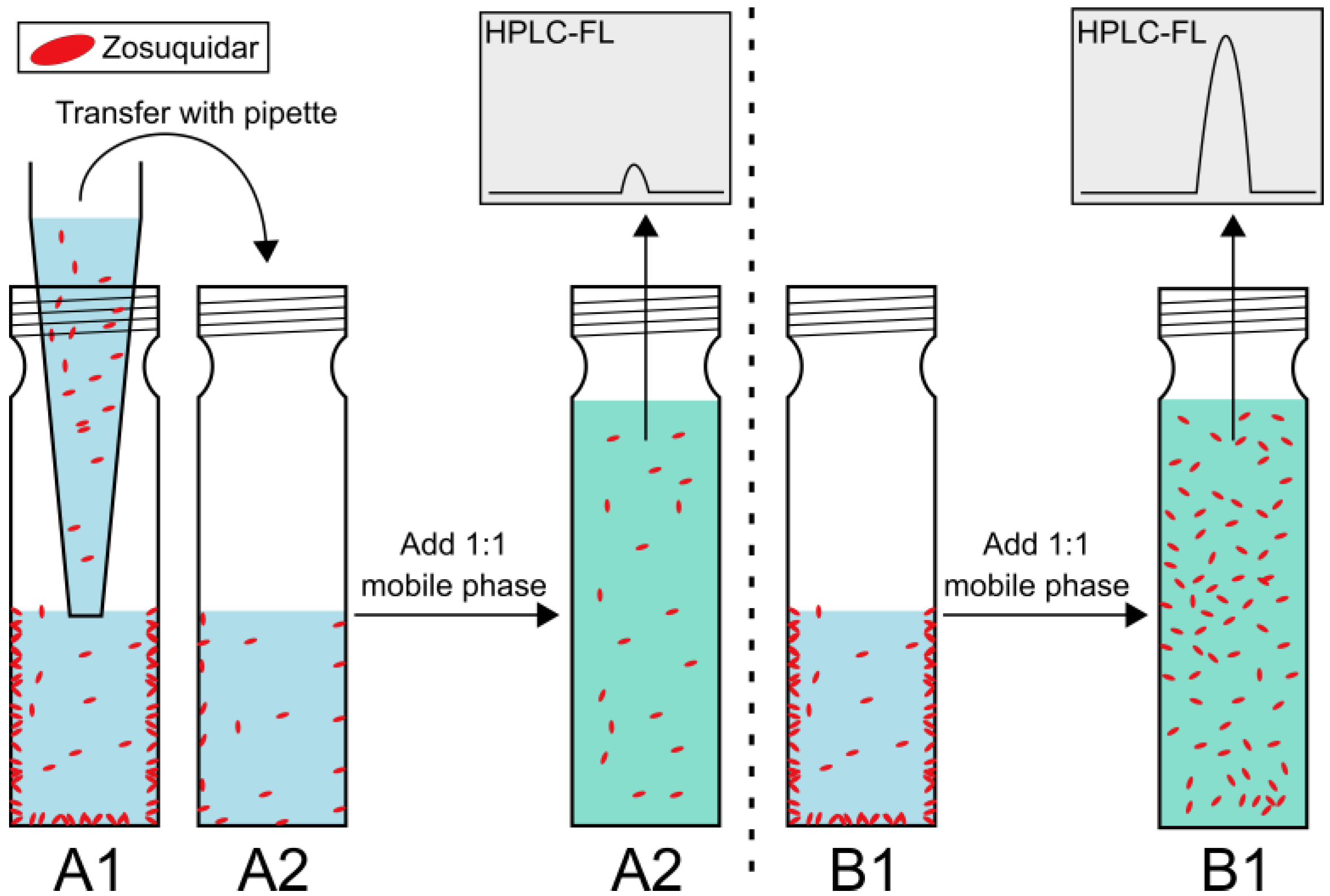
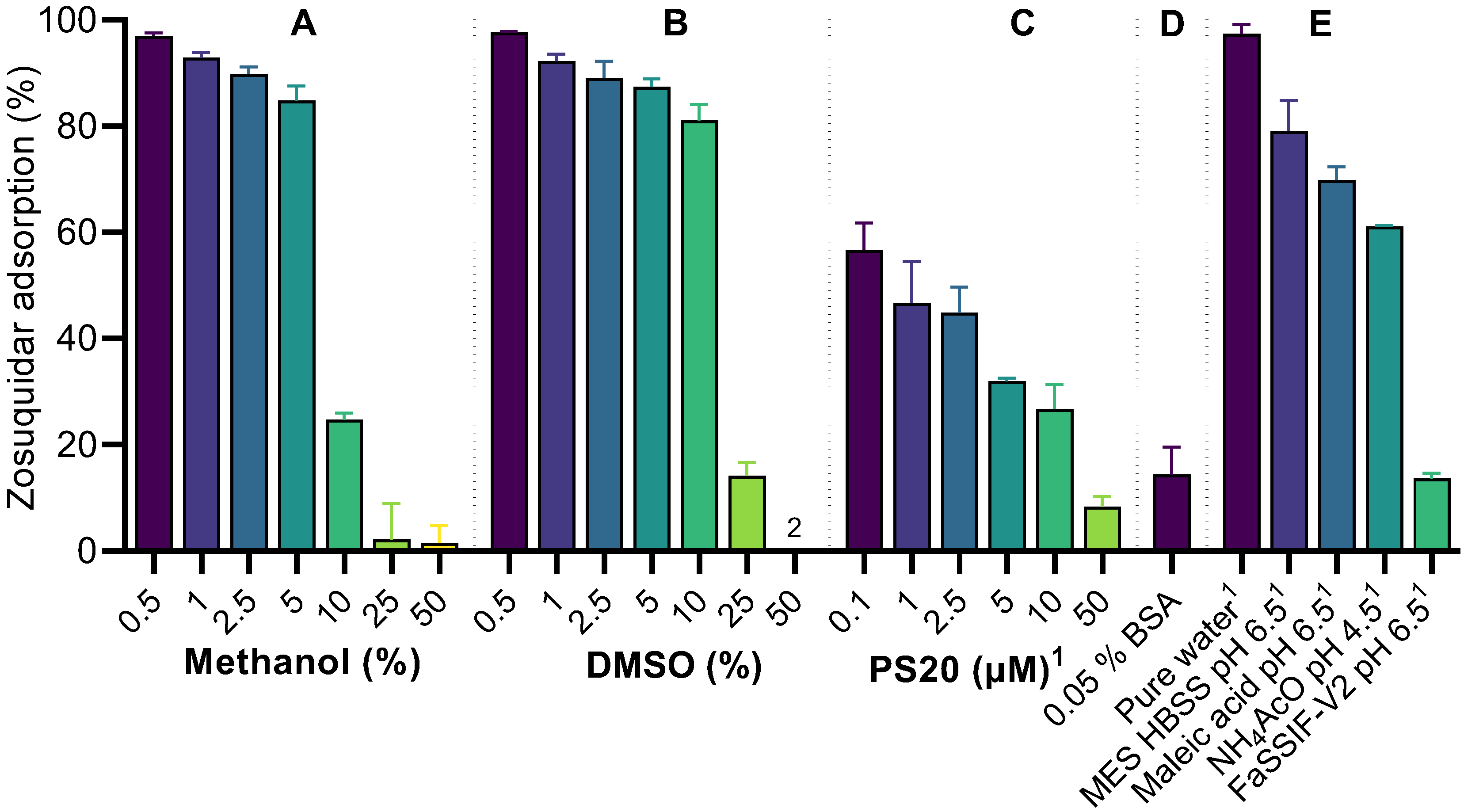
2.3. Nonspecific Zosuquidar Adsorption
2.4. Cell Cultivation
2.5. Transcellular Etoposide Permeability across Cell Monolayers
2.5.1. Assessment of Cell Monolayer Integrity
2.5.2. Data Analysis
2.6. Calcein-AM Assay
2.7. Etoposide and Zosuquidar Quantification by HPLC-FL
2.8. Pharmacokinetic Study in Sprague Dawley Rats
2.8.1. Study Design, Dosing, and Sampling
2.8.2. Data Analysis
3. Results and Discussion
3.1. Zosuquidar pKa and Solubility
3.2. Zosuquidar Adsorbed to Various Lab Equipment
3.3. Transcellular Etoposide Permeability Studies
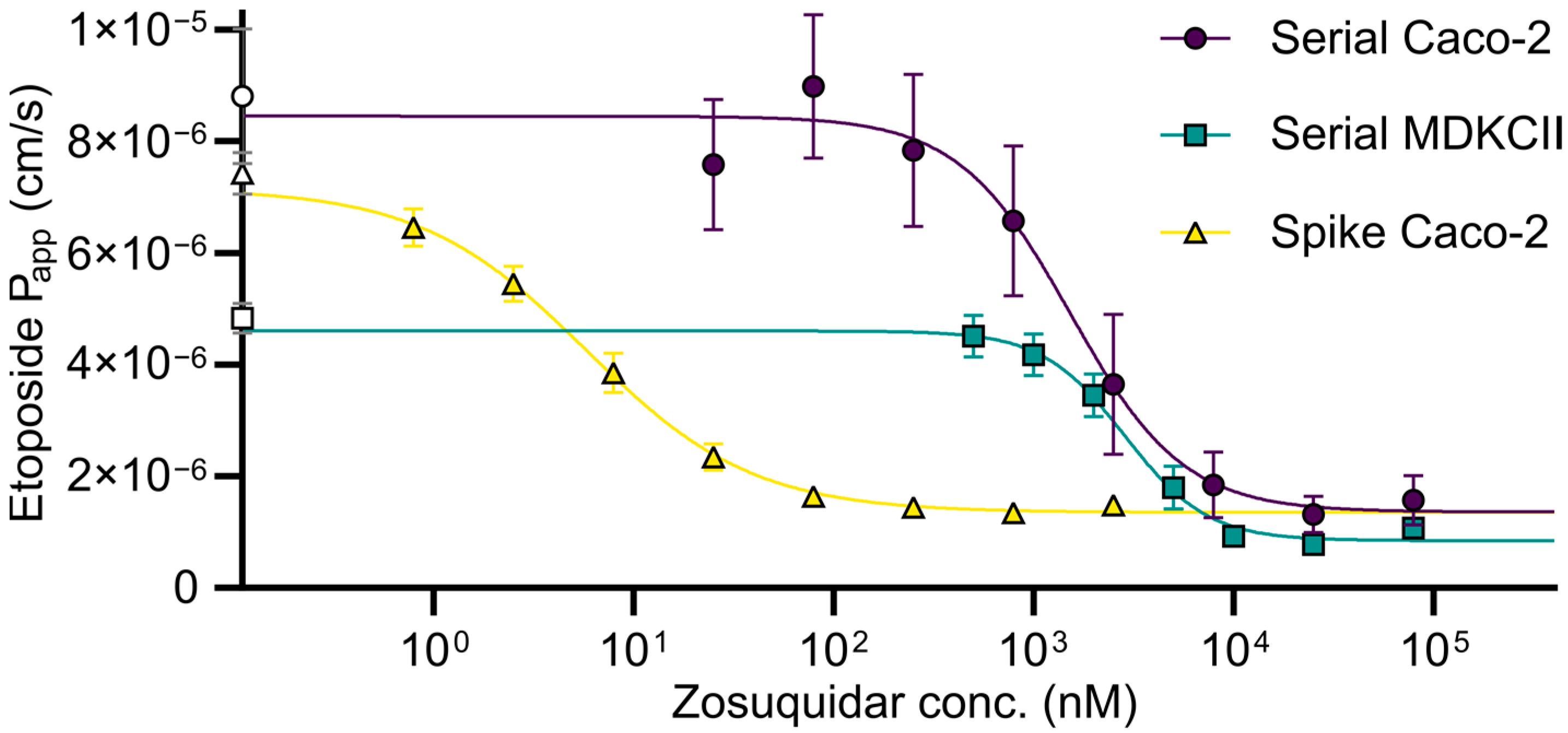
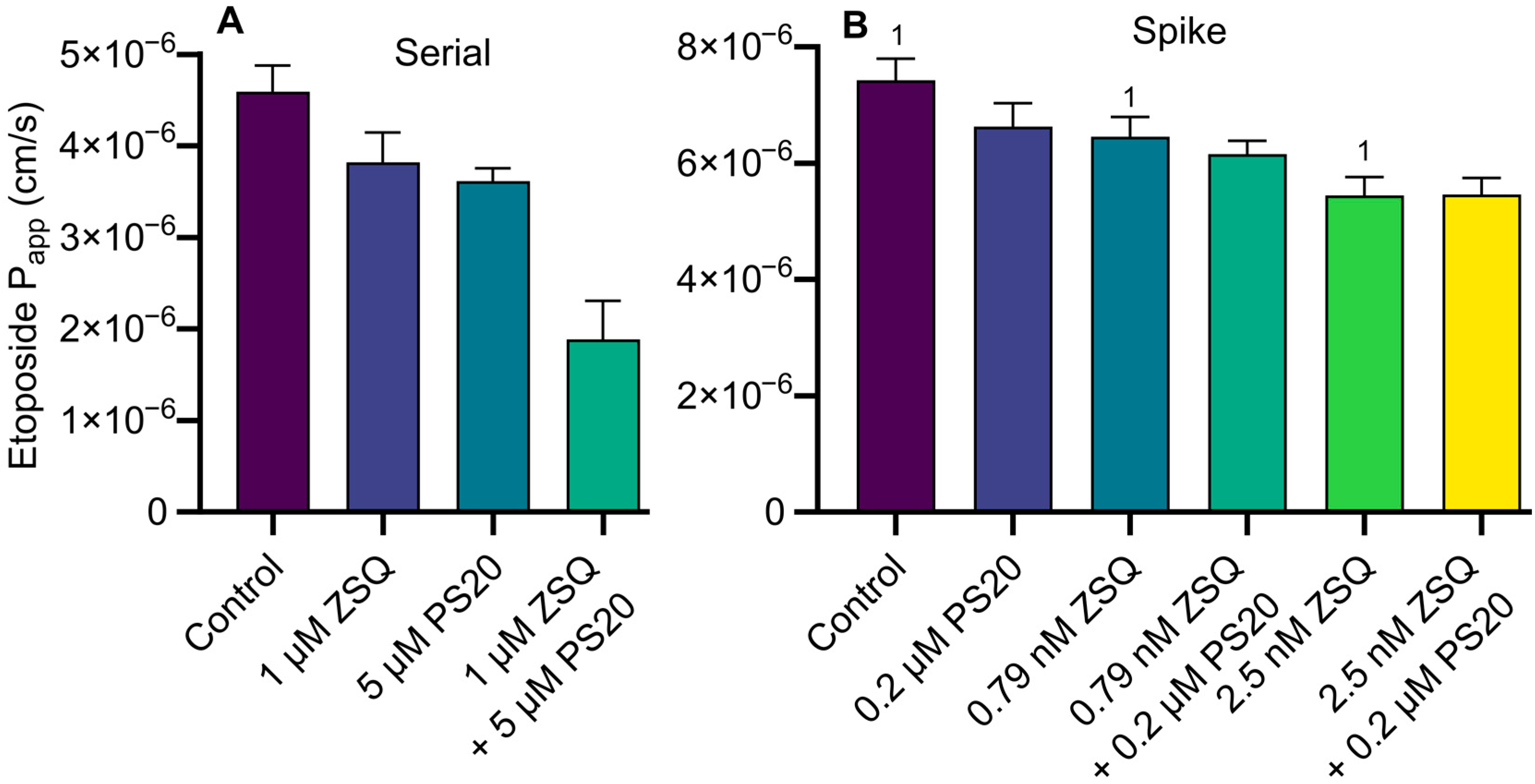
3.4. Calcein-AM Assay: Serial Dilution vs. Spike Method and Combinational Inhibition
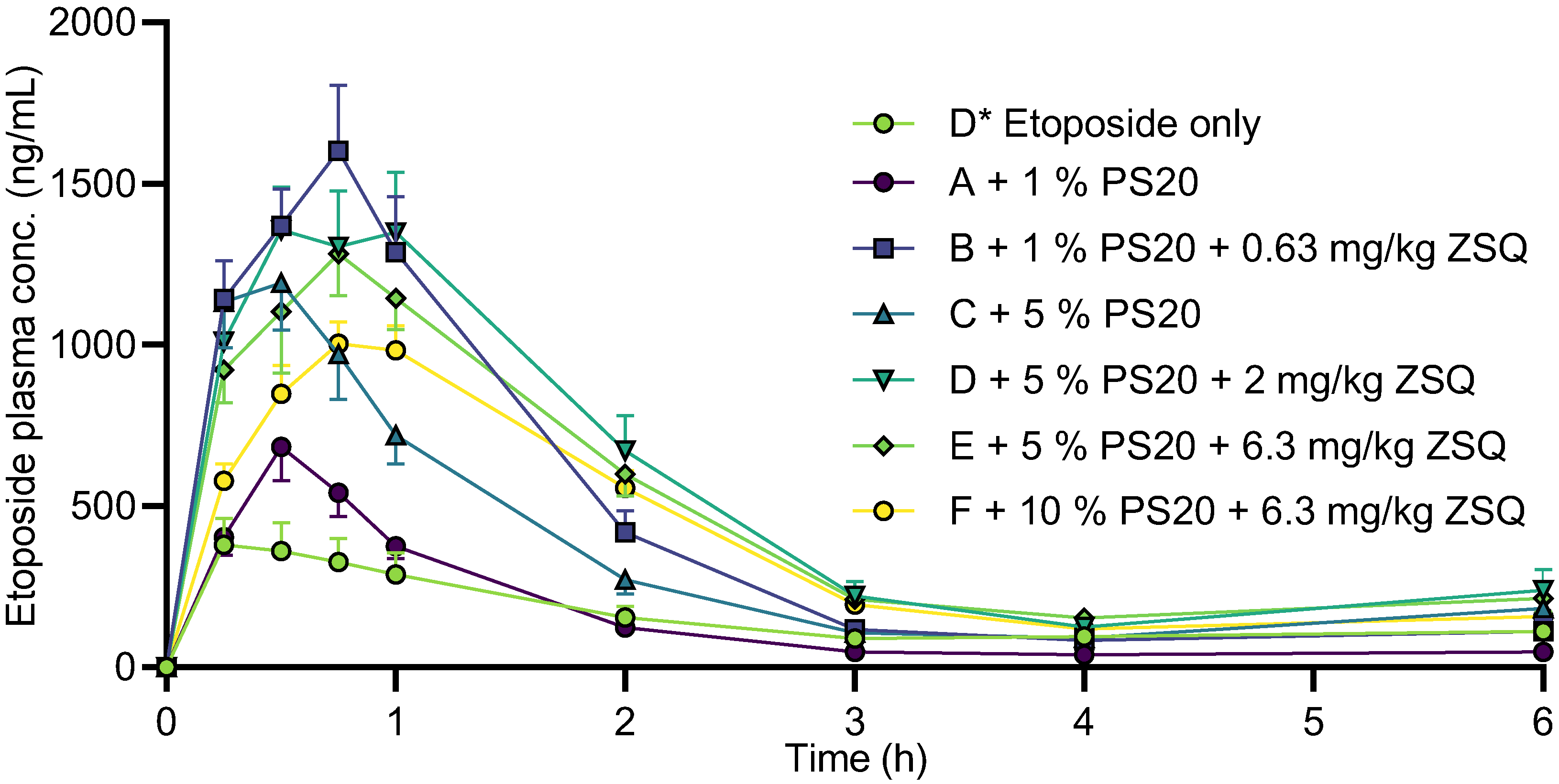
3.5. Etoposide Pharmacokinetics
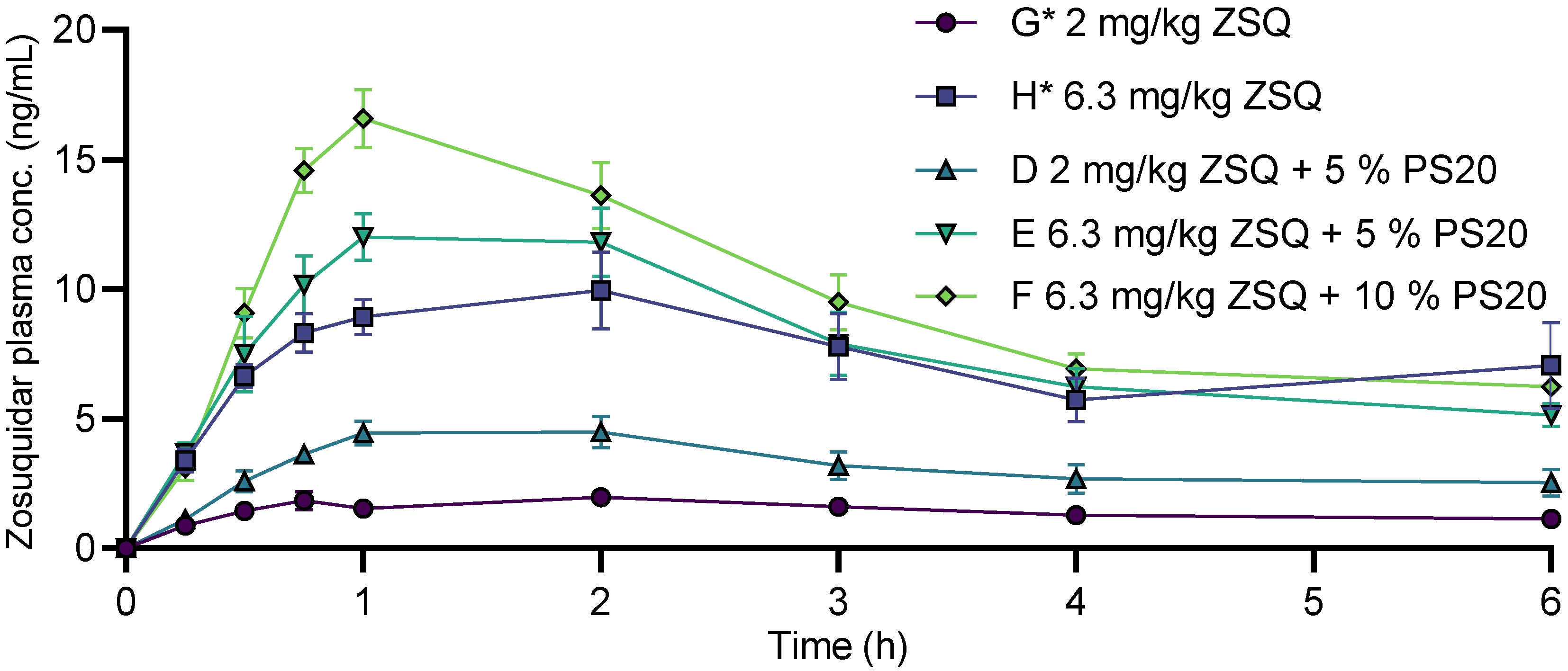
3.6. Zosuquidar Pharmacokinetics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smolinski, M.P.; Urgaonkar, S.; Pitzonka, L.; Cutler, M.; Lee, G.; Suh, K.H.; Lau, J.Y.N. Discovery of Encequidar, First-in-Class Intestine Specific P-glycoprotein Inhibitor. J. Med. Chem. 2021, 64, 3677–3693. [Google Scholar] [CrossRef]
