
The More the Better—Investigation of Polymethoxylated N-Carboranyl Quinazolines as Novel Hybrid Breast Cancer Resistance Protein Inhibitors
 , , , , ,
, , , , ,
Abstract
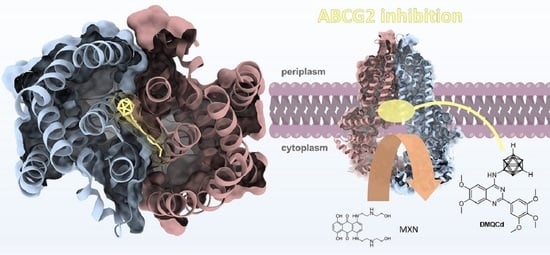
:
1. Introduction

2. Materials and Methods
2.1. Synthetic Procedures and Analytical Data
2.2. Biological Methods
2.2.1. Cultivation of MDCKII Cells
2.2.2. Determination of Cell Viability by WST-1 Cell Proliferation Assay
2.2.3. Determination of ABCG2 Interaction with Hoechst 33342 Accumulation Assay
2.2.4. Determination of Autofluorescence
2.2.5. Reversal of Multidrug Resistance
2.3. Molecular Docking/Computational Methods
3. Results and Discussion
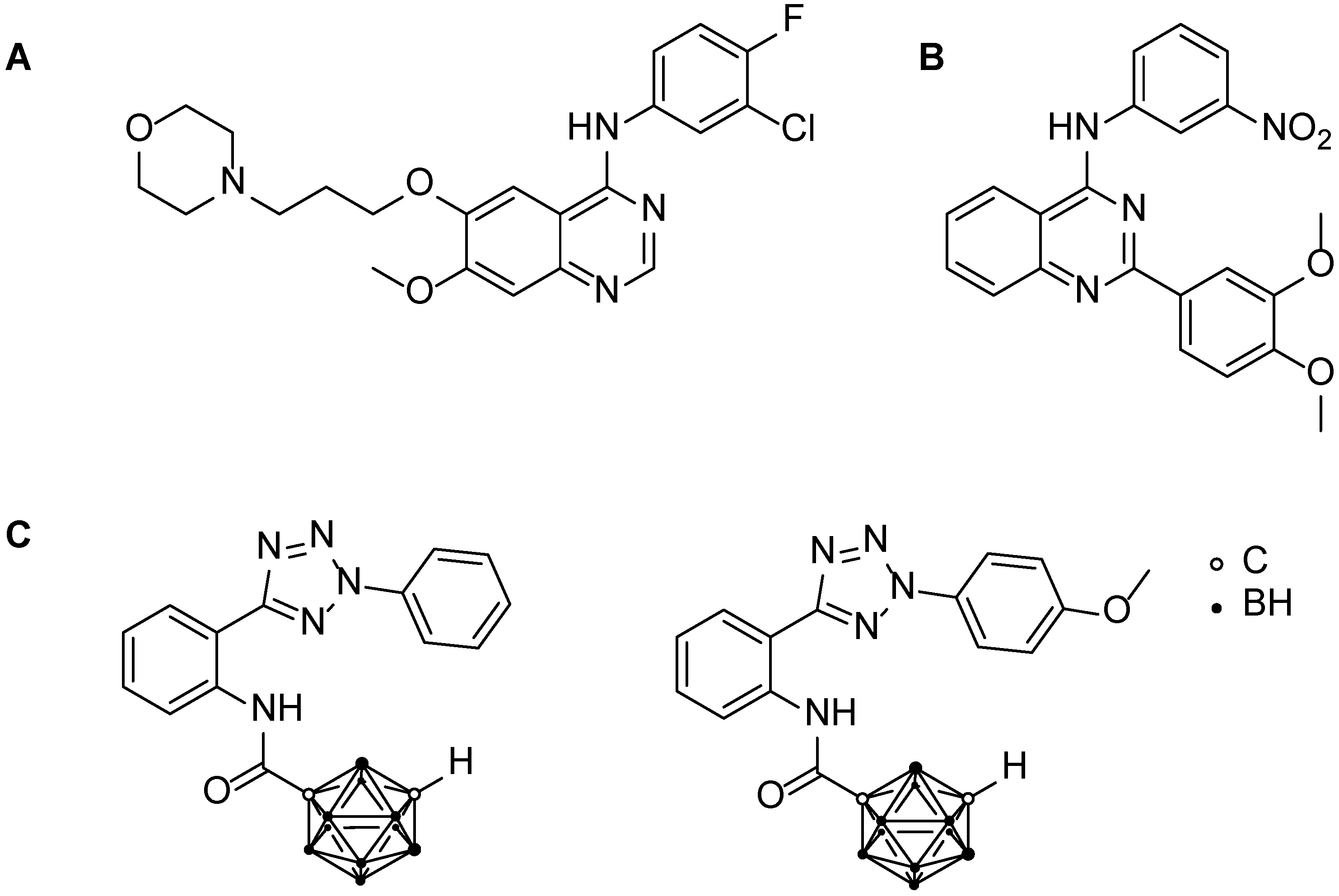
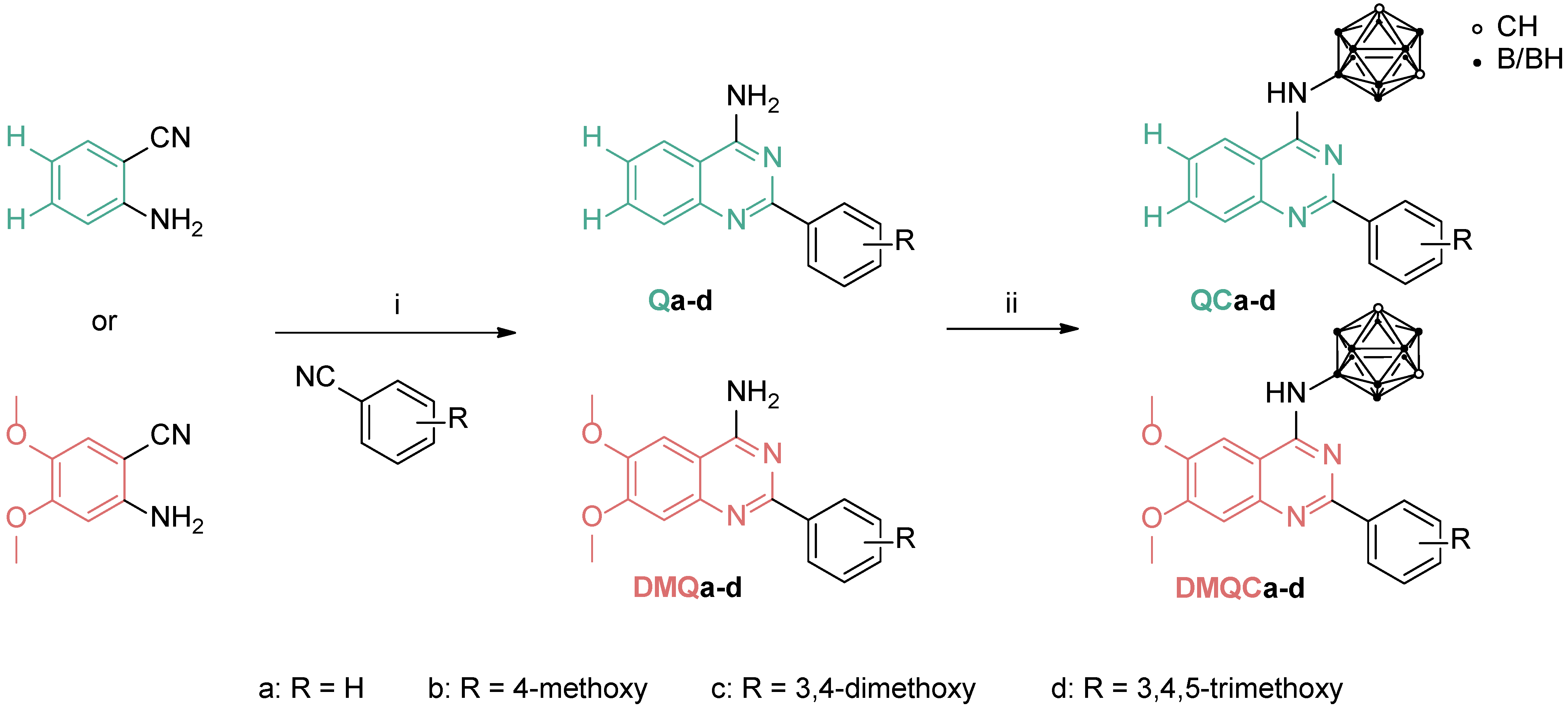
3.1. Chemistry
3.2. Biological Investigations
3.2.1. Determination of Cytotoxicity in MDCKII-hABCG2 and MDCKII Wild-Type Cells
3.2.2. Hoechst 33342 Assay-Based Evaluation of the Inhibitory Potential toward MDCKII-hABCG2 Cells
