
Recent Advances in Mesoporous Silica Nanoparticle-Mediated Drug Delivery for Breast Cancer Treatment

Abstract
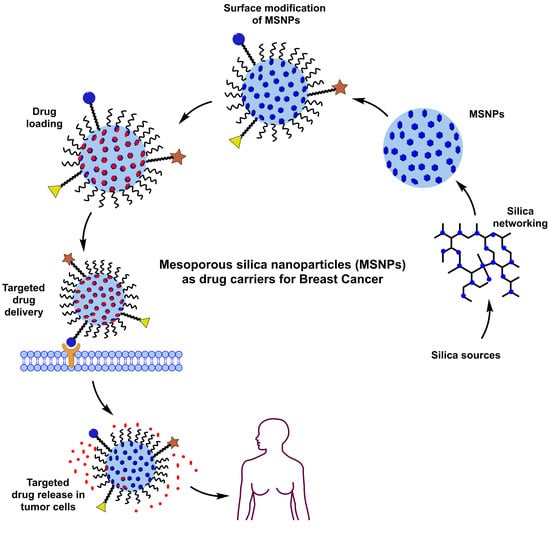
:
1. Introduction
2. Exclusive Terminologies and Synthesis Methods of Mesoporous Silica Nanoparticles
2.1. Synthesis Methods
2.1.1. Sol–Gel Method



2.1.2. Hydrothermal Method
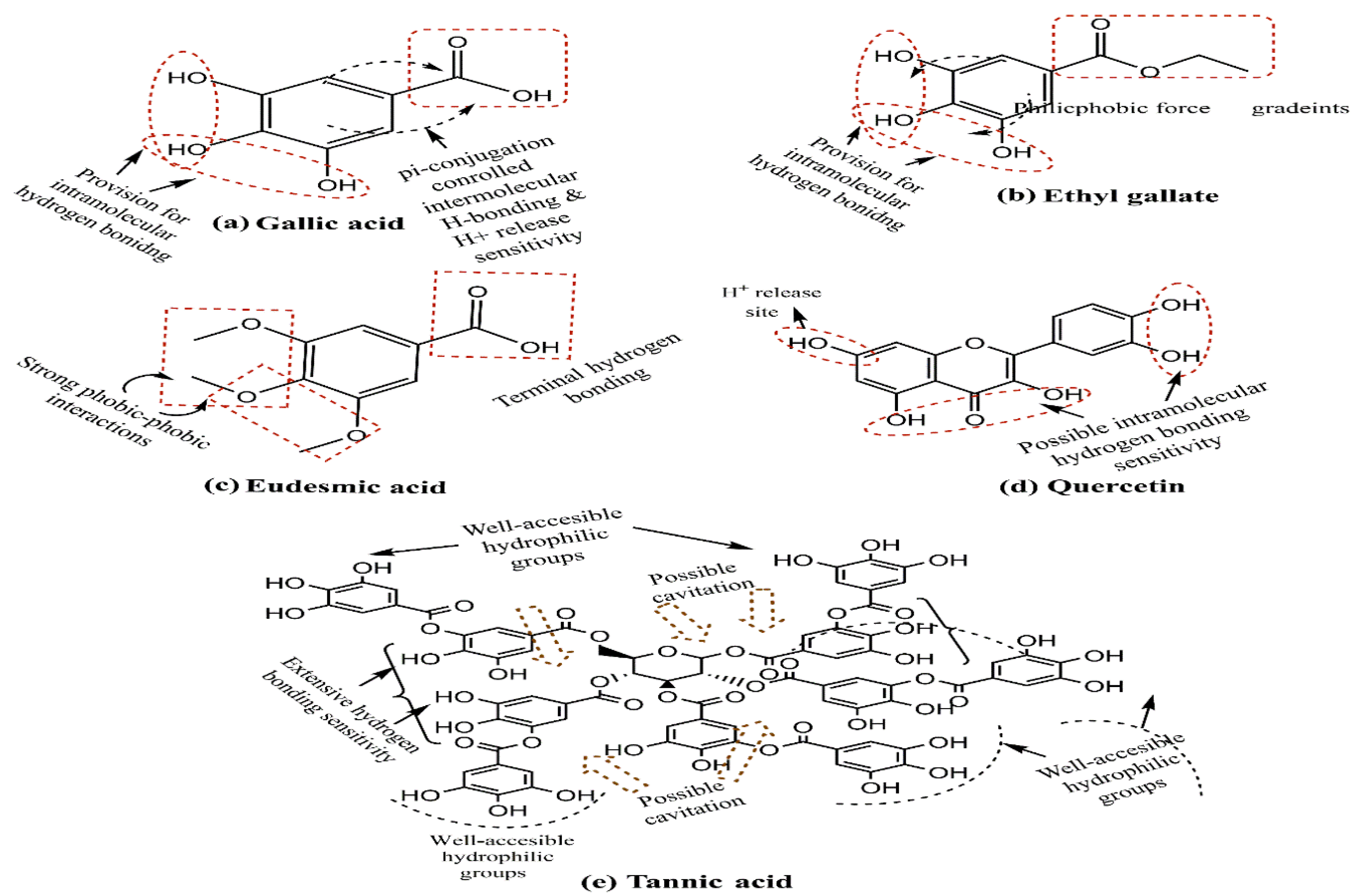
2.1.3. Green Synthesis
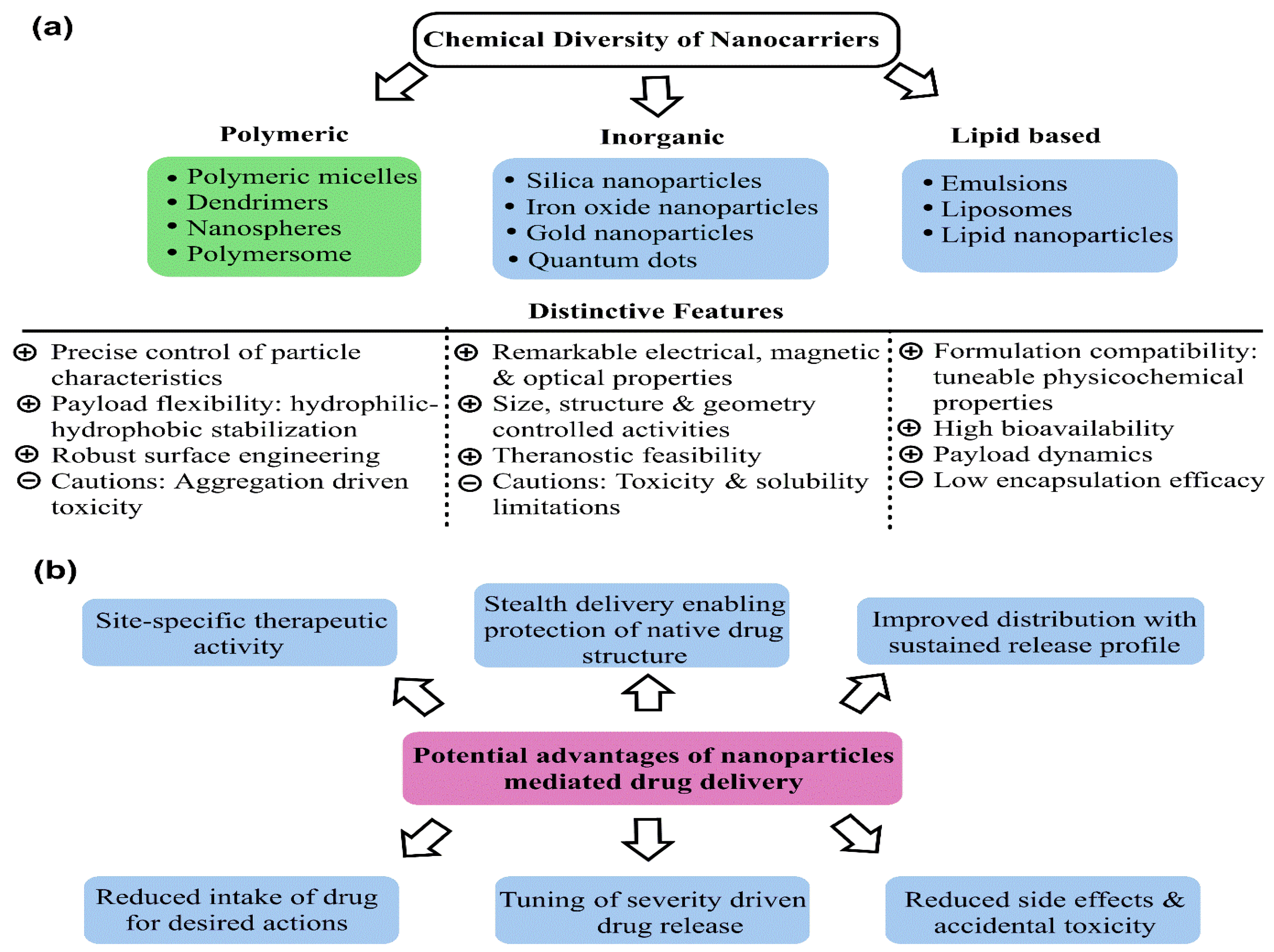
3. Promising Aspects of Mesoporous Silica Nanoparticles as Drug Vehicles
3.1. Surface Chemistry
3.2. Effect of Varying Sizes
3.3. Morphology or Particle Shape
3.4. Pore Diameter
3.5. The Gatekeeping Concept
3.6. The Flexibility of Drug Loading
3.7. Drug Release from MSNPs
3.8. Improved Drug Pharmacokinetics with Mesoporous Silica Nanoparticles
4. Drug Release Mechanisms of Mesoporous Silica Nanoparticles
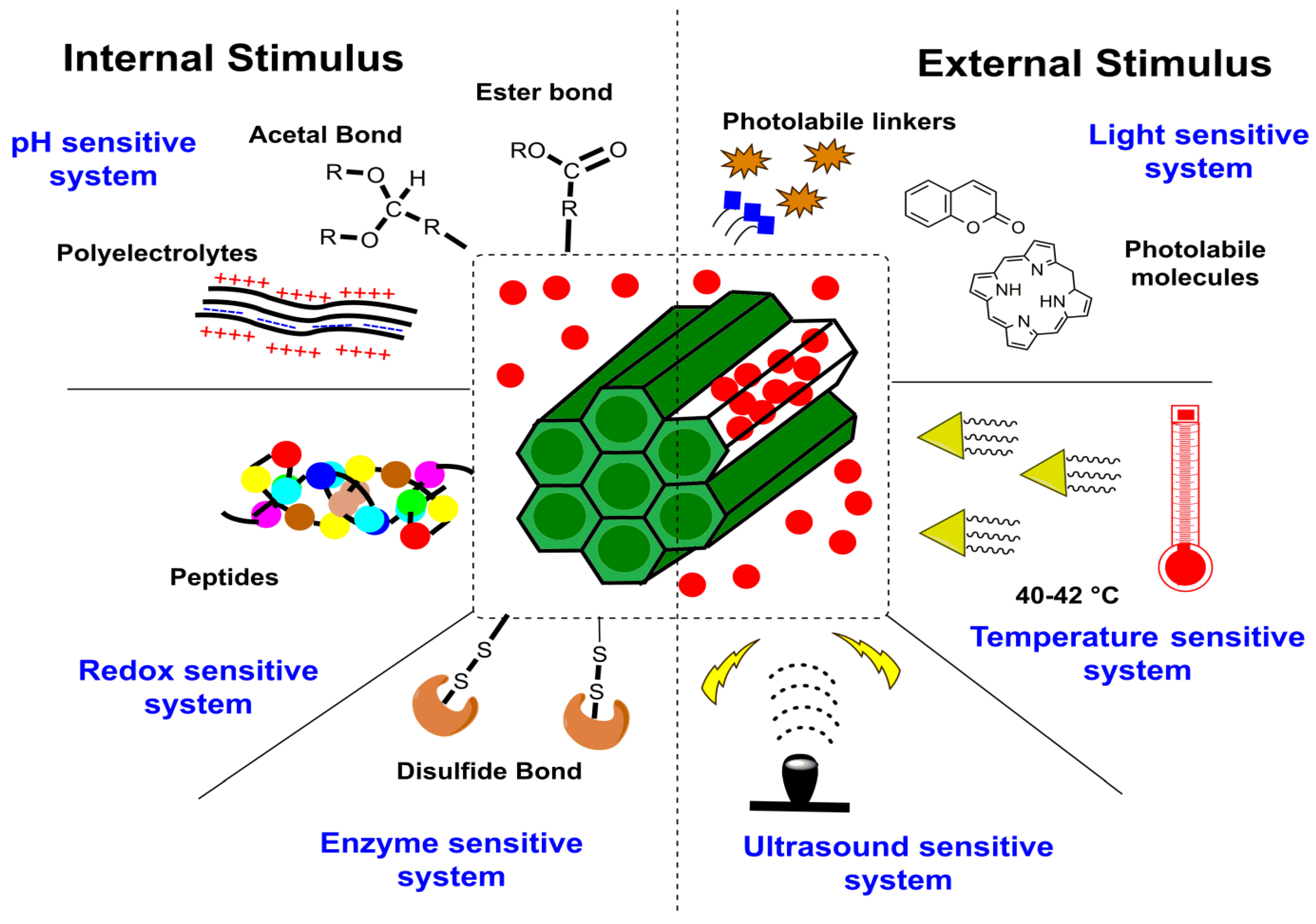
4.1. Stimulus Response
4.1.1. Internal Stimulus
- (i)
- pH stimuli-responsive system
- (ii)
- Redox stimuli-responsive system
- (iii)
- Enzyme stimuli-responsive system
4.1.2. External Stimulus
- (i)
- Light stimuli-responsive system
- (ii)
- Temperature stimuli-responsive system
- (iii)
- Ultrasound stimuli-responsive system
4.2. Dual Stimuli-Responsive System
4.3. Multiple Stimuli-Responsive System
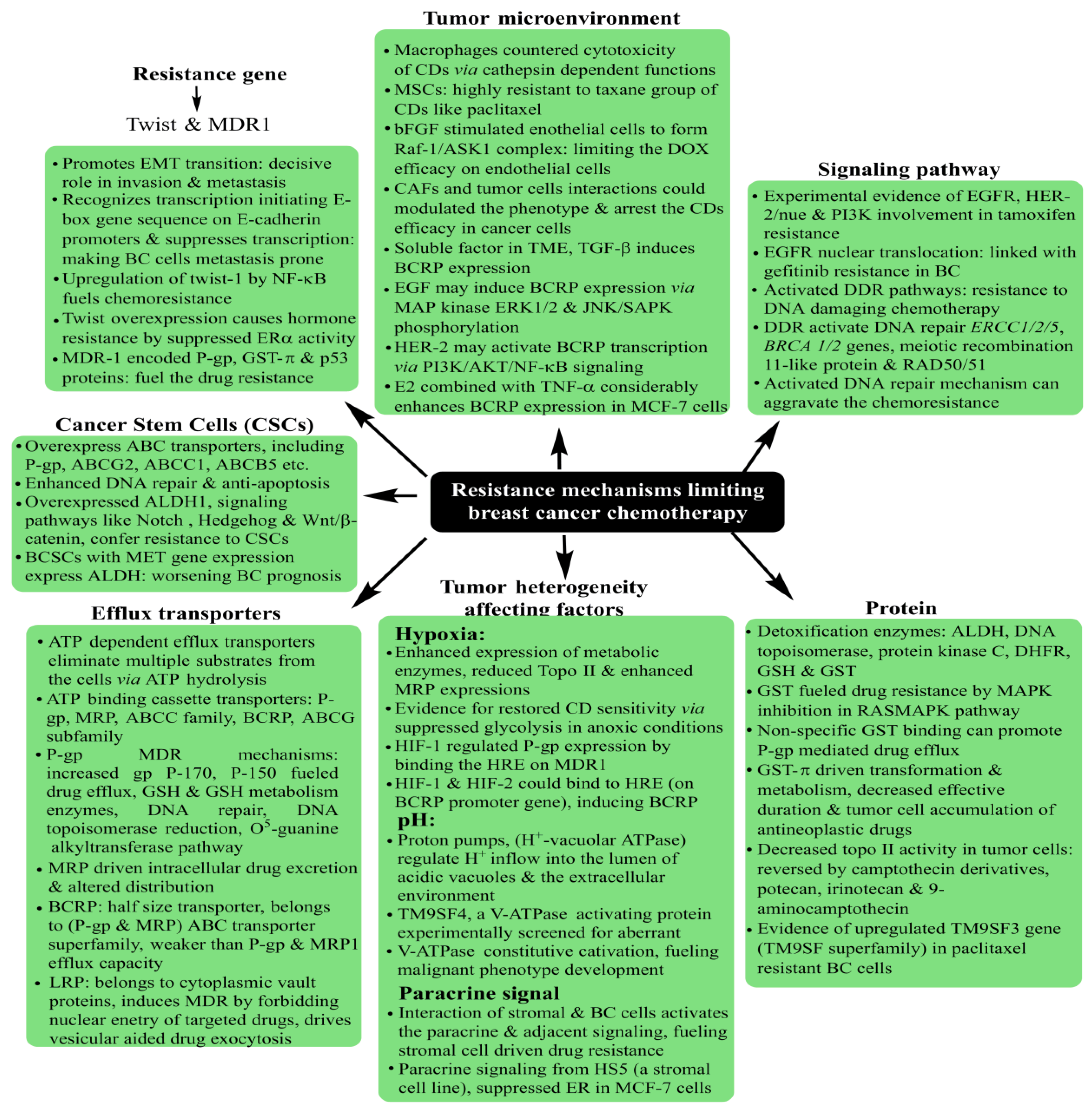
5. Breast Cancer: Pathophysiology, Mortality and Chemoresistance
6. Discussion of Recent Attempts Using Mesoporous Silica Nanoparticles for Breast Cancer Treatment
6.1. Singular Drug Delivery Using Mesoporous Silica Nanoparticles
6.1.1. Distinguished Research Attempts from 2018
6.1.2. Salient Attempts from 2019
6.1.3. Major Research Attempts from 2020
6.1.4. Major Studies Reported in 2021
6.1.5. Significant Research Attempts from 2022
6.2. Combinatorial Drug Delivery Using Mesoporous Silica Nanoparticles
6.2.1. Research Attempts from 2021
6.2.2. Select Research Attempts from 2022
7. Future Challenges in the Therapeutic Usage of Mesoporous Silica Nanoparticles
8. Summary and Takeaway Message
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tao, Z.; Shi, A.; Lu, C.; Song, T.; Zhang, Z.; Zhao, J. Breast cancer: Epidemiology and etiology. Cell Biochem. Biophys. 2015, 72, 333–338. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Prim. 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; de la Garza Salazar, J.; Pritchard, K.; Amadori, D.; Haidinger, R.; Hudis, C.A.; Khaled, H.; Liu, M.C.; Martin, M.; Namer, M.; et al. The global breast cancer burden: Variations in epidemiology and survival. Clin. Breast Cancer 2005, 6, 391–401. [Google Scholar]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, X.; Lu, Y.; Tian, H.; Meng, X.; Wei, M.; Cho, W.C. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed. Pharmacother. 2019, 114, 108800. [Google Scholar] [CrossRef]
- Muley, H.; Fad, R.; Rodriguez-Rodriguez, R.; Casals, N. Drug uptake-based chemoresistance in breast cancer treatment. Biochem. Phamacol. 2020, 177, 113959. [Google Scholar] [CrossRef]
- Lainetti, P.d.F.; Leis-Filho, A.F.; Laufer-Amorim, R.; Battazza, A.; Fonseca-Alves, C.E. Mechanisms of resistance to chemotherapy in breast cancer and possible targets in drug delivery systems. Pharmaceutics 2020, 12, 1193. [Google Scholar] [CrossRef]
- Izci, M.; Maksoudian, C.; Manshian, B.B.; Soenen, S.J. The use of alternative strategies for enhanced nanoparticle delivery to solid tumors. Chem. Rev. 2021, 121, 1746–1803. [Google Scholar] [CrossRef]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-based drug delivery in cancer therapy and its role in overcoming drug resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef]
- Singh, A.P.; Biswas, A.; Shukla, A.; Maiti, P. Targeted therapy in chronic diseases using nanomaterial-based drug delivery vehicles. Signal Transduct. Target. Ther. 2019, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Din, F.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva-Candal, A.; Brown, T.; Krishnan, V.; Lopez-Loureiro, I.; Ávila-Gómez, P.; Pusuluri, A.; Pérez-Díaz, A.; Correa-Paz, C.; Hervella, P.; Castillo, J.; et al. Shape effect in active targeting of nanoparticles to inflamed cerebral endothelium under static and flow conditions. J. Control. Release 2019, 309, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Uhl, C.G.; Gao, Y.; Zhou, S.; Liu, Y. The shape effect on polymer nanoparticle transport in a blood vessel. RSC Adv. 2018, 8, 8089–8100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallet-Regí, M.; Balas, F.; Arcos, D. Mesoporous materials for drug delivery. Angew. Chem. Int. Ed. 2007, 46, 7548–7558. [Google Scholar] [CrossRef]
- Rosenholm, J.M.; Sahlgren, C.; Linden, M. Multifunctional mesoporous silica nanoparticles for combined therapeutic, diagnostic and targeted action in cancer treatment. Curr. Drug Targets 2011, 12, 1166–1186. [Google Scholar] [CrossRef]
- Baeza, A.; Colilla, M.; Vallet-Regí, M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery. Expert Opin. Drug Deliv. 2015, 12, 319–337. [Google Scholar] [CrossRef]
- Weaver, J.L.; Tobin, G.A.; Ingle, T.; Bancos, S.; Stevens, D.; Rouse, R.; Howard, D.; Goodwin, K.E.; Knapton, A.; Li, X.; et al. Evaluating the potential of gold, silver, and silica nanoparticles to saturate mononuclear phagocytic system tissues under repeat dosing conditions. Part. Fibre Toxicol. 2017, 14, 25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, C.; Bai, J.; Wu, C.; Xiao, Y.; Li, Y.; Zheng, J.; Yang, R.; Tan, W. Silver nanoparticle gated, mesoporous silica coated gold nanorods (AuNR@MS@AgNPs): Low premature release and multifunctional cancer theranostic platform. ACS Appl. Mater. Interfaces 2015, 7, 6211–6219. [Google Scholar] [CrossRef]
- Kumar, R.; Mondal, K.; Panda, P.K.; Kaushik, A.; Abolhassani, R.; Ahuja, R.; Rubahne, H.G.; Mishra, Y.K. Core-shell nanostructures: Perspectives towards drug delivery applications. J. Mater. Chem. B 2020, 8, 8992–9027. [Google Scholar] [CrossRef]
- Malik, P.; Gupta, R.; Malik, V.; Ameta, R.K. Emerging nanomaterials for improved biosensing. Meas. Sens. 2021, 16, 10050. [Google Scholar] [CrossRef]
- Malik, P.; Ameta, R.K. Recent Progress in Au nanoparticle treatment of lung cancers. In Biomedical Engineering and Its Applications in Healthcare; Basu, S., Ed.; Springer Nature Publications: Berlin/Heidelberg, Germany, 2019; ISBN 978-981-13-3705-5. [Google Scholar]
- Rani, R.; Sethi, K.; Singh, G. Nanomaterials and their applications in bioimaging. In Plant Nanobionics; Prasad, R., Ed.; Nanotechnology in the Life Sciences; Springer: Cham, Switzerland, 2019; pp. 429–450. ISBN 978-3-030-16379-2. [Google Scholar]
- Huo, Q.; Margolese, D.I.; Stucky, G.D. Stucky Surfactant control of phases in the synthesis of mesoporous silica-based materials. Chem. Mater. 1996, 8, 1147–1160. [Google Scholar] [CrossRef]
- Trewyn, B.G.; Slowing, I.I.; Giri, S.; Chen, H.T.; Lin, V.S.Y. Synthesis and functionalization of a mesoporous silica nanoparticle based on the sol-gel process and applications in controlled release. Acc. Chem. Res. 2007, 40, 846–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayan, R.; Nayak, U.Y.; Raichur, A.M.; Garg, S. Mesoporous silica nanoparticles: A comprehensive review on synthesis and recent advances. Pharmaceutics 2018, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.; Singh, R.K.; Chrzanowski, W. Silica-based mesoporous nanoparticles for controlled drug delivery. J. Tissue Eng. 2013, 4, 2041731413503357. [Google Scholar] [CrossRef] [Green Version]
- Costa, J.A.S.; de Jesus, R.A.; Santos, D.O.; Mano, J.F.; Romao, L.P.C.; Paranhos, C.M. Recent progresses in the adsorption of organic, inorganic, and gas compounds by MCM-41-based mesoporous materials. Microporous Mesoporous Mater. 2020, 291, 109698. [Google Scholar] [CrossRef]