- Jackson, C.G.C.A.; Hung, T.; Segelov, E.; Barlow, P.; Prenen, H.; McLaren, B.; Hung, N.A.; Clarke, K.; Chao, T.-Y.; Dai, M.-S.; et al. Oral paclitaxel with encequidar compared to intravenous paclitaxel in patients with advanced cancer: A randomised crossover pharmacokinetic study. Br. J. Clin. Pharmacol. 2021, 87, 4670–4680. [Google Scholar] [CrossRef]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three Decades of P-gp Inhibitors: Skimming Through Several Generations and Scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef]
- Karthika, C.; Sureshkumar, R.; Zehravi, M.; Akter, R.; Ali, F.; Ramproshad, S.; Mondal, B.; Tagde, P.; Ahmed, Z.; Khan, F.S.; et al. Multidrug Resistance of Cancer Cells and the Vital Role of P-Glycoprotein. Life 2022, 12, 897. [Google Scholar] [CrossRef]
- Nielsen, R.B.; Holm, R.; Pijpers, I.; Snoeys, J.; Nielsen, U.G.; Nielsen, C.U. Oral etoposide and zosuquidar bioavailability in rats: Effect of co-administration and in vitro-in vivo correlation of P-glycoprotein inhibition. Int. J. Pharm. X 2021, 3, 100089. [Google Scholar] [CrossRef]