3.2.3. Determination of Autofluorescence
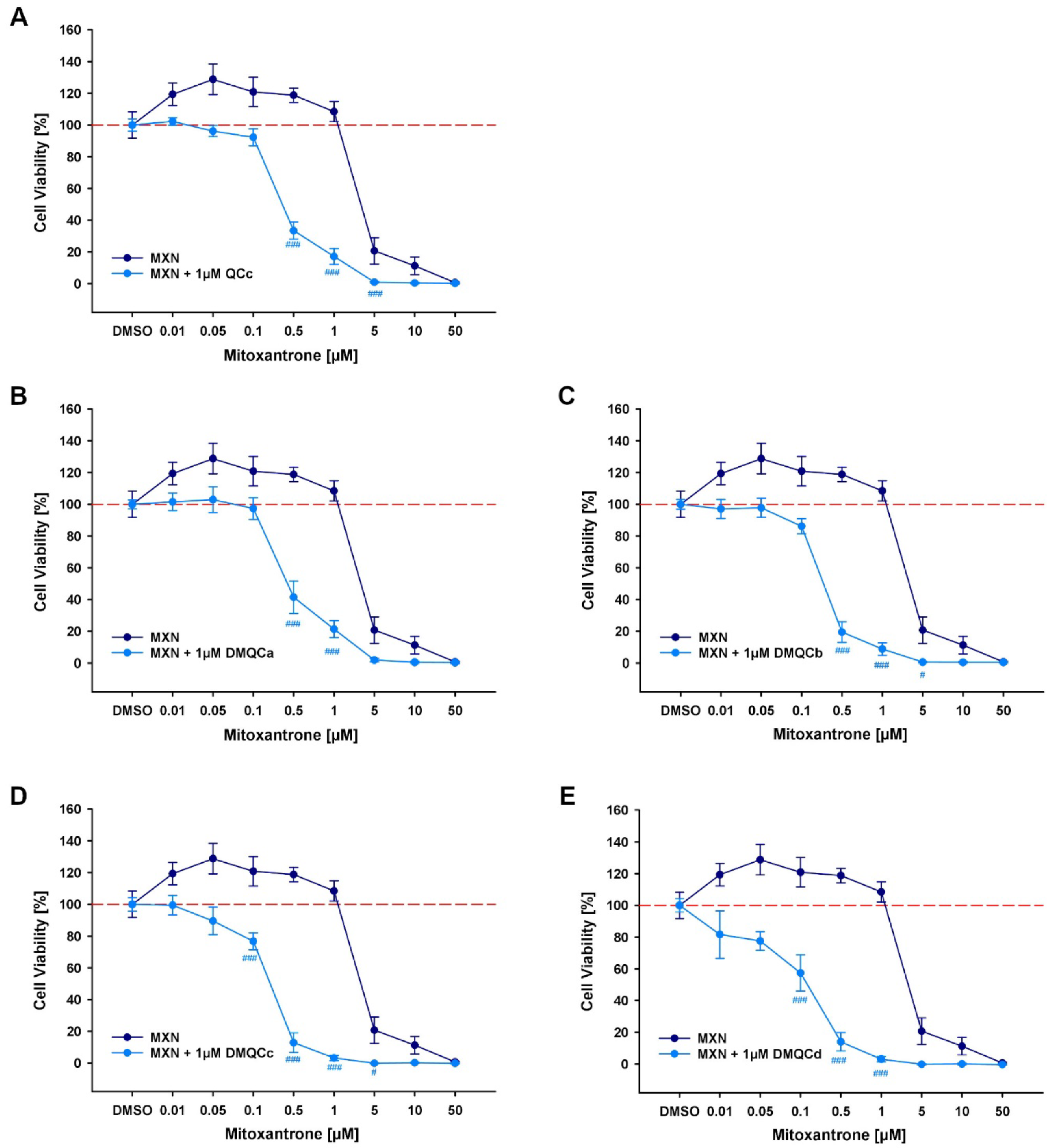
3.2.4. Reversal of ABCG2-Mediated MDR
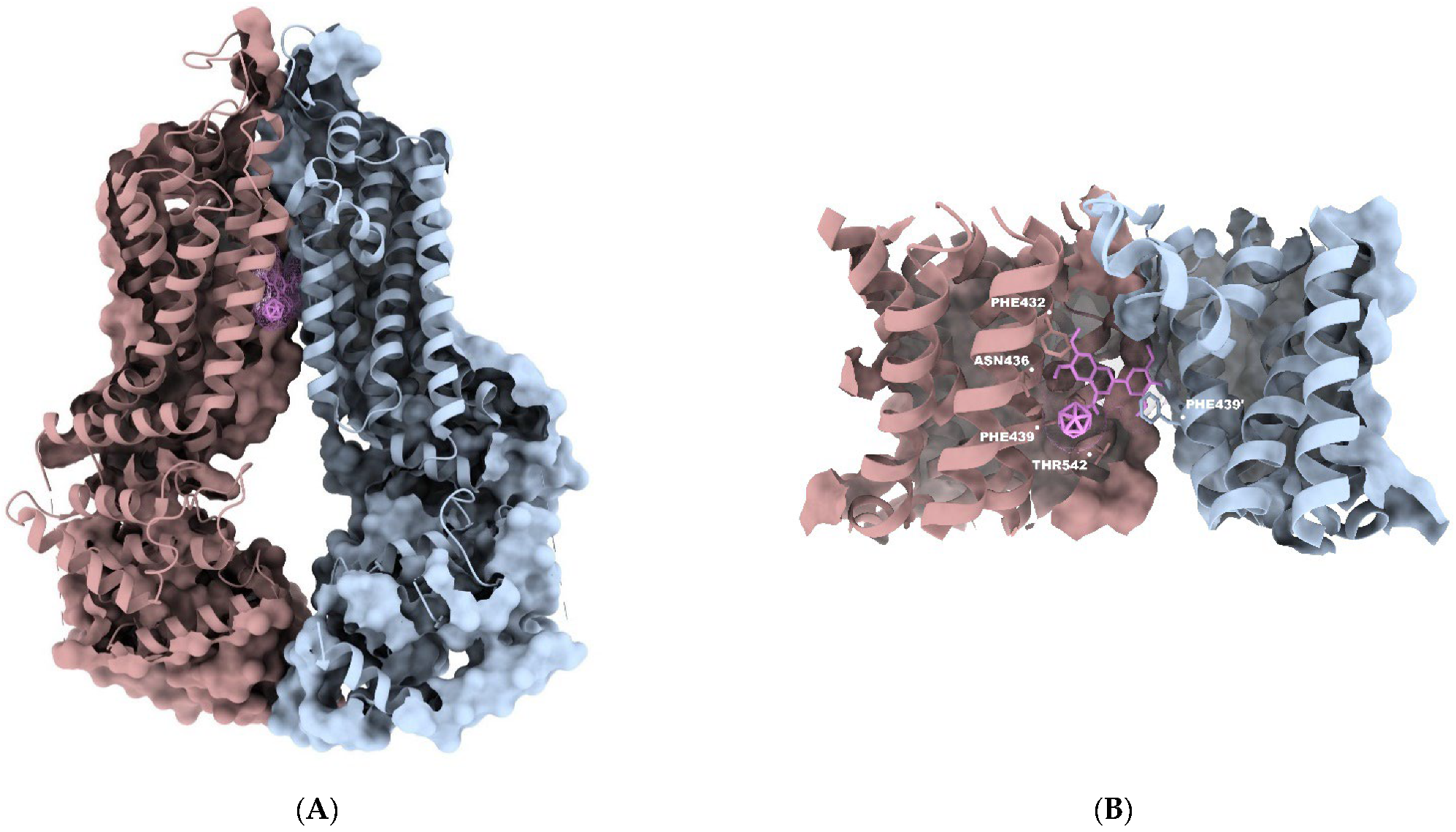
3.3. In Silico Investigations—Molecular Docking Simulations and Investigation of Mode of Inhibition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
Appendix A
- A.