- Oye, G.; Sjöblom, J.; Stöcker, M. Synthesis, characterization and potential applications of new materials in the mesoporous range. Adv. Colloid Interface Sci. 2001, 89–90, 439–466. [Google Scholar] [CrossRef]
- Naono, H.; Hakuman, M.; Tsunehisa, T.; Tamura, N.; Nakai, K. Formation process of MCM-41 precursor and porous texture of MCM-41. J. Colloid Interface Sci. 2000, 224, 358–365. [Google Scholar] [CrossRef]
- Huang, X.; Townley, H.E. An Assessment of mesoporous silica nanoparticle architectures as antigen carriers. Pharmaceutics 2020, 12, 294. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wu, D.; Sun, Y.; Zhong, B. The synthesis of MCM-48 with high yields. Mater. Res. Bullet. 2001, 36, 1717–1720. [Google Scholar] [CrossRef]
- Nandiyanto, A.B.D.; Kim, S.G.; Iskandar, F.; Okuyama, K. Synthesis of spherical mesoporous silica nanoparticles with nanometer-size controllable pores and outer diameters. Microporous Mesoporous Mater. 2009, 120, 447–453. [Google Scholar] [CrossRef]
- Heikkilä, T.; Salonen, J.; Tuura, J.; Hamdy, M.S.; Mul, G.; Kumar, N.; Salmi, T.; Murzin, D.Y.; Laitinen, L.; Kaukonen, A.M.; et al. Mesoporous silica material TUD-1 as a drug delivery system. Int. J. Pharm. 2007, 331, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Kalbasi, J.R.; Zirakbash, A. Synthesis, characterization and drug release studies of poly(2-hydroxyethyl methacrylate)/KIT-5 nanocomposite as an innovative organic–inorganic hybrid carrier system. RSC Adv. 2015, 5, 12463–12471. [Google Scholar] [CrossRef]
- Chircov, C.; Spoială, A.; Păun, C.; Cracium, L.; Ficai, D.; Ficai, A.; Andronescu, E.; Turcule, S.C. Mesoporous silica platforms with potential applications in release and adsorption of active agents. Molecules 2020, 25, 3814. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Luo, Z.S.; Pang, W.Q.; Fan, Y.W.; Chen, X.H.; Cui, F.Z. Dilute solution routes to various controllable morphologies of MCM-41 silica with a basic medium. Chem. Mater. 2001, 13, 258–263. [Google Scholar] [CrossRef]
- Fowler, C.E.; Khushalani, D.; Lebeau, B.; Mann, S. Nanoscale materials with mesostructured interiors. Adv. Mater. 2001, 13, 649–652. [Google Scholar] [CrossRef]
- Nooney, R.I.; Thirunavukkarasu, D.; Chen, Y.M.; Josephs, R.; Ostafin, A.E. Synthesis of nanoscale mesoporous silica spheres with controlled particle size. Chem. Mater. 2002, 14, 4721–4728. [Google Scholar] [CrossRef]
- Lai, C.Y.; Trewyn, B.G.; Jeftinija, D.M.; Jeftinija, K.; Xu, S.; Jeftinija, S.; Lin, V.S.Y. A Mesoporous Silica Nanosphere-Based Carrier System with Chemically Removable CdS Nanoparticle Caps for Stimuli-Responsive Controlled Release of Neurotransmitters and Drug Molecules. J. Am. Chem. Soc. 2003, 125, 4451–4459. [Google Scholar] [CrossRef]
- Lin, Y.S.; Haynes, C.L. Impacts of mesoporous silica nanoparticle size, pore ordering, and pore integrity on hemolytic activity. J. Am. Chem. Soc. 2010, 132, 4834–4842. [Google Scholar] [CrossRef]
- Williams, S.; Neumann, A.; Bremer, I.; Su, Y.; Dräger, G.; Kasper, C.; Behrens, P. Nanoporous silica nanoparticles as biomaterials: Evaluation of different strategies for the functionalization with polysialic acid by step-by-step cytocompatibility testing. J. Mater. Sci. Mater. Med. 2015, 26, 125. [Google Scholar] [CrossRef]
- Qiao, Z.A.; Zhang, L.; Guo, M.; Liu, Y.; Huo, Q. Synthesis of mesoporous silica nanoparticles via controlled hydrolysis and condensation of silicon alkoxide. Chem. Mater. 2009, 21, 3823–3829. [Google Scholar] [CrossRef]
- Blin, J.L.; Michaux, F.; Stébé, M.J. Nanostuctured mesoporous materials from different silica sources using fluorinated surfactants as templates. Colloids Surf. A Physicochem. Eng. Asp. 2016, 510, 104–112. [Google Scholar] [CrossRef]
- Brevet, D.; Jouannin, C.; Tourné-Péteilh, C.; Devoisselle, J.M.; Vioux, A.; Viau, L. Self-encapsulation of a drug-containing ionic liquid into mesoporous silica monoliths or nanoparticles by a sol-gel process. RSC Adv. 2016, 6, 82916–82923. [Google Scholar] [CrossRef]
- Siddiqui, B.; Rehman, A.; Haq, I.; Al-Dossary, A.A.; Elaissari, A.; Ahmed, N. Exploiting recent trends for the synthesis and surface functionalization of mesoporous silica nanoparticles towards biomedical applications. Int. J. Pharm. X 2022, 4, 10016. [Google Scholar] [CrossRef]
- Wang, Y. Synthesis and formation of hierarchical mesoporous silica network in acidic aqueous solutions of sodium silicate and cationic surfactant. Colloid J. 2010, 72, 737–742. [Google Scholar] [CrossRef]
- Das, D.; Yang, Y.; O’Brien, J.S.; Breznan, D.; Nimesh, S.; Bernatchez, S.; Hill, M.; Sayari, A.; Vincent, R.; Kumarathasan, P. Synthesis and physicochemical characterization of mesoporous SiO2 nanoparticles. J. Nanomater. 2014, 2014, 62. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Bastakoti, B.P.; Imura, M.; Tang, J.; Aldalbahi, A.; Torad, N.L.; Yamauchi, Y. Dual soft-template system based on colloidal chemistry for the synthesis of hollow mesoporous silica nanoparticles. Chem. Eur. J. 2015, 21, 6375–6380. [Google Scholar] [CrossRef] [PubMed]
- Gai, F.; Zhou, T.; Chu, G.; Li, Y.; Liu, Y.; Huo, Q.; Akhtar, F. Mixed anionic surfactant-templated mesoporous silica nanoparticles for fluorescence detection of Fe3+. Dalton Trans. 2016, 45, 508–514. [Google Scholar] [CrossRef] [Green Version]
- She, X.; Chen, L.; Velleman, L.; Li, C.; Zhu, H.; He, C.; Wang, T.; Shigdar, S.; Duan, W.; Kong, L. Fabrication of high specificity hollow mesoporous silica nanoparticles assisted by Eudragit for targeted drug delivery. J. Colloid Interface Sci. 2015, 445, 151–160. [Google Scholar] [CrossRef]
- Um, K.; Chang, H.; Lee, K. Facile synthesis of hollow mesoporous zinc silicate nanoparticles using a dual surfactant system. RSC Adv. 2016, 6, 98717–98721. [Google Scholar] [CrossRef]
- Owens, G.J.; Singh, R.K.; Foroutan, F.; Alqaysi, M.; Han, C.M.; Mahapatra, C.; Kim, H.W.; Knowles, J.C. Sol-gel based materials for biomedical applications. Prog. Mater. Sci. 2016, 77, 1–79. [Google Scholar] [CrossRef]
- Zhou, C.; Yan, C.; Zhao, J.; Wang, H.; Zhou, Q.; Luo, W. Rapid synthesis of morphology-controlled mesoporous silica nanoparticles from silica fume. J. Taiwan Inst. Chem. Eng. 2016, 62, 307–312. [Google Scholar] [CrossRef]
- Sharma, R.K.; Wang, S.C.; Maity, J.P.; Banerjee, P.; Dey, G.; Huang, Y.H.; Bundschuh, J.; Hsiao, P.G.; Chen, T.H.; Chen, C.Y. A novel BMSN (biologically synthesized mesoporous silica nanoparticles) material: Synthesis using a bacteria-mediated biosurfactant and characterization. RSC Adv. 2021, 11, 32906–32916. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Lee, J.H.; Chun, J. Facile approach for the synthesis of spherical mesoporous silica nanoparticles from sodium silicate. Mater. Lett. 2021, 283, 128765. [Google Scholar] [CrossRef]
- Khalil, M.; Amanda, A.; Yunarti, R.T.; Jan, B.M.; Irawan, S. Synthesis and application of mesoporous silica nanoparticles as gas migration control additive in oil and gas cement. J. Pet. Sci. Eng. 2020, 195, 107660. [Google Scholar] [CrossRef]
- Lu, F.; Qian, W.; Zhan, C.; Wang, Q.; Shen, Q.; Zhong, J.; Fan, Q.; Huang, W. Facile synthesis of hollow mesoporous silica nanoparticles with in-situ formed CuS templates. Mater. Lett. 2019, 250, 25–29. [Google Scholar] [CrossRef]
- Fulvio, P.F.; Pikus, S.; Jaroniec, M. Tailoring properties of SBA-15 materials by controlling conditions of hydrothermal synthesis. J. Mater. Chem. 2005, 15, 5049–5053. [Google Scholar] [CrossRef]
- Soares, D.C.F.; Soares, L.M.; de Goes, A.M.; Melo, E.M.; de Barros, A.L.B.; Bicalho, T.C.A.S.; Leao, N.M.; Tebaldi, M.L. Mesoporous SBA-16 silica nanoparticles as a potential vaccine adjuvant against Paracoccidioides brasiliensis. Microporous Mesoporous Mater. 2020, 291, 109676. [Google Scholar] [CrossRef]