- Al-Ali, A.A.A.; Nielsen, R.B.; Steffansen, B.; Holm, R.; Nielsen, C.U. Nonionic surfactants modulate the transport activity of ATP-binding cassette (ABC) transporters and solute carriers (SLC): Relevance to oral drug absorption. Int. J. Pharm. 2019, 566, 410–433. [Google Scholar] [CrossRef]
- Al-Ali, A.A.A.; Quach, J.R.C.; Bundgaard, C.; Steffansen, B.; Holm, R.; Nielsen, C.U. Polysorbate 20 alters the oral bioavailability of etoposide in wild type and mdr1a deficient Sprague-Dawley rats. Int. J. Pharm. 2018, 543, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Beig, A.; Fine-Shamir, N.; Porat, D.; Lindley, D.; Miller, J.M.; Dahan, A. Concomitant solubility-permeability increase: Vitamin E TPGS vs. amorphous solid dispersion as oral delivery systems for etoposide. Eur. J. Pharm. Biopharm. 2017, 121, 97–103. [Google Scholar] [CrossRef]
- Van Maanen, J.; Retel, J.; De Vries, J.; Pinedo, H. Mechanism of action of antitumor drug etoposide: A review. JNCI J. Natl. Cancer Inst. 1988, 80, 1526–1533. [Google Scholar] [CrossRef]
- Al-Ali, A.A.A.; Sandra, L.; Versweyveld, D.; Pijpers, I.; Dillen, L.; Vermeulen, A.; Snoeys, J.; Holm, R.; Nielsen, C.U. High-dose etoposide formulations do not saturate intestinal P-glycoprotein: Development, stability, and pharmacokinetics in Sprague-Dawley rats. Int. J. Pharm. 2020, 583, 119399. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef] [Green Version]