- General procedure for preparation of quinazolin-4-amines
- B.
- General procedure for preparation of N-carboranyl 2-phenylquinazolin-4-amines
References
- Natarajan, K.; Xie, Y.; Baer, M.R.; Ross, D.D. Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem. Pharmacol. 2012, 83, 1084–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukal, S.; Guin, D.; Rawat, C.; Bora, S.; Mishra, M.K.; Sharma, P.; Paul, P.R.; Kanojia, N.; Grewal, G.K.; Kukreti, S.; et al. Multidrug efflux transporter ABCG2: Expression and regulation. Cell. Mol. Life Sci. 2021, 78, 6887–6939. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Zheng, Y.; Ma, L.; Tian, L.; Sun, Q. Clinically-Relevant ABC Transporter for Anti-Cancer Drug Resistance. Front. Pharmacol. 2021, 12, 648407. [Google Scholar] [CrossRef] [PubMed]
- Stacy, A.E.; Jansson, P.J.; Des Richardson, R. Molecular pharmacology of ABCG2 and its role in chemoresistance. Mol. Pharmacol. 2013, 84, 655–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Li, H.; Li, J.; Zhu, Z.; Yin, S.; Hao, X.; Yao, M.; Zheng, S.; Gu, J. Analysis of ABCG2 expression and side population identifies intrinsic drug efflux in the HCC cell line MHCC-97L and its modulation by Akt signaling. Carcinogenesis 2008, 29, 2289–2297. [Google Scholar] [CrossRef] [Green Version]
- Kurimchak, A.M.; Herrera-Montávez, C.; Montserrat-Sangrà, S.; Araiza-Olivera, D.; Hu, J.; Neumann-Domer, R.; Kuruvilla, M.; Bellacosa, A.; Testa, J.R.; Jin, J.; et al. The drug efflux pump MDR1 promotes intrinsic and acquired resistance to PROTACs in cancer cells. Sci. Signal. 2022, 15, eabn2707. [Google Scholar] [CrossRef]
- Theile, D.; Wizgall, P. Acquired ABC-transporter overexpression in cancer cells: Transcriptional induction or Darwinian selection? Naunyn Schmiedebergs. Arch. Pharmacol. 2021, 394, 1621–1632. [Google Scholar] [CrossRef]
- Xia, C.Q.; Smith, P.G. Drug efflux transporters and multidrug resistance in acute leukemia: Therapeutic impact and novel approaches to mediation. Mol. Pharmacol. 2012, 82, 1008–1021. [Google Scholar] [CrossRef] [Green Version]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Wilkens, S. Structure and mechanism of ABC transporters. F1000Prime Rep. 2015, 7, 14. [Google Scholar] [CrossRef]
- George, A.M. ABC Transporters—40 Years on; Springer: Cham, Switzerland, 2016; ISBN 978-3-319-23476-2. [Google Scholar]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Matsuo, K.; Eno, M.L.; Ahn, E.H.; Shahzad, M.M.K.; Im, D.D.; Rosenshein, N.B.; Sood, A.K. Multidrug resistance gene (MDR-1) and risk of brain metastasis in epithelial ovarian, fallopian tube, and peritoneal cancer. Am. J. Clin. Oncol. 2011, 34, 488–493. [Google Scholar] [CrossRef]
- Damiani, D.; Tiribelli, M.; Calistri, E.; Geromin, A.; Chiarvesio, A.; Michelutti, A.; Cavallin, M.; Fanin, R. The prognostic value of P-glycoprotein (ABCB) and breast cancer resistance protein (ABCG2) in adults with de novo acute myeloid leukemia with normal karyotype. Haematologica 2006, 91, 825–828. [Google Scholar]
- Sun, S.; Cai, J.; Yang, Q.; Zhu, Y.; Zhao, S.; Wang, Z. Prognostic Value and Implication for Chemotherapy Treatment of ABCB1 in Epithelial Ovarian Cancer: A Meta-Analysis. PLoS ONE 2016, 11, e0166058. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, Y.; Takada, T.; Suzuki, H. Inhibitors of Human ABCG2: From Technical Background to Recent Updates with Clinical Implications. Front. Pharmacol. 2019, 10, 208. [Google Scholar] [CrossRef] [Green Version]
- Moinul, M.; Amin, S.A.; Jha, T.; Gayen, S. Updated chemical scaffolds of ABCG2 inhibitors and their structure-inhibition relationships for future development. Eur. J. Med. Chem. 2022, 241, 114628. [Google Scholar] [CrossRef]