- Li, Q.; Wang, W.; Hu, G.; Cui, X.; Sun, D.; Jin, Z.; Zhao, K. Evaluation of chitosan derivatives modified mesoporous silica nanoparticles as delivery carrier. Molecules 2021, 26, 2490. [Google Scholar] [CrossRef]
- Mohamed Isa, E.D.; Ahmad, H.; Abdul Rahman, M.B.; Gill, M.R. Progress in mesoporous silica nanoparticles as drug delivery agents for cancer treatment. Pharmaceutics 2021, 13, 152. [Google Scholar] [CrossRef]
- Mehmood, S.; Mahmood, M.; Núñez-Delgado, A.; Alatalo, J.M.; Elrys, A.S.; Rizwan, M.; Weng, J.; Li, W.; Ahmed, W. A green method for removing chromium (VI) from aqueous systems using novel silicon nanoparticles: Adsorption and interaction mechanisms. Environ. Res. 2022, 213, 113614. [Google Scholar] [CrossRef] [PubMed]
- Abburi, A.; Ali, M.; Moriya, P.V. Synthesis of mesoporous silica nanoparticles from waste hexafluorosilicic acid of fertilizer industry. J. Mater. Res. Technol. 2020, 9, 8074–8080. [Google Scholar] [CrossRef]
- Mohamad, D.F.; Osman, N.S.; Nazri, M.K.H.M.; Mazlan, A.A.; Hanafi, M.F.; Esa, Y.A.M.; Rafi, M.I.I.M.; Zailani, M.N.; Rahman, N.N.; Abd Rahman, A.H.; et al. Synthesis of mesoporous silica nanoparticle from banana peel ash for removal of phenol and methyl orange in aqueous solution. Mater. Today Proc. 2019, 19, 1119–1125. [Google Scholar] [CrossRef]
- Usgodaarachchi, L.; Thambiliyagodage, C.; Wijesekera, R.; Bakker, M.G. Synthesis of mesoporous silica nanoparticles derived from rice husk and surface-controlled amine functionalization for efficient adsorption of methylene blue from aqueous solution. Curr. Opin. Green Sustain. Chem. 2021, 4, 100116. [Google Scholar] [CrossRef]
- Imoisili, P.E.; Ukoba, K.O.; Jen, T.C. Green technology extraction and characterisation of silica nanoparticles from palm kernel shell ash via sol-gel. J. Mater. Res. Technol. 2020, 9, 307–313. [Google Scholar] [CrossRef]
- Naseem, T.; Baig, M.M.; Warsi, M.F.; Hussain, R.; Agboola, P.O.; Waseem, M. Mesoporous silica prepared via a green route: A comparative study for the removal of crystal violet from wastewater. Mater. Res. Express 2020, 8, 15005. [Google Scholar] [CrossRef]
- Venezia, V.; Sannino, F.; Costantini, A.; Silvestri, B.; Cimino, S.; Califano, V. Mesoporous silica nanoparticles for β-glucosidase immobilization by templating with a green material: Tannic acid. Microporous Mesoporous Mater. 2020, 302, 110203. [Google Scholar] [CrossRef]
- Napierska, D.; Thomassen, L.C.; Lison, D.; Martens, J.A.; Hoet, P.H. The nanosilica hazard: Another variable entity. Part. Fibre Toxicol. 2010, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Martin, K.R. The chemistry of silica and its potential health benefits. J. Nutr. Health Aging 2007, 11, 94–97. [Google Scholar]
- Croissant, J.G.; Fatieiev, Y.; Khashab, N.M. Degradability and clearance of silicon, organosilica, silsesquioxane, silica mixed oxide, and mesoporous silica nanoparticles. Adv. Mater. 2017, 29, 1604634. [Google Scholar] [CrossRef]
- Slowing, I.I.; Wu, C.W.; Vivero-Escoto, J.L.; Lin, V.S.Y. Mesoporous silica nanoparticles for reducing hemolytic activity towards mammalian red blood cells. Small 2009, 5, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Breznan, D.; Das, D.D.; MacKinnon-Roy, C.; Bernatchez, S.; Sayari, A.; Hill, M.; Renaud Vincent, R.; Kumarathasan, P. Physicochemical properties can be key determinants of mesoporous silica nanoparticle potency in vitro. ACS Nano 2018, 12, 12062–12079. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Zhang, Z.; Gao, F.; Li, Y.; Shi, J. In vivo biodistribution and urinary excretion of mesoporous silica nanoparticles: Effects of particle size and PEGylation. Small 2011, 7, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Malugin, A.; Ghandehari, H. Impact of silica nanoparticle design on cellular toxicity and hemolytic activity. ACS Nano 2011, 5, 5717–5728. [Google Scholar] [CrossRef] [PubMed]
- Townson, J.L.; Lin, Y.S.; Agola, J.O.; Carnes, E.C.; Leong, H.S.; Lewis, J.D.; Haynes, C.L.; Brinker, C.J. Re-examining the size/charge paradigm: Differing in vivo characteristics of size- and charge-matched mesoporous silica nanoparticles. J. Am. Chem. Soc. 2013, 135, 16030–16033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahikkala, A.; Pereira, S.A.P.; Figueiredo, P.; Passos, M.L.C.; Araújo, A.R.T.S.; Saraiva, M.L.M.F.S.; Santos, H.A. Mesoporous Silica Nanoparticles for Targeted and Stimuli-Responsive Delivery of Chemotherapeutics: A Review. Adv. Biosyst. 2018, 2, 1800020. [Google Scholar] [CrossRef]
- Ortega, E.; Ruiz, M.A.; Peralta, S.; Russo, G.; Morales, M.E. Improvement of mesoporous silica nanoparticles: A new approach in the administration of NSAIDS. J. Drug Deliv. Sci. Technol. 2020, 58, 101833. [Google Scholar] [CrossRef]
- Haddick, L.; Zhang, W.; Reinhard, S.; Moller, K.; Engelke, H.; Wagner, E.; Bein, T. Particle-size-dependent delivery of antitumoral miRNA using targeted mesoporous silica nanoparticles. Pharmaceutics 2020, 12, 505. [Google Scholar] [CrossRef] [PubMed]
- Möller, K.; Bein, T. Degradable drug carriers: Vanishing mesoporous silica nanoparticles. Chem. Mater. 2019, 31, 4364–4378. [Google Scholar] [CrossRef]
- Lv, X.; Zhang, L.; Xing, F.; Lin, H. Controlled synthesis of monodispersed mesoporous silica nanoparticles: Particle size tuning and formation mechanism investigation. Microporous Mesoporous Mater. 2016, 225, 238–244. [Google Scholar] [CrossRef]
- Yismaw, S.; Kohns, R.; Schneider, D.; Poppitz, D.; Ebbinghaus, S.G.; Gläser, R.; Tallarek, U.; Enke, D. Particle size control of monodispersed spherical nanoparticles with MCM-48-type mesostructure via novel rapid synthesis procedure. J. Nanopart. Res. 2019, 21, 258. [Google Scholar] [CrossRef]
- Ribeiro, T.; Rodrigues, A.S.; Calderon, S.; Fidalgo, A.; Gonçalves, J.L.M.; André, V.; Duarte, M.T.; Ferreira, P.J.; Farinha, J.P.S.; Baleizão, C. Silica nanocarriers with user-defined precise diameters by controlled template self-assembly. J. Colloid Interface Sci. 2020, 561, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Catalano, F.; Pompa, P.P. Design Rules for Mesoporous Silica toward the Nanosize: A Systematic Study. ACS Appl. Mater. Interfaces 2019, 11, 47237–47246. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Yoon, S.; Lee, J.H. Facile large-scale synthesis of mesoporous silica nanoparticles at room temperature in a monophasic system with fine size control. Microporous Mesoporous Mater. 2019, 288, 109595. [Google Scholar] [CrossRef]
- Luo, J.; Panzarasa, G.; Osypova, A.; Sorin, F.; Spano, F.; Rossi, R.M.; Sadeghpour, A.; Boesel, L.F. Polyphenols as morphogenetic agents for the controlled synthesis of mesoporous silica nanoparticles. Chem. Mater. 2019, 31, 3192–3200. [Google Scholar] [CrossRef]
- Xu, C.; Lei, C.; Yu, C. Mesoporous silica nanoparticles for protein protection and delivery. Front. Chem. 2019, 7, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousavi Elyerdi, S.M.; Sarvi, M.N.; O’Connor, A.J. Synthesis of ultra small nanoparticles (<50 nm) of mesoporous MCM-48 for bio-adsorption. J. Porous Mater. 2019, 26, 839–846. [Google Scholar]
- Mohamed Isa, E.D.; Ahmad, H.; Abdul Rahman, M.B. Optimization of synthesis parameters of mesoporous silica nanoparticles based on ionic liquid by experimental design and its application as a drug delivery agent. J. Nanomater. 2019, 2019, 4982054. [Google Scholar] [CrossRef] [Green Version]
- Ya-Dong, C.; Hong-Yuan, L.; Sin-Yen, L.; Shy-Guey, W.; Yusuke, Y.; Kevin, C.W. Controlling particle size and structural properties of mesoporous silica nanoparticles using the Taguchi method. Phys. Chem. C 2011, 115, 13158–13165. [Google Scholar]