- Alam, A.; Kung, R.; Kowal, J.; McLeod, R.A.; Tremp, N.; Broude, E.V.; Roninson, I.B.; Stahlberg, H.; Locher, K.P. Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc. Natl. Acad. Sci. USA 2018, 115, E1973–E1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef]
- Seelig, A.; Gerebtzoff, G. Enhancement of drug absorption by noncharged detergents through membrane and P-glycoprotein binding. Expert Opin. Drug Metab. Toxicol. 2006, 2, 733–752. [Google Scholar] [CrossRef] [PubMed]
- Rege, B.D.; Kao, J.P.; Polli, J.E. Effects of nonionic surfactants on membrane transporters in Caco-2 cell monolayers. Eur. J. Pharm. Sci. 2002, 16, 237–246. [Google Scholar] [CrossRef]
- Wei, Z.; Yuan, S.; Hao, J.; Fang, X. Mechanism of inhibition of P-glycoprotein mediated efflux by Pluronic P123/F127 block copolymers: Relationship between copolymer concentration and inhibitory activity. Eur. J. Pharm. Biopharm. 2013, 83, 266–274. [Google Scholar] [CrossRef]
- Moesgaard, L.; Reinholdt, P.; Nielsen, C.U.; Kongsted, J. Mechanism behind Polysorbates’ Inhibitory Effect on P-Glycoprotein. Mol. Pharm. 2022, 19, 2248–2253. [Google Scholar] [CrossRef]