- Zattoni, I.F.; Delabio, L.C.; de Paula Dutra, J.; Kita, D.H.; Scheiffer, G.; Hembecker, M.; Da Pereira, G.S.; Moure, V.R.; Valdameri, G. Targeting breast cancer resistance protein (BCRP/ABCG2): Functional inhibitors and expression modulators. Eur. J. Med. Chem. 2022, 237, 114346. [Google Scholar] [CrossRef]
- Silbermann, K.; Li, J.; Namasivayam, V.; Baltes, F.; Bendas, G.; Stefan, S.M.; Wiese, M. Superior Pyrimidine Derivatives as Selective ABCG2 Inhibitors and Broad-Spectrum ABCB1, ABCC1, and ABCG2 Antagonists. J. Med. Chem. 2020, 63, 10412–10432. [Google Scholar] [CrossRef]
- Stefan, S.M. Multi-target ABC transporter modulators: What next and where to go? Future Med. Chem. 2019, 11, 2353–2358. [Google Scholar] [CrossRef]
- Galetti, M.; Petronini, P.G.; Fumarola, C.; Cretella, D.; La Monica, S.; Bonelli, M.; Cavazzoni, A.; Saccani, F.; Caffarra, C.; Andreoli, R.; et al. Effect of ABCG2/BCRP Expression on Efflux and Uptake of Gefitinib in NSCLC Cell Lines. PLoS ONE 2015, 10, e0141795. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhang, X.; Wang, F.; Wang, X.; Yang, K.; Xu, M.; To, K.K.W.; Li, Q.; Fu, L. Effect of ceritinib (LDK378) on enhancement of chemotherapeutic agents in ABCB1 and ABCG2 overexpressing cells in vitro and in vivo. Oncotarget 2015, 6, 44643–44659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juvale, K.; Gallus, J.; Wiese, M. Investigation of quinazolines as inhibitors of breast cancer resistance protein (ABCG2). Bioorg. Med. Chem. 2013, 21, 7858–7873. [Google Scholar] [CrossRef] [PubMed]
- Kraege, S.; Stefan, K.; Juvale, K.; Ross, T.; Willmes, T.; Wiese, M. The combination of quinazoline and chalcone moieties leads to novel potent heterodimeric modulators of breast cancer resistance protein (BCRP/ABCG2). Eur. J. Med. Chem. 2016, 117, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Krapf, M.K.; Gallus, J.; Namasivayam, V.; Wiese, M. 2,4,6-Substituted Quinazolines with Extraordinary Inhibitory Potency toward ABCG2. J. Med. Chem. 2018, 61, 7952–7976. [Google Scholar] [CrossRef]
- Krapf, M.K.; Gallus, J.; Wiese, M. 4-Anilino-2-pyridylquinazolines and -pyrimidines as Highly Potent and Nontoxic Inhibitors of Breast Cancer Resistance Protein (ABCG2). J. Med. Chem. 2017, 60, 4474–4495. [Google Scholar] [CrossRef]
- Krapf, M.K.; Gallus, J.; Wiese, M. Synthesis and biological investigation of 2,4-substituted quinazolines as highly potent inhibitors of breast cancer resistance protein (ABCG2). Eur. J. Med. Chem. 2017, 139, 587–611. [Google Scholar] [CrossRef]
- Krapf, M.K.; Wiese, M. Synthesis and Biological Evaluation of 4-Anilino-quinazolines and -quinolines as Inhibitors of Breast Cancer Resistance Protein (ABCG2). J. Med. Chem. 2016, 59, 5449–5461. [Google Scholar] [CrossRef]
- Stockmann, P.; Gozzi, M.; Kuhnert, R.; Sárosi, M.B.; Hey-Hawkins, E. New keys for old locks: Carborane-containing drugs as platforms for mechanism-based therapies. Chem. Soc. Rev. 2019, 48, 3497–3512. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Du, F.; Tang, L.; Xu, J.; Zhao, Y.; Wu, X.; Li, M.; Shen, J.; Wen, Q.; Cho, C.H.; et al. Carboranes as unique pharmacophores in antitumor medicinal chemistry. Mol. Ther. Oncolytics 2022, 24, 400–416. [Google Scholar] [CrossRef]
- Marfavi, A.; Kavianpour, P.; Rendina, L.M. Carboranes in drug discovery, chemical biology and molecular imaging. Nat. Rev. Chem. 2022, 6, 486–504. [Google Scholar] [CrossRef]
- Köhler, S.C.; Vahdati, S.; Scholz, M.S.; Wiese, M. Structure activity relationships, multidrug resistance reversal and selectivity of heteroarylphenyl ABCG2 inhibitors. Eur. J. Med. Chem. 2018, 146, 483–500. [Google Scholar] [CrossRef]