- Kachbouri, S.; Mnasri, N.; Elaloui, E.; Moussaoui, Y. Tuning particle morphology of mesoporous silica nanoparticles for adsorption of dyes from aqueous solution. J. Saudi Chem. Soc. 2018, 22, 405–415. [Google Scholar] [CrossRef]
- Song, J.C.; Xue, F.F.; Zhang, X.X.; Lu, Z.Y.; Sun, Z.Y. Synthesis of yolk-shell mesoporous silica nanoparticles via a facile one-pot approach. Chem. Commun. 2017, 53, 3761–3764. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; Wang, P.Y.; Tang, X.T.; Elzatahry, A.A.; Wang, S.W.; Al-Dahyan, D.; Zhao, M.Y.; Yao, C.; Hung, C.T.; Zhu, X.H.; et al. Facile synthesis of uniform virus-like mesoporous silica nanoparticles for enhanced cellular internalization. ACS Cent. Sci. 2017, 3, 839–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed Isa, E.D.; Abdul Rahman, M.B.; Ahmad, H. Monodispersed mesoporous silica nanospheres based on pyridinium ionic liquids. J. Porous Mater. 2018, 25, 1439–1446. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhou, X.; He, C. Mesoporous silica nanoparticles for tissue-engineering applications. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11, e1573. [Google Scholar] [CrossRef]
- Hong, X.Y.; Zhong, X.F.; Du, G.S.; Hou, Y.Y.; Zhang, Y.T.; Zhang, Z.R.; Gong, T.; Zhang, L.; Sun, X. The pore size of mesoporous silica nanoparticles regulates their antigen delivery efficiency. Sci. Adv. 2020, 6, eaaz4462. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ru, J.; Sun, S.; Teng, Z.; Dong, H.; Song, P.; Yang, Y.; Guo, H. Uniform dendrimer-like mesoporous silica nanoparticles as a nano-adjuvant for foot-and-mouth disease virus-like particle vaccine. J. Mater. Chem. B 2019, 7, 3446–3454. [Google Scholar] [CrossRef]
- He, Y.; Li, J.; Long, M.; Liang, S.; Xu, H. Tuning pore size of mesoporous silica nanoparticles simply by varying reaction parameters. J. Non-Cryst. Solids 2017, 457, 9–12. [Google Scholar] [CrossRef]
- Ryu, J.; Kim, W.; Yun, J.; Lee, K.; Lee, J.; Yu, H.; Kim, J.H.; Kim, J.J.; Jang, J. Fabrication of uniform wrinkled silica nanoparticles and their application to abrasives in chemical mechanical planarization. ACS Appl. Mater. Interfaces 2018, 10, 11843–11851. [Google Scholar] [CrossRef]
- Saikia, D.; Deka, J.R.; Wu, C.E.; Yang, Y.C.; Kao, H.M. pH responsive selective protein adsorption by carboxylic acid functionalized large pore mesoporous silica nanoparticles SBA-1. Mater. Sci. Eng. C 2019, 94, 344–356. [Google Scholar] [CrossRef]
- Xu, B.; Su, Y.; Chen, L.; Cai, J.; Huang, B. Preparation of mesoporous silica nanoparticles with controlled pore size, particle diameter, morphology, and structure by two-step process of chlorosilane residue. Ceram. Int. 2018, 44, 22241–22248. [Google Scholar] [CrossRef]
- Yamamoto, E.; Mori, S.; Shimojima, A.; Wada, H.; Kuroda, K. Fabrication of colloidal crystals composed of pore-expanded mesoporous silica nanoparticles prepared by a controlled growth method. Nanoscale 2017, 9, 2464–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Song, H.; Yu, M.; Xu, C.; Liu, Y.; Tang, J.; Yang, Y.; Yu, C. Room temperature synthesis of dendritic mesoporous silica nanoparticles with small sizes and enhanced mRNA delivery performance. J. Mater. Chem. B 2018, 6, 4089–4095. [Google Scholar] [CrossRef]
- Eskandari, P.; Bigdeli, B.; Daryasari, M.P.; Baharifar, H.; Bazri, B.; Shourian, M.; Amani, A.; Sadighi, A.; Goliaei, B.; Khoobi, M.; et al. Gold-capped mesoporous silica nanoparticles as an excellent enzyme-responsive nanocarrier for controlled doxorubicin delivery. J. Drug Target. 2019, 27, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, K.; Gupta, S.; Gnanadhas, D.P.; Ramamurthy, P.C.; Chakravortty, D.; Raichur, A.M. Protamine-capped mesoporous silica nanoparticles for biologically triggered drug release. Part. Part. Syst. Charact. 2014, 31, 449–458. [Google Scholar] [CrossRef]
- He, Y.; Su, Z.; Xue, L.; Xua, H.; Zhang, C. Co-delivery of erlotinib and doxorubicin by pH sensitive charge conversion nanocarrier for synergistic therapy. J. Control. Release 2016, 229, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Xue, M.; Zink, J.I. Functioning of nanovalves on polymer coated mesoporous silica nanoparticles. Nanoscale 2013, 5, 10300–10306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montoto, A.H.; Llopis-Lorente, A.; Gorbe, M.; Terrés, J.M.; Cao-Milán, R.; de Greñu, B.D.; Alfonso, M.; Ibañez, J.; Marcos, M.D.; Orzáez, M.; et al. Janus gold nanostars-mesoporous silica nanoparticles for NIR light-triggered drug delivery. Chem. Eur. J. 2019, 25, 8471–8478. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Z.; Yao, X.; Chen, X.; Chen, X. Intracellular pH-operated mechanized mesoporous silica nanoparticles as potential drug carries. Microporous Mesoporous Mater. 2015, 201, 169–175. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Zhao, M.; Lei, Z.; Guo, H.; Tang, Y.; Yan, H. Redox-responsive hollow mesoporous silica nanoparticles constructed via host-guest interactions for controllable drug release. J. Biomater. Sci. Polym. Ed. 2020, 31, 472–490. [Google Scholar] [CrossRef]
- Yao, X.; Tian, Z.; Liu, J.; Zhu, Y.; Hanagata, N. Mesoporous silica nanoparticles capped with graphene quantum dots for potential chemo-photothermal synergistic cancer therapy. Langmuir 2017, 33, 591–599. [Google Scholar] [CrossRef]
- Sun, R.; Wang, W.; Yongqiang, W.; Zhang, X. Recent advance on mesoporous silica nanoparticles-based controlled release system: Intelligent switches open up new horizon. Nanomaterials 2015, 5, 2019–2053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Hu, Y.; Xu, L.; Liu, M.; Wu, H.; He, B. Dual pH and glucose sensitive gel gated mesoporous silica nanoparticles for drug delivery. Chin. Chem. Lett. 2018, 29, 1795–1798. [Google Scholar] [CrossRef]
- Zhang, Z.; Runa, A.; Wu, J.; Zhang, H.; Li, X.; He, Z. Bioresponsivenanogated ensemble based on structure-switchable aptamer directed assembly and disassembly of gold nanoparticles from mesoporous silica supports. Chin. Chem. Lett. 2019, 30, 779–782. [Google Scholar] [CrossRef]
- She, X.; Chen, L.; Li, C.; He, C.; He, L.; Kong, L. Functionalization of hollow mesoporous silica nanoparticles for improved 5-FU Loading. J. Nanomater. 2015, 16, 108. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Y.; Wang, J.; Yang, Y.; Li, Y.; Yuan, Y.; Liu, C. Charge-reversal APTES-modified mesoporous silica nanoparticles with high drug loading and release controllability. ACS Appl. Mater. Interfaces 2016, 8, 17166–17175. [Google Scholar] [CrossRef] [PubMed]
- Bouchoucha, M.; Gaudreault, R.C.; Fortin, M.A.; Kleitz, F. Mesoporous silica nanoparticles: Selective surface functionalization for optimal relaxometric and drug loading performances. Adv. Funct. Mater. 2014, 24, 5911–5923. [Google Scholar] [CrossRef]
- Sharma, J.; Polizos, G. Hollow silica particles: Recent progress and future perspectives. Nanomaterials 2020, 10, 1599. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Lopes, A.; Xu, D.; Liu, B. Multimetallic Hollow Mesoporous nanospheres with synergistically structural and compositional effects for highly efficient ethanol electrooxidation. ACS Cent. Sci. 2018, 4, 1412–1419. [Google Scholar] [CrossRef] [Green Version]
- Palanikumar, L.; Kim, H.Y.; Oh, J.Y.; Thomas, A.P.; Choi, E.S.; Jeena, M.T.; Joo, S.H.; Ryu, J.H. Noncovalent surface locking of mesoporous silica nanoparticles for exceptionally high hydrophobic drug loading and enhanced colloidal stability. Biomacromolecules 2015, 16, 2701–2714. [Google Scholar] [CrossRef] [PubMed]
- Rahman, Z.U.; Wei, N.; Li, Z.; Sun, W.; Wang, D. Preparation of hollow mesoporous silica nanospheres: Controllable template synthesis and their application in drug delivery. New J. Chem. 2017, 41, 14122–14129. [Google Scholar] [CrossRef]