- Li, N.; Wang, D.D.; Sui, Z.G.; Qi, X.Y.; Ji, L.Y.; Wang, X.L.; Yang, L. Development of an Improved Three-Dimensional In Vitro Intestinal Mucosa Model for Drug Absorption Evaluation. Tissue Eng. Part C-Methods 2013, 19, 708–719. [Google Scholar] [CrossRef]
- Yasuda, M. Dissociation constants of some carboxylic acids in mixed aqueous solvents. Bull. Chem. Soc. Jpn. 1959, 32, 429–432. [Google Scholar] [CrossRef] [Green Version]
- Shedlovsky, T.; Kay, R.L. The ionization constant of acetic acid in water-methanol mixtures at 25-degrees from conductance measurements. J. Phys. Chem. 1956, 60, 151–155. [Google Scholar] [CrossRef]
- Al-Ali, A.A.A.; Steffansen, B.; Holm, R.; Nielsen, C.U. Nonionic surfactants increase digoxin absorption in Caco-2 and MDCKII MDR1 cells: Impact on P-glycoprotein inhibition, barrier function, and repeated cellular exposure. Int. J. Pharm. 2018, 551, 270–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drugbank. Zosuquidar. Available online: https://go.drugbank.com/drugs/DB06191 (accessed on 13 January 2023).
- Patist, A.; Bhagwat, S.S.; Penfield, K.W.; Aikens, P.; Shah, D.O. On the measurement of critical micelle concentrations of pure and technical-grade nonionic surfactants. J. Surfactants Deterg. 2000, 3, 53–58. [Google Scholar] [CrossRef]