- Bruno, N.C.; Niljianskul, N.; Buchwald, S.L. N-substituted 2-aminobiphenylpalladium methanesulfonate precatalysts and their use in C-C and C-N cross-couplings. J. Org. Chem. 2014, 79, 4161–4166. [Google Scholar] [CrossRef] [Green Version]
- Dziedzic, R.M.; Saleh, L.M.A.; Axtell, J.C.; Martin, J.L.; Stevens, S.L.; Royappa, A.T.; Rheingold, A.L.; Spokoyny, A.M. B-N, B-O, and B-CN Bond Formation via Palladium-Catalyzed Cross-Coupling of B-Bromo-Carboranes. J. Am. Chem. Soc. 2016, 138, 9081–9084. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Granger, P. NMR Nomenclature: Nuclear Spin Properties and Conventions for Chemical Shifts. IUPAC Recommendations 2001. Solid State Nucl. Magn. Reson. 2002, 22, 458–483. [Google Scholar] [CrossRef] [Green Version]
- Kuhnert, L.; Giantin, M.; Dacasto, M.; Halwachs, S.; Honscha, W. AhR-activating pesticides increase the bovine ABCG2 efflux activity in MDCKII-bABCG2 cells. PLoS ONE 2020, 15, e0237163. [Google Scholar] [CrossRef]
- Wassermann, L.; Halwachs, S.; Lindner, S.; Honscha, K.U.; Honscha, W. Determination of functional ABCG2 activity and assessment of drug-ABCG2 interactions in dairy animals using a novel MDCKII in vitro model. J. Pharm. Sci. 2013, 102, 772–784. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, 73–78. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Taylor, N.; Manolaridis, I.; Jackson, S.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Structure of an ABC transporter: Complete Structure. 2017. Available online: https://www.wwpdb.org/pdb?id=pdb_00005nj3 (accessed on 1 December 2022).
- Orlando, B.J.; Liao, M. Structure of ABCG2 Bound to Mitoxantrone. 2020. Available online: https://www.wwpdb.org/pdb?id=pdb_00006vxi (accessed on 1 December 2022).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhnert, R.; Sárosi, M.-B.; George, S.; Lönnecke, P.; Hofmann, B.; Steinhilber, D.; Murganic, B.; Mijatovic, S.; Maksimovic-Ivanic, D.; Hey-Hawkins, E. CarbORev-5901: The First Carborane-Based Inhibitor of the 5-Lipoxygenase Pathway. ChemMedChem 2017, 12, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Van Muijlwijk-Koezen, J.E.; Timmerman, H.; van der Goot, H.; Menge, W.M.; Frijtag Von Drabbe Künzel, J.; de Groote, M.; IJzerman, A.P. Isoquinoline and quinazoline urea analogues as antagonists for the human adenosine A(3) receptor. J. Med. Chem. 2000, 43, 2227–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modi, A.; Roy, D.; Sharma, S.; Vishnoi, J.R.; Pareek, P.; Elhence, P.; Sharma, P.; Purohit, P. ABC transporters in breast cancer: Their roles in multidrug resistance and beyond. J. Drug Target. 2022, 30, 927–947. [Google Scholar] [CrossRef]
- Mo, W.; Zhang, J.-T. Human ABCG2: Structure, function, and its role in multidrug resistance. Int. J. Biochem. Mol. Biol. 2012, 3, 1–27. [Google Scholar]
- Yu, Q.; Ni, D.; Kowal, J.; Manolaridis, I.; Jackson, S.M.; Stahlberg, H.; Locher, K.P. Structures of ABCG2 under turnover conditions reveal a key step in the drug transport mechanism. Nat. Commun. 2021, 12, 4376. [Google Scholar] [CrossRef]
- Orlando, B.J.; Liao, M. ABCG2 transports anticancer drugs via a closed-to-open switch. Nat. Commun. 2020, 11, 2264. [Google Scholar] [CrossRef]
- Gose, T.; Shafi, T.; Fukuda, Y.; Das, S.; Wang, Y.; Allcock, A.; Gavan McHarg, A.; Lynch, J.; Chen, T.; Tamai, I.; et al. ABCG2 requires a single aromatic amino acid to “clamp” substrates and inhibitors into the binding pocket. FASEB J. 2020, 34, 4890–4903. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Altmann, K.H.; Locher, K.P. Structure of Inhibitor-Bound ABCG2. 2018. Available online: https://www.wwpdb.org/pdb?id=pdb_00006feq (accessed on 1 December 2022).