- Kao, K.C.; Mou, C.Y. Pore-expanded mesoporous silica nanoparticles with alkanes/ethanol as pore expanding agent. Microporous Mesoporous Mater. 2013, 169, 7–15. [Google Scholar] [CrossRef]
- Kim, M.H.; Na, H.K.; Kim, Y.K.; Ryoo, S.R.; Cho, H.S.; Lee, K.E.; Jeon, H.; Ryoo, R.; Min, D.H. Facile synthesis of monodispersed mesoporous silica nanoparticles with ultra-large pores and their application in gene delivery. ACS Nano 2011, 5, 3568–3576. [Google Scholar] [CrossRef] [PubMed]
- Nieto, A.; Colilla, M.; Balas, F.; Vallet-Regí, M. Surface Electrochemistry of mesoporous silicas as a key factor in the design of tailored delivery devices. Langmuir 2010, 26, 5038–5049. [Google Scholar] [CrossRef] [PubMed]
- Almomen, A.; El-Toni, A.M.; Badran, M.; Alhowyan, A.; Abul Kalam, M.; Alshamsan, A.; Alkholief, M. The design of anionic surfactant-based amino-functionalized mesoporous silica nanoparticles and their application in transdermal drug delivery. Pharmaceutics 2020, 12, 1035. [Google Scholar] [CrossRef] [PubMed]
- Datt, A.; El-Maazawi, I.; Larsen, S.C. Aspirin loading and release from MCM-41 functionalized with aminopropyl groups via co-condensation or post synthesis modification methods. J. Phys. Chem. C 2012, 116, 18358–18366. [Google Scholar] [CrossRef]
- Song, S.W.; Hidajat, K.; Kawi, S. Functionalized SBA-15 materials as carriers for controlled drug delivery: Influence of surface properties on matrix-drug interactions. Langmuir 2005, 21, 9568–9575. [Google Scholar] [CrossRef]
- Baumann, F.; Paul, T.; Wassersleben, S.; Regenthal, R.; Enke, D.; Aigner, A. Characterization of drug release from mesoporous sio2-based membranes with variable pore structure and geometry. Pharmaceutics 2022, 14, 1184. [Google Scholar] [CrossRef]
- Bukara, K.; Schueller, L.; Rosier, J.; Martens, M.A.; Daems, T.; Verheyden, L.; Eelen, S.; Speybroeck, M.V.; Libanati, C.; Martens, J.A.; et al. Ordered mesoporous silica to enhance the bioavailability of poorly water-soluble drugs: Proof of concept in man. Eur. J. Pharm. Biopharm. 2016, 108, 220–225. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Bai, X.; Jiang, T.; Zhang, Q.; Wang, S. Mesoporous silica nanoparticles for increasing the oral bioavailability and permeation of poorly water soluble drugs. Mol. Pharm. 2012, 9, 505–513. [Google Scholar] [CrossRef]
- Thomas, M.J.K.; Slipper, I.; Walunj, A.; Jain, A.; Favretto, M.E.; Kallinteri, P.; Douroumis, D. Inclusion of poorly soluble drugs in highly ordered mesoporous silica nanoparticles. Int. J. Pharm. 2010, 387, 272–277. [Google Scholar] [CrossRef]
- Lee, N.K.; Park, S.S.; Ha, C.S. pH-sensitive drug delivery system based on mesoporous silica modified with Poly-L-Lysine (PLL) as a Gatekeeper. J. Nanosci. Nanotechnol. 2020, 20, 6925–6934. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Hu, J.; Bian, C.; Zhu, C.; Chen, C.; Guo, Z.; Zhang, Z.; Agyekum, G.A.; Zhang, Z.; Cao, X. pH-responsive and biodegradable ZnO-capped mesoporous silica composite nanoparticles for drug delivery. Materials 2020, 13, 3950. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ma, T.; Tang, W.; Wang, X.; Wang, Y.; Zhuang, J.; Zhu, Y.; Wang, P. Reversibly-regulated drug release using poly (tannic acid) fabricated nanocarriers for reduced secondary side effects in tumor therapy. Nanoscale Horiz. 2020, 5, 986–998. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Qin, Z.; Liang, X.; He, X.; Zhu, B.; Lu, Z.; Wei, Q.; Zheng, L. A pH-responsive mesoporous silica nanoparticles-based drug delivery system with controlled release of andrographolide for OA treatment. Regen. Biomater. 2021, 8, rbab020. [Google Scholar] [CrossRef]
- Wagner, J.; Gößl, D.; Ustyanovska, N.; Xiong, M.; Hauser, D.; Zhuzhgova, O.; Hočevar, S.; Taskoparan, B.; Poller, L.; Datz, S.; et al. Mesoporous silica nanoparticles as pH-responsive carrier for theimmune-activating drug resiquimod enhance the local immune response in mice. ACS Nano 2021, 15, 4450–4466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.B.; Chen, X.J.; Fan, X.D.; Zhu, J.J.; Wei, Y.H.; Zheng, H.S.; Zheng, H.Y.; Wang, B.H.; Piao, J.G.; Li, F.Z. Lipid/PAA-coated mesoporous silica nanoparticles for dual-pH-responsive codelivery of arsenic trioxide/paclitaxel against breast cancer cells. Acta Pharmacol. Sin. 2021, 42, 832–842. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, F.; Al-Ammari, A.; Sun, D. An optimized mesoporous silica nanosphere-based carrier system with chemically removable Au nanoparticle caps for redox-stimulated and targeted drug delivery. Nanotechnology 2020, 31, 475102. [Google Scholar] [CrossRef]
- Li, L.; Lan, S.; Ma, D. Ultrastable and versatile layer-by-layer coating based on kinetically trapped host-guest complexation for mesoporous silica nanoparticles. Part. Part. Syst. Charact. 2020, 37, 2000075. [Google Scholar] [CrossRef]
- Xu, J.; Liu, Y.; Li, G.; Peng, M.; Xu, S.; Liu, H. A reduction-triggered nanocarrier based on host-guest interaction between pillar[5]arene derivative and viologen on MSN for intracellular delivery. J. Drug Deliv. Sci. Technol. 2022, 68, 103055. [Google Scholar]
- Jiang, H.; Shi, X.; Yu, X.; He, X.; An, Y.; Lu, H. Hyaluronidase enzyme-responsive targeted nanoparticles for effective delivery of 5-fluorouracil in colon cancer. Pharm. Res. 2018, 35, 73. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, M.; Ying, H.; Su, D.; Zhang, H.; Lu, G.; Chen, J. Extracellular matrix component shelled nanoparticles as dual enzyme-responsive drug delivery vehicles for cancer therapy. ACS Biomater. Sci. Eng. 2018, 4, 2404–2411. [Google Scholar] [CrossRef]
- Cai, D.; Han, C.; Liu, C.; Ma, X.; Qian, J.; Zhou, J.; Li, Y.; Sun, Y.; Zhang, C.; Zhu, W. Chitosan-capped enzyme-responsive hollow mesoporous silica nanoplatforms for colon-specific drug delivery. Nanoscale Res. Lett. 2020, 15, 123. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, T.; Wang, D.; Jiang, W.; Fu, J. Acid and light stimuli-responsive mesoporous silica nanoparticles for controlled release. J. Mater. Sci. 2019, 54, 6199–6211. [Google Scholar] [CrossRef]
- Salinas, Y.; Brüggemann, O.; Monkowius, U.; Teasdale, I. Visible light photocleavable ruthenium-based molecular gates to reversibly control release from mesoporous silica nanoparticles. Nanomaterials 2020, 10, 1030. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, H.; Wang, H.; Han, Y.; Zheng, Z.; Liu, X.; Feng, B.; Zhang, H. Light-responsive dual-functional biodegradable mesoporous silica nanoparticles with drug delivery and lubrication enhancement for the treatment of osteoarthritis. Nanoscale 2021, 13, 6394–6399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, X.; Shi, H.; Dong, G.; Zhou, M.; Wang, T.; Xin, H. Thermo-responsive mesoporous silica/lipid bilayer hybrid nanoparticles for doxorubicin on-demand delivery and reduced premature release. Colloids Surf. B 2017, 160, 527–534. [Google Scholar] [CrossRef]
- Castillo, R.R.; Lozano, D.; González, B.; Manzano, M.; Izquierdo-Barba, I.; Vallet-Regí, M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery: An update. Expert Opin. Drug Deliv. 2019, 16, 415–439. [Google Scholar] [CrossRef]
- Raza, A.; Rasheed, T.; Nabeel, F.; Hayat, U.; Bilal, M.; Iqbal, H.M. Endogenous and exogenous stimuli-responsive drug delivery systems for programmed site-specific release. Molecules 2019, 24, 1117. [Google Scholar] [CrossRef] [Green Version]
- Peralta, M.E.; Jadhav, S.A.; Magnacca, G.; Scalarone, D.; Mártire, D.O.; Parolo, M.E.; Carlos, L. Synthesis and in vitro testing of thermoresponsive polymer-grafted core-shell magnetic mesoporous silica nanoparticles for efficient controlled and targeted drug delivery. J. Colloid Interface Sci. 2019, 544, 198–205. [Google Scholar] [CrossRef]