- Rangel-Yagui, C.O.; Pessoa, A., Jr.; Tavares, L.C. Micellar solubilization of drugs. J. Pharm. Pharm. Sci. 2005, 8, 147–165. [Google Scholar]
- Mizutani, T.; Mizutani, A. Estimation of Adsorption of Drugs and Proteins on Glass Surfaces with Controlled Pore Glass as a Reference. J. Pharm. Sci. 1978, 67, 1102–1105. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.J.; Wang, L.; Antonucci, V.; Hartman, R.; Diamandopoulos, P.; Gong, X.Y. Effect of Sample Diluents on the Quantitation of Basic Compounds by High Performance Liquid Chromaography. Chromatographia 2018, 81, 1631–1639. [Google Scholar] [CrossRef]
- Ikeda, K.; Ikawa, K.; Kozumi, T.; Yokoshige, S.; Horikawa, S.; Morikawa, N. Development and validation of a GC-EI-MS method with reduced adsorption loss for the quantification of olanzapine in human plasma. Anal. Bioanal. Chem. 2012, 403, 1823–1830. [Google Scholar] [CrossRef]
- Murakoshi, M.; Fukuzawa, K.; Sato, Y.; Asakawa, N. Adsorption Phenomenon and Development of Low Adsorption Vials for LC and LC/MS. Available online: https://www.shimadzu.com/an/sites/shimadzu.com.an/files/pim/pim_document_file/others/11099/adsorption_phenomenon.pdf (accessed on 13 January 2023).