- Kowal, J.; Ni, D.; Jackson, S.M.; Manolaridis, I.; Stahlberg, H.; Locher, K.P. Structural Basis of Drug Recognition by the Multidrug Transporter ABCG2. J. Mol. Biol. 2021, 433, 166980. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | IC50 ± SEM [µM] MDCKII-WT | IC50 ± SEM [µM] MDCKII-hABCG2 |
|---|---|---|---|
| QCa | H | 43.230 ± 0.159 | >10 |
| QCb | 4-methoxy | 44.167 ± 0.174 | >10 |
| QCc | 3,4-dimethoxy | >50 | >50 |
| QCd | 3,4,5-trimethoxy | >50 | >50 |
| DMQCa | H | >50 | >50 |
| DMQCb | 4-methoxy | >25 * | >25 * |
| DMQCc | 3,4-dimethoxy | >50 | >50 |
| DMQCd | 3,4,5-trimethoxy | >50 | >50 |
| Treatment of MDCKII-hABCG2 | IC50 [µM] | Left-Shift Factor | Comparison to MXN * |
|---|---|---|---|
| MXN | 2.649 ± 0.594 | ||
| MXN + 1 µM QCc | 0.346 ± 0.033 | 5.4-fold | *** |
| MXN + 1 µM DMQCa | 0.426 ± 0.057 | 4.3-fold | *** |
| MXN + 1 µM DMQCb | 0.254 ± 0.031 | 7.2-fold | *** |
| MXN + 1 µM DMQCc | 0.184 ± 0.025 | 9.8-fold | *** |
| MXN + 1 µM DMQCd | 0.144 ± 0.033 | 11.6-fold | *** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stockmann, P.; Kuhnert, L.; Leinung, W.; Lakoma, C.; Scholz, B.; Paskas, S.; Mijatović, S.; Maksimović-Ivanić, D.; Honscha, W.; Hey-Hawkins, E. The More the Better—Investigation of Polymethoxylated N-Carboranyl Quinazolines as Novel Hybrid Breast Cancer Resistance Protein Inhibitors. Pharmaceutics 2023, 15, 241. https://doi.org/10.3390/pharmaceutics15010241
Stockmann P, Kuhnert L, Leinung W, Lakoma C, Scholz B, Paskas S, Mijatović S, Maksimović-Ivanić D, Honscha W, Hey-Hawkins E. The More the Better—Investigation of Polymethoxylated N-Carboranyl Quinazolines as Novel Hybrid Breast Cancer Resistance Protein Inhibitors. Pharmaceutics. 2023; 15(1):241. https://doi.org/10.3390/pharmaceutics15010241
Chicago/Turabian StyleStockmann, Philipp, Lydia Kuhnert, Wencke Leinung, Cathleen Lakoma, Birte Scholz, Svetlana Paskas, Sanja Mijatović, Danijela Maksimović-Ivanić, Walther Honscha, and Evamarie Hey-Hawkins. 2023. "The More the Better—Investigation of Polymethoxylated N-Carboranyl Quinazolines as Novel Hybrid Breast Cancer Resistance Protein Inhibitors" Pharmaceutics 15, no. 1: 241. https://doi.org/10.3390/pharmaceutics15010241