- Shi, Z.; Yang, C.; Li, R.; Ruan, L. Microwave thermal-triggered drug delivery using thermosensitive peptide-coated core-shell mesoporous silica nanoparticles. J. Mater. Sci. 2020, 55, 6118–6129. [Google Scholar] [CrossRef]
- Tu, L.; Liao, Z.; Luo, Z.; Wu, Y.L.; Herrmann, A.; Huo, S. Ultrasound-controlled drug release and drug activation for cancer therapy. In Exploration; Wiley Online Library: Hoboken, NJ, USA, 2021; Volume 1, p. 20210023. [Google Scholar]
- Manzano, M.; Vallet-Regí, M. Ultrasound responsive mesoporous silica nanoparticles for biomedical applications. Chem. Commun. 2019, 55, 2731–2740. [Google Scholar] [CrossRef] [Green Version]
- Vallet-Regí, M.; Colilla, M.; Izquierdo-Barba, I.; Manzano, M. Mesoporous silica nanoparticles for drug delivery: Current insights. Molecules 2017, 23, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paris, J.L.; de la Torre, P.; Cabañas, M.V.; Manzano, M.; Grau, M.; Flores, A.I.; Vallet-Regí, M. Vectorization of ultrasound-responsive nanoparticles in placental mesenchymal stem cells for cancer therapy. Nanoscale 2017, 4, 5528–5537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.A.; Chen, W.; Zhang, L.; Wu, H.H.; Zink, J.I. A responsive mesoporous silica nanoparticle platform for magnetic resonance imaging-guided high-intensity focused ultrasound-stimulated cargo delivery with controllable location, time, and dose. J. Am. Chem. Soc. 2019, 141, 17670–17684. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.U.; Ali, S.; Tariq, I.; Ali, M.Y.; Pinnapreddy, S.R.; Preis, E.; Wolk, C.; Harvey, R.D.; Hause, G.; Brubler, J.; et al. Ultrasound-responsive smart drug delivery system of lipid coated mesoporous silica nanoparticles. Pharmaceutics 2021, 13, 1396. [Google Scholar] [CrossRef]
- Li, T.; Shi, S.; Goel, S.; Shen, X.; Xie, X.; Chen, Z.; Zhang, H.; Li, S.; Qin, X.; Yang, H.; et al. Recent advancements in mesoporous silica nanoparticles towards therapeutic applications for cancer. Acta Biomater. 2019, 89, 1–13. [Google Scholar] [CrossRef]
- Huang, L.; Liu, J.; Gao, F.; Cheng, Q.; Lu, B.; Zheng, H.; Xu, H.; Xu, P.; Zhang, X.; Zeng, X. Dual-responsive, hyaluronic acid targeted drug delivery system based on hollow mesoporous silica nanoparticles for cancer therapy. J. Mater. Chem. B 2018, 6, 4618–4629. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.L.; Xiong, W.Y.; Ma, J.B.; Gao, T.F.; Peng, S.Y.; Xiao, W. Design of dual-responsive nanocarries with high drug loading capacity based on hollow mesoporous organosilica nanoparticles. Mater. Chem. Phys. 2019, 233, 230–235. [Google Scholar] [CrossRef]
- Song, Y.; Cai, L.; Tian, Z.; Wu, Y.; Chen, J. Phytochemical curcumin-coformulated, silver-decorated melanin-like polydopamine/mesoporous silica composites with improved antibacterial and chemotherapeutic effects against drug-resistant cancer cells. ACS Omega 2020, 5, 15083–15094. [Google Scholar] [CrossRef]
- Zhang, R.Q.; Liu, Z.Q.; Luo, Y.L.; Xu, F.; Chen, Y.S. Tri-stimuli responsive carbon nanotubes covered by mesoporous silica graft copolymer multifunctional materials for intracellular drug delivery. J. Ind. Eng. Chem. 2019, 80, 431–443. [Google Scholar] [CrossRef]
- Salve, R.; Kumar, P.; Ngamcherdtrakul, W.; Gajbhiye, V.; Yantasee, W. Stimuli-responsive mesoporous silica nanoparticles: A custom-tailored next generation approach in cargo delivery. Mater. Sci. Eng. C 2021, 124, 112084. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C. Donald Maxwell Parkin Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.; Mukherjee, T.K. Recent Progress in gold and silver nanoparticles biomedical attributes towards lung and breast cancer treatment. Int. J. Pharm. 2018, 553, 483–509. [Google Scholar] [CrossRef] [PubMed]
- Rizwanullah, M.; Ahmad, M.Z.; Ghoneim, M.M.; Alshehri, S.; Imam, S.S.; Md, S.; Alhakamy, N.A.; Jain, K.; Ahmad, J. Receptor-mediated targeted delivery of surface-modified nanomedicine in breast cancer: Recent update and challenges. Pharmaceutics 2021, 13, 2039. [Google Scholar] [CrossRef] [PubMed]
- Duo, Y.; Li, Y.; Chen, C.; Liu, B.; Wang, X.; Zeng, X.; Chen, H. DOX-loaded pH-sensitive mesoporous silica nanoparticles coated with PDA and PEG induce pro-death autophagy in breast cancer. RSC Adv. 2017, 7, 39641–39650. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Tambe, P.; Paknikar, K.M.; Gajbhiye, V. Folate/N-acetyl glucosamine conjugated mesoporous silica nanoparticles for targeting breast cancer cells: A comparative study. Colloids Surf. B 2017, 156, 203–212. [Google Scholar] [CrossRef]
- Liu, Z.; Tao, Z.; Zhang, Q.; Wan, S.; Zhang, F.; Zhang, Y.; Wu, G.; Wang, J. YSA-conjugated mesoporous silica nanoparticles effectively target EphA2-overexpressing breast cancer cells. Cancer Chemother. Pharmacol. 2018, 81, 687–695. [Google Scholar] [CrossRef]
- Li, N.; Wang, Z.; Zhang, Y.; Zhang, K.; Xie, J.; Liu, Y.; Li, W.; Feng, N. Curcumin-loaded redox-responsive mesoporous silica nanoparticles for targeted breast cancer therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, 921–935. [Google Scholar] [CrossRef] [Green Version]
- Portilho, F.L.; Pinto, S.R.; da Silva de Barros, A.O.; Neto, E.H.; dos Santos, S.N.; Bernardes, E.S.; Ozdemir, D.I.; Asikoglu, M.; Magalhães, L.; Alencar, R.; et al. In loco retention effect of magnetic core mesoporous silica nanoparticles doped with trastuzumab as intralesional nanodrug for breast cancer. Artif. Cells Nanomed. Biotechnol. 2018, 46, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Sengar, P.; Juárez, P.; Meza, A.V.; Arellano, D.L.; Jain, A.; Chauhan, K.; Hirata, G.A.; Fournier, P.G.J. Development of a functionalized UV-emitting nanocomposite for the treatment of cancer using indirect photodynamic therapy. J. Nanobiotechnol. 2018, 16, 1–19. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hayama, K.; Sasagawa, I.; Okada, Y.; Kawase, T.; Tsubokawa, N.; Tsuchimochi, M. HER2-targeted multifunctional silica nanoparticles specifically enhance the radiosensitivity of HER2-overexpressing breast cancer cells. Int. J. Mol. Sci. 2018, 19, 908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmani, S.; Budimir, J.; Sejalon, M.; Daurat, M.; Aggad, D.; Vives, E.; Raehm, L.; Garcia, M.; Lichon, L.; Bobo, M.G.; et al. Large pore mesoporous silica and organosilica nanoparticles for pepstatin a delivery in breast cancer cells. Molecules 2019, 24, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tambe, P.; Kumar, P.; Paknikar, K.M.; Gajbhiye, V. Decapeptide functionalized targeted mesoporous silica nanoparticles with doxorubicin exhibit enhanced apoptotic effect in breast and prostate cancer cells. Int. J. Nanomed. 2018, 13, 7669–7680. [Google Scholar] [CrossRef] [Green Version]
- Kundu, M.; Chatterjee, S.; Ghosh, N.; Manna, P.; Das, J.; Sil, P.C. Tumor targeted delivery of umbelliferone via a smart mesoporous silica nanoparticles controlled-release drug delivery system for increased anticancer efficiency. Mater. Sci. Eng. C 2018, 116, 111239. [Google Scholar] [CrossRef]
- Pallares, R.M.; Agbo, P.; Liu, X.; An, D.; Gauny, S.S.; Zeltmann, S.; Minor, A.M.; Abergel, R.J. Engineering mesoporous silica nanoparticles for targeted alpha therapy against breast cancer. ACS Appl. Mater. Interface 2020, 36, 40078–40084. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Zhang, F.; Chen, F.; Zheng, X.; Hu, H.; Yang, C.; Tu, Z.; Wang, Z.; Chang, Z.; Lu, J.; et al. Biomimetic diselenide-bridged mesoporous organosilica nanoparticles as an x-ray-responsive biodegradable carrier for chemo-immunotherapy. Adv. Mater. 2020, 32, 2004385. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; You, Y.; Li, X.; Liu, L.; Guo, F.; Zhang, Q.; Liu, D.; Tong, Y.; Ding, S.; Wang, J. Preparation of RGD peptide/folate acid double-targeted mesoporous silica nanoparticles and its application in human breast cancer MCF-7 cells. Front. Pharmacol. 2020, 11, 898. [Google Scholar] [CrossRef]