- Ludescher, C.; Eisterer, W.; Hilbe, W.; Hofmann, J.; Thaler, J. Decreased potency of MDR-modulators under serum conditions determined by a functional assay. Br. J. Haematol. 1995, 91, 652–657. [Google Scholar] [CrossRef]
- Lehnert, M.; de Giuli, R.; Kunke, K.; Emerson, S.; Dalton, W.S.; Salmon, S.E. Serum can inhibit reversal of multidrug resistance by chemosensitisers. Eur. J. Cancer 1996, 32, 862–867. [Google Scholar] [CrossRef]
- Ingels, F.M.; Augustijns, P.F. Biological, pharmaceutical, and analytical considerations with respect to the transport media used in the absorption screening system, Caco-2. J. Pharm. Sci. 2003, 92, 1545–1558. [Google Scholar] [CrossRef]
- Hait, S.K.; Moulik, S.P. Determination of critical micelle concentration (CMC) of nonionic surfactants by donor-acceptor interaction with iodine and correlation of CMC with hydrophile-lipophile balance and other parameters of the surfactants. J. Surfactants Deterg. 2001, 4, 303–309. [Google Scholar] [CrossRef]
- Mahmood, M.E.; Al-Koofee, D.A. Effect of temperature changes on critical micelle concentration for tween series surfactant. Glob. J. Sci. Front. Res. Chem. 2013, 13, 1–7. [Google Scholar]
- Mohajeri, E.; Noudeh, G.D. Effect of temperature on the critical micelle concentration and micellization thermodynamic of nonionic surfactants: Polyoxyethylene sorbitan fatty acid esters. E-J. Chem. 2012, 9, 2268–2274. [Google Scholar] [CrossRef]
- McConnell, E.L.; Basit, A.W.; Murdan, S. Measurements of rat and mouse gastrointestinal pH fluid and lymphoid tissue, and implications for in-vivo experiments. J. Pharm. Pharmacol. 2008, 60, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Bankstahl, J.P.; Bankstahl, M.; Romermann, K.; Wanek, T.; Stanek, J.; Windhorst, A.D.; Fedrowitz, M.; Erker, T.; Muller, M.; Loscher, W.; et al. Tariquidar and Elacridar Are Dose-Dependently Transported by P-Glycoprotein and Bcrp at the Blood-Brain Barrier: A Small-Animal Positron Emission Tomography and In Vitro Study. Drug Metab. Dispos. 2013, 41, 754–762. [Google Scholar] [CrossRef]










{kind=link}
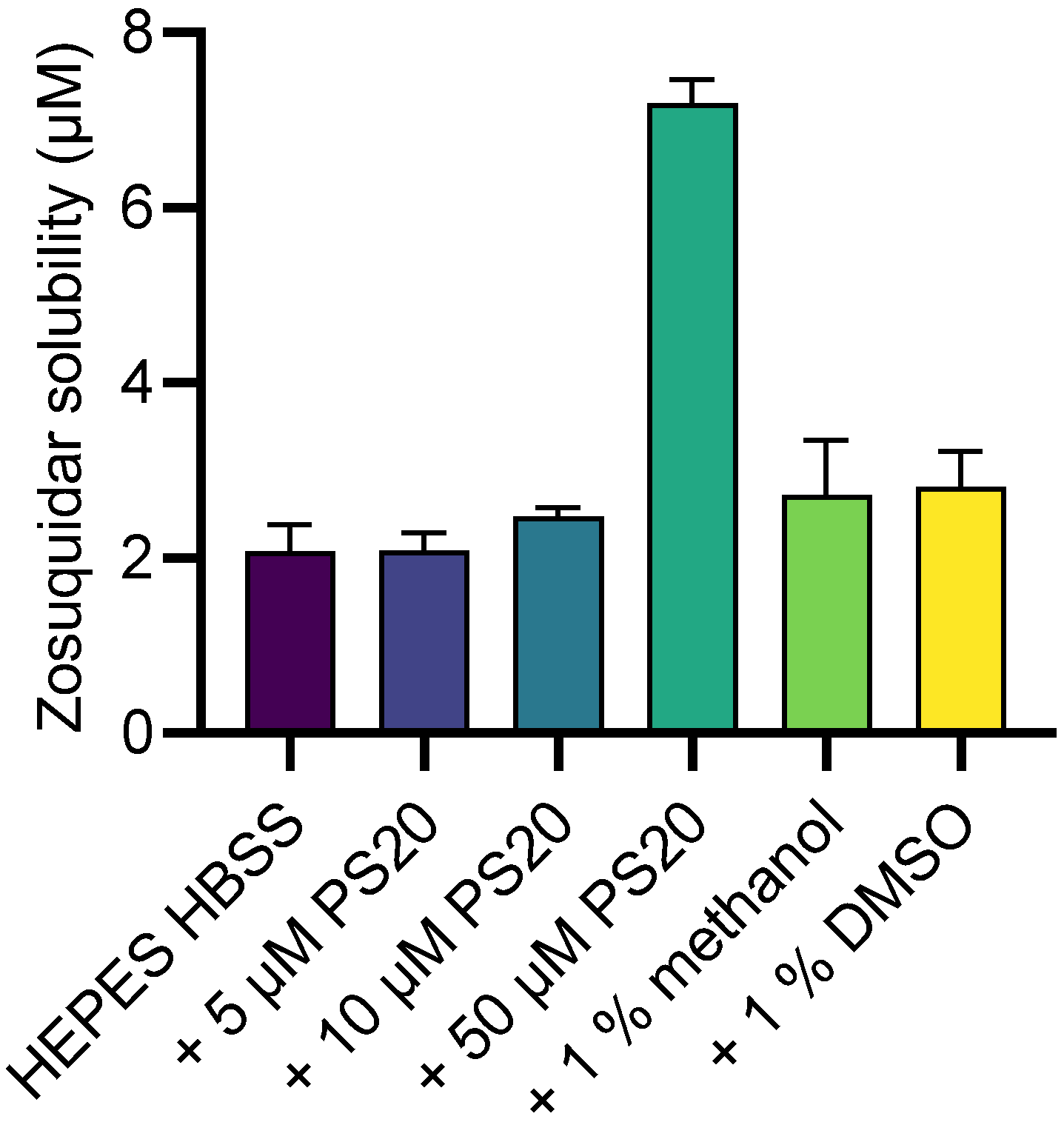{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment Series | 1 | 2 | 3 |
|---|---|---|---|
| Method | Conventional serial dilution | Conventional serial dilution | Spike method |
| Cell line | Caco-2 | MDCKII-MDR1 | Caco-2 |
| No. of cell passages | 4 | 4 | 4 |
| Direction of permeation | A–B + B–A | B–A | A–B + B–A |
| Cell layer integrity | TEER + 3H-mannitol permeability (post study) | TEER + Lucifer yellow permeability | TEER + Lucifer yellow permeability |
| [5] | Present Study | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Group Name | D* | F* | G* | H* | A | B | C | D | E | F |