- Day, C.M.; Sweetman, M.J.; Hickey, S.M.; Song, Y.; Liu, Y.; Zhang, N.; Plush, S.E.; Garg, S. Concept Design, development and preliminary physical and chemical characterization of tamoxifen-guided-mesoporous silica nanoparticles. Molecules 2021, 26, 219. [Google Scholar] [CrossRef]
- Murugan, B.; Krishnan, U.M. Differently sized drug-loaded mesoporous silica nanoparticles elicit differential gene expression in MCF-7 cancer cells. Nanomedicine 2021, 16, 1017–1034. [Google Scholar] [CrossRef]
- Nia, M.H.; Koshani, R.; Munguia-Lopez, J.G.; Kiasat, A.R.; Kinsella, J.M.; van de Ven, T.G.M. Biotemplated hollow mesoporous silica particles as efficient carriers for drug delivery. ACS Appl. Biomater. 2021, 4, 4201–4214. [Google Scholar]
- Porrang, S.; Rahemi, N.; Davaran, S.; Mahdavi, M.; Hassanzadeh, B. Preparation and in-vitro evaluation of mesoporous biogenic silica nanoparticles obtained from rice and wheat husk as a biocompatible carrier for anti-cancer drug delivery. Eur. J. Pharm. Sci. 2021, 163, 105866. [Google Scholar] [CrossRef] [PubMed]
- Laranjeira, M.S.; Ribeiro, T.P.; Magalhães, A.I.; Silva, P.C.; Santos, J.A.M.; Monteiro, F.J. Magnetic mesoporous silica nanoparticles as a theranostic approach for breast cancer: Loading and release of the poorly soluble drug exemestane. Int. J. Pharm. 2022, 619, 121711. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Kundu, M.; Dutta, S.; Mahalanobish, S.; Ghosh, N.; Das, J.; Sil, P.C. Enhancement of anti-neoplastic effects of cuminaldehyde against breast cancer via mesoporous silica nanoparticle based targeted drug delivery system. Life Sci. 2022, 298, 120525. [Google Scholar] [CrossRef] [PubMed]
- Parnian, J.; Ma’mani, L.; Bakhtiari, M.R.; Safavi, M. Overcoming the non-kinetic activity of EGFR1 using multi-functionalized mesoporous silica nanocarrier for in vitro delivery of siRNA. Sci. Rep. 2022, 12, 17208. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Choi, M.; Kim, H.E.; Jin, M.; Jeon, W.J.; Jung, M.; Yoo, H.; Won, J.H.; Na, Y.G.; Lee, J.Y.; et al. Mannosylatedpoly(acrylic acid)-coated mesoporous silica nanoparticles for anticancer therapy. J. Control. Release 2022, 349, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Lohiya, G.; Katti, D.S. Carboxylated chitosan-mediated improved efficacy of mesoporous silica nanoparticle-based targeted drug delivery system for breast cancer therapy. Carbohydr. Polym. 2022, 277, 118822. [Google Scholar] [CrossRef]
- Chang, J.; Mo, L.; Song, J.; Wang, X.; Liu, H.; Meng, C.; Wu, Y. A pH-responsive mesoporous silica nanoparticle based drug delivery system for targeted breast cancer therapy. J. Mater. Chem. B 2022, 10, 3375–3385. [Google Scholar] [CrossRef]
- Ren, W.; Iqbal, M.Z.; Zeng, L.; Chen, T.; Pan, Y.; Zhao, J.; Yin, H.; Zhang, L.; Zhang, J.; Li, A.; et al. Black TiO2 based core–shell nanocomposites as doxorubicin carriers for thermal imaging guided synergistic therapy of breast cancer. Nanoscale 2017, 9, 11195–11204. [Google Scholar] [CrossRef]
- Shen, Y.; Li, M.; Liu, T.; Liu, J.; Xie, Y.; Zhang, J.; Xu, S.; Liu, H. A dual-functional HER2 aptamer-conjugated, pH-activated mesoporous silica nanocarrier-based drug delivery system provides in vitro synergistic cytotoxicity in HER2-positive breast cancer cells. Int. J. Nanomed. 2019, 14, 4029–4044. [Google Scholar] [CrossRef] [Green Version]
- Ali, O.M.; Bekhit, A.A.; Khattab, S.N.; Helmy, M.W.; Abdel-Ghany, Y.S.; Teleb, M.; Elzoghby, A.O. Synthesis of lactoferrin mesoporous silica nanoparticles for pemetrexed/ellagic acid synergistic breast cancer therapy. Colloids Surf. B 2020, 188, 110824. [Google Scholar] [CrossRef]
- Xu, P.; Yao, J.; Li, Z.; Wang, M.; Zhou, L.; Zhong, G.; Zheng, Y.; Li, N.; Zhai, Z.; Yang, S.; et al. Therapeutic effect of doxorubicin-chlorin E6-loaded mesoporous silica nanoparticles combined with ultrasound on triple-negative breast cancer. Int. J. Nanomed. 2020, 15, 2659–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moodley, T.; Singh, M. Polymeric mesoporous silica nanoparticles for combination drug delivery in vitro. Biointerface Res. Appl. Chem. 2021, 11, 11905–11919. [Google Scholar]
- Zhuang, J.; Chen, S.; Hu, Y.; Yang, F.; Huo, Q.; Xie, N. Tumour-targeted and redox-responsive mesoporous silica nanoparticles for controlled release of doxorubicin and an siRNA against metastatic breast cancer. Int. J. Nanomed. 2021, 16, 1961–1976. [Google Scholar] [CrossRef] [PubMed]
- Lohiya, G.; Katti, D.S. Mesoporous silica nanoparticle-based combination of niclosamide and doxorubicin: Effect of treatment regimens on breast cancer subtypes. ACS Appl. Bio Mater. 2021, 4, 7811–7824. [Google Scholar] [CrossRef]
- Viswanathan, T.M.; Krishnakumar, V.; Senthilkumar, D.; Chitradevi, K.; Vijayabhaskar, R.; Kannan, V.R.; Kumar, N.S.; Sundar, K.; Kunjiappan, S.; Babkiewicz, E.; et al. Combinatorial delivery of Gallium (III) nitrate and curcumin complex-loaded hollow mesoporous silica nanoparticles for breast cancer treatment. Nanomaterials 2022, 12, 1472. [Google Scholar] [CrossRef]
- Maktedar, S.; Malik, P.; Avashthi, G.; Singh, M. Dispersion enhancing effect of sonochemically functionalized graphene oxide for catalysing antioxidant efficacy of curcumin. Ultrason. Sonochem. 2017, 39, 208–217. [Google Scholar] [CrossRef]
- Malik, P.; Maktedar, S.; Avashthi, G.; Mukherjee, T.K.; Singh, M. Robust curcumin-mustard oil emulsions for pro to anti-oxidant modulation of graphene oxide. Arab. J. Chem. 2020, 13, 4606–4628. [Google Scholar] [CrossRef]
- Malik, P.; Singh, M.; Inwati, G.; Mukherjee, T.K.; Singh, S. Green silver nanoparticle and Tween-20 modulated pro-oxidant to antioxidant curcumin transformation in aqueous CTAB stabilized peanut oil emulsions. J. Mol. Liq. 2019, 291, 111252. [Google Scholar] [CrossRef]
- Malik, P.; Mukherjee, T.K. Structure-Function elucidation of antioxidative and prooxidative activities of the polyphenolic compound curcumin. Chin. J. Biol. 2014, 2014, 396708. [Google Scholar] [CrossRef] [Green Version]
- Malik, P.; Hoidal, J.R.; Mukherjee, T.K. Recent advances in curcumin treated non-small cell lung cancers: An impetus of pleiotropic traits and nanocarrier aided delivery. Curr. Med. Chem. 2020, 27, 1–45. [Google Scholar] [CrossRef]
- Predarska, I.; Saoud, M.; Drača, D.; Morgan, I.; Komazec, T.; Eichhorn, T.; Mihajlovic, E.; Dunderovic, D.; Mijatovic, S.; Maksimovic´-Ivanic, D.; et al. Mesoporous silica nanoparticles enhance the anticancer efficacy of platinum(iv)-phenolate conjugates in breast cancer cell lines. Nanomaterials 2022, 12, 3767. [Google Scholar] [CrossRef] [PubMed]
- Ramezani-Aliakbari, M.; Varshosaz, J.; Mirian, M.; Khodarahmi, G.; Rostami, M. pH-responsive glucosamine anchored polydopamine coated mesoporous silica nanoparticles for delivery of Anderson-type polyoxomolybdate in breast cancer. J. Microencapsul. 2022, 39, 433–451. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhang, W.; Song, P.; Li, W.; Chen, X.; Ge, F.; Gui, L.; Yang, K.; Tao, Y.; Guocheng, D. Redox-responsive mesoporous silica nanoparticles for chemophotodynamic combination cancer therapy. Mater. Res. Express 2022, 9, 45401. [Google Scholar] [CrossRef]
- Lohiya, G.; Katti, D.S. A synergistic combination of niclosamide and doxorubicin as an efficacious therapy for all clinical subtypes of breast cancer. Cancers 2021, 13, 3299. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, Y.; Kilchrist, K.V.; Li, J.; Duvall, C.L.; Oupický, D. Endosomolytic and tumor-penetrating mesoporous silica nanoparticles for siRNA/miRNA combination cancer therapy. ACS Appl. Mater. Interfaces 2020, 12, 4308–4322. [Google Scholar] [CrossRef] [PubMed]
- Manjusha, V.; Reshma, L.R.; Anirudhan, T.S. Mesoporous silica gated mixed micelle for the targeted co-delivery of doxorubicin and paclitaxel. J. Drug Deliv. Sci. Technol. 2022, 26, 104032. [Google Scholar] [CrossRef]
- Karaman, D.S.; Kaasalainen, M.; Kettiger, H.; Rosenholm, J.M. Opportunities and challenges of silicon-based nanoparticles for drug delivery and imaging. In Characterization of Pharmaceutical Nano- and Microsystems, 1st ed.; Peltonen, L., Ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2021; Chapter 9. [Google Scholar]
- Sha, X.; Dai, Y.; Song, X.; Liu, S.; Zhang, S.; Li, J. The opportunities and challenges of silica nanomaterial for atherosclerosis. Int. J. Nanomed. 2021, 16, 701–714. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
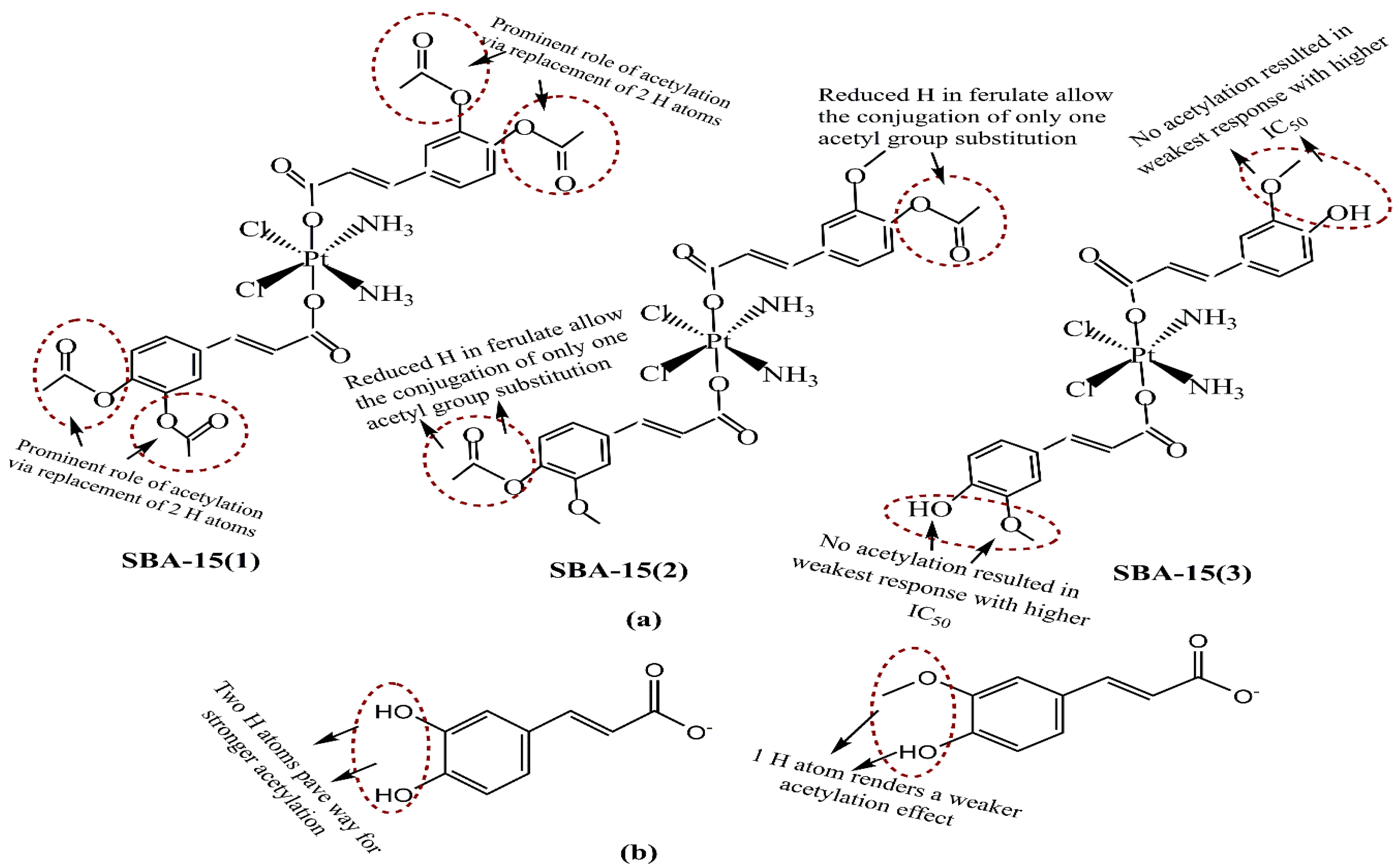{kind=link}
{kind=link}
| Name of Receptor | Physiological Distinctions | Potential in Breast Cancer Treatment |
|---|---|---|
| ErbB tyrosine kinase receptors | Widely investigated growth factor receptors in BC that consist of four homolog receptors: ErbB1 (HER1/EGFR), ErbB2 (HER2/neu), ErbB3 (HER3) and ErbB4 (HER4). EGFR and HER2 are overexpressed in 15–20% and 20–25% of BC cells. The BC cells overexpressing ErbB receptors demonstrate aggressive clinical behavior. | Enhanced cytotoxicity of CURC delivered via EGFR targeting GE11 peptide-conjugated CURC-loaded PLGA-PEG NPs (in MCF-7 cells). HER2 Ab-conjugated PTX and rapamycin-loaded glycerol monooleate-coated magnetic NPs exhibited enhanced uptake in MCF-BC cells with 24-fold reduced toxicity and 3-fold lower IC50 extent (for PTX). For rapamycin, 71-fold reduced IC50 than anative drug with targeted magnetic NPs. EGFR-conjugated immuno NPs exhibited ~13-fold higher uptake and antiproliferative activity than unconjugated NPs in MCF-7 BC cells. HER2-targeted PLGA/montmorillonite-trastuzumab NPs exhibited 12.74 and 13.11-fold larger therapeutic efficacy than untargeted NPs. |
| Folate receptor | Binds to water-soluble, low-molecular-weight FA (vitamin B9), essential in normal mitotic cell division;, binds to the folate ligand and then internalizes the complexes via receptor-mediated endocytosis. | Studied for high anticancer efficacy of PTX (in 4T1 BC cells), cisplatin (DDP) and docetaxel (in MDA-MB-231 BC cells), CURC (in MCF-7 cells), enhanced cellular uptake for deoxycholic acid-O-carboxymethylated CTS and vincristine sulfate-loaded PLGA-PEG NPs |
| Estrogen receptors | Belong to thenuclear hormone receptor superfamily, differentially overexpressed in 60–80% of BC cells, internalized in the cancer cells upon binding to ERs | Enhanced cellular uptake of PEG-conjugated PTX-epirubicin co-loaded liposomal NPs, TAM surface-grafted liposomes loaded with DOX (in MCF-7 cells), DOX and DOX + TAM-loaded liposomes (MCF-7 cells) |
| CD44/Hyaluronan receptor | A natural component of ECM, hyaluronan is critically involved in cell proliferation, migration and invasion. CD44 regulates lymphocyte adhesion to endothelial cells during lymphocyte migration (a process equivalent to metastasis in solid tumors). CD44 prevails as an HA receptorandprevails sparsely on the surface of epithelial, hematopoietic and neuronal cells. Recent studies have screened CD44 as a BC stem cell marker. | HA-functionalized CTS lipoic acid NPs loaded with 17α-methyltestosterone exhibited enhanced internalization in CD44 overexpressing BT-20 BC cells than CD44-negative MCF-7 cells. Enhanced uptake and cytotoxicity of PLGA NPs in MDA-MB-231 cells. Higher docetaxel uptake for self-assembled PLGA-HA block copolymers (in MDA-MB-231 cells) than untargeted NPs. HA matrix NPs with intrinsic CD-44 tropism and loaded rapamycin exhibited a 3.2-fold enhanced uptake in CD44-positive MDA-MB-468 cells. HA-lysine-lipoic acid NPs loaded with DOX exhibited a 20-fold enhanced uptake in MCF-7/ADR cells. |
| Luteinizing hormone-releasing hormone receptor (LHRH) | A hormonal decapeptide produced by the hypothalamus, LHRH is also known as a gonadotropin-releasing hormone. Plays a key role in regulating the pituitary–gonadal axis and reproduction. Overexpressed in 50% of BC cells. | LHRH-conjugated PTX showed enhanced specificity for targeting MDA-MB-231 cells. LHRH-CTS-conjugated NPs enhanced cellular uptake with two-fold reduced IC50in LHR overexpressing MCF-7 cells than non-targeted NPs. DDP-loaded LHRH-modified dextran NPs exhibited a higher uptake with reduced nephrotoxicity of DDP than non-targeted NPs (in 4T1 BC cells). |
| Chosen Drug(s), Examined Cell Line, Animal Model | Functionalizing Moiety on MSNPs, Prominent Physicochemicaldistinctions and Other Observations | Outcomes of Therapeutic Efficacy |
|---|---|---|
| Studies in singular mode | ||
| Anderson-type manganese polyoxomolybdate (POMo), MDA-MB-231 cells | PDA coating and glucosamine anchoring, Hydrodynamic size (HS): 195 nm, ζ-potential: −18.9 mV, 45% loading | pH-sensitive release profile enhanced anticancer activity compared to the free POMo with the highest cellular uptake and apoptotic extents |
| DOX, 4T1 murine mammary carcinoma cells, and BALB/c mice implanted with 4T1 tumors, (1–20) μg·mL−1 for 6, 12 and 24 h | Disulfide bonds (SS) as redox-sensitive linkers and HA as capping and targeting agents, (242–250) nm hydrodynamic diameter, <0.09 as PDI over 7 days, glutathione (10 mM, redox sensitivity) and hyaluronidase (150 U·mL−1, enzyme sensitivity) presence catalyzed a 58.39% DOX release at <7 pH | Higher uptake and lower IC50 of NC delivered DOX in 4T1 (0.39 μg·mL−1) than 293T cells (2.83 μg·mL−1). SS and HA coating revealed 1.45 (6 h), 1.04 (12 h) and 2.65 (24 h)-fold higher fluorescence than uncoated and non-SS hollow MSNPs |
| Combinatorial delivery mode | ||
| DOX and chlorine Ce6 (Ce6) co-loaded into the pores of -SS-surface conjugated MSNPs. The final coating withcarboxymethylCTS gave DOX/Ce6@MSC NCs, 20 mg DOX with 20 mg Ce6 (1.1:1) administered to MCF-7 human BC cells, monitored for 2, 6 and 12 h for uptake studies | Average PS of MSNPs was 158.07 nm, while for DOX/Ce6@MSC NCs, the ζ-potential changes from −37 mV (MSNPs) to 19.1 mv (-NH2-functionalized state) and again −32.3 mV for DOX + Ce6-loaded state, implyinga modification, high SA (799.12 m2·g−1) and PV (0.27 cm3·g−1) for MSNPs were reduced to 91.72 m2·g−1and 0.09 cm3·g−1 on DOX + Ce6 loading. H&E staining showed no major tissue damage in healthy mice until 30 days, non-toxicity of NC | Reduced viability for DOX + Ce6-loaded MSNPs with laser irradiation than DOX alone, DOX + Ce6 delivered via SS-COOH-functionalized and neat NCs. 58.32% survival for DOX + Ce6 delivery but for laser combination, it was only 39.59%. ROS generation was showedby the bright green fluorescence andflow cytometry studies. Successful replication in subcutaneous mice xenografts screened via the highest TV reduction |
| ATO andPTX, MCF-7 cells, mice bearing MCF-7-derived tumors, 1 mg·kg−1 ATO and 0.5 mg·kg−1 PTX were administered to MCF-7 tumor-bearing mice, via tail vein every 2 days. Effects were monitored viaTV and body weight measurement on alternate days untilthe 24-day treatment | Coating with PAA and pH-sensitive lipids, tumor-targeting peptide F56, spherical morphology and 117.7 ± 1.51 nm PS (via TEM), which increased to 124.3 ± 1.53 nm for –NH2 functionalization, which also increased ζ-potential from −30.3 ± 1.15 to 37.3 ± 1.16 mV.3 nm pore size, 1.088 cm3·g−1, PV and 1203.453 m2·g−1 SA, reduced on –NH2 functionalization and PAA grafting, 76% ATO and 97% PTX release at <5 pH | 1:1, 1:2 and 2:1 PTX + ATO exhibited synergism with <1 CI, 29.5% and 29.9% cell cycle arrest at G2/M phase, 21.45 and 61.48% post 24 h apoptosis for 1:2 and 2:1 proportions, significantly higher than PTX (7.41%) andATO (7.41%), greater Bcl-2inhibition, caspase