| Etoposide dose (mg/kg) | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Zosuquidar dose (mg/kg) | - | 0.63 | 2.0 | 6.3 | - | 0.63 | - | 2.0 | 6.3 | 6.3 |
| Polysorbate 20 conc. (% v/v) | - | - | - | - | 1.0 | 1.0 | 5.0 | 5.0 | 5.0 | 10 |
| Etoposide pharmacokinetics | ||||||||||
| AUC0–6h (µg/mL × h) | 0.947 ± 0.156 | 2.47 ± 0.44 | 3.86 ± 0.50 | 4.27 ± 0.47 | 0.921 ± 0.084 | 2.61 ± 0.24 | 1.97 ± 0.24 | 3.08 ± 0.38 | 2.80 ± 0.20 | 2.31 ± 0.16 |
| Bioavailability (%) | 5.51 ± 0.91 | 14.4 ± 2.5 | 22.4 ± 2.9 | 24.8 ± 2.7 | 5.35 ± 0.49 | 15.2 ± 1.4 | 11.5 ± 1.4 | 17.9 ± 2.2 | 16.3 ± 1.2 | 13.4 ± 1.0 |
| C15min (µg/mL) | 0.381 ± 0.082 | 1.29 ± 0.16 | 1.62 ± 0.26 | 1.69 ± 0.18 | 0.404 ± 0.058 | 1.14 ± 0.12 | 1.13 ± 0.14 | 1.01 ± 0.10 | 0.923 ± 0.103 | 0.579 ± 0.052 |
| Cmax (µg/mL) | 0.390 ± 0.084 | 1.59 ± 0.29 | 2.24 ± 0.54 | 2.11 ± 0.20 | 0.711 ± 0.086 | 1.75 ± 0.16 | 1.22 ± 0.14 | 1.43 ± 0.18 | 1.30 ± 0.13 | 1.02 ± 0.07 |
| tmax (min) | 15 [15;30] | 45 [15;45] | 38 [26;60] | 45 [15;60] | 30 [30;34] | 45 [30;49] | 30 [26;30] | 38 [30;60] | 45 [41;49] | 45 [41;60] |
| t½ (min) | 80.8 ± 13.2 | 39.3 ± 3.0 | 51.4 ± 2.4 | 56.5 ± 4.8 | 42.4 ± 2.4 | 37.2 ± 1.2 | 43.3 ± 2.0 | 52.9 ± 2.8 | 54.7 ± 4.5 | 56.4 ± 2.2 |
| Zosuquidar pharmacokinetics | ||||||||||
| AUC0–6h (ng/mL × h) | BLQ | 8.67 ± 0.77 | 43.6 ± 5.4 | BLQ | 18.9 ± 2.8 | 47.0 ± 3.7 | 56.8 ± 4.2 | |||
| Bioavailability (%) | 2.58 ± 0.23 | 4.12 ± 0.51 | 5.64 ± 0.83 | 4.45 ± 0.35 | 5.38 ± 0.40 | |||||
| Cmax (ng/mL) | 2.10 ± 0.27 | 10.9 ± 1.2 | 4.8 ± 0.5 | 13.2 ± 1.0 | 16.6 ± 1.1 | |||||
| tmax (min) | 120 [45;135] | 90 [45;120] | 120 [56;120] | 60 [56;120] | 60 [56;75] | |||||
| t½ (min) | 217 ± 27 | 161 ± 13 | 180 ± 23 | 152 ± 20 | 147 ± 10 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nielsen, R.B.; Holm, R.; Pijpers, I.; Snoeys, J.; Nielsen, U.G.; Nielsen, C.U. Combinational Inhibition of P-Glycoprotein-Mediated Etoposide Transport by Zosuquidar and Polysorbate 20. Pharmaceutics 2023, 15, 283. https://doi.org/10.3390/pharmaceutics15010283
Nielsen RB, Holm R, Pijpers I, Snoeys J, Nielsen UG, Nielsen CU. Combinational Inhibition of P-Glycoprotein-Mediated Etoposide Transport by Zosuquidar and Polysorbate 20. Pharmaceutics. 2023; 15(1):283. https://doi.org/10.3390/pharmaceutics15010283
Chicago/Turabian StyleNielsen, Rasmus Blaaholm, René Holm, Ils Pijpers, Jan Snoeys, Ulla Gro Nielsen, and Carsten Uhd Nielsen. 2023. "Combinational Inhibition of P-Glycoprotein-Mediated Etoposide Transport by Zosuquidar and Polysorbate 20" Pharmaceutics 15, no. 1: 283. https://doi.org/10.3390/pharmaceutics15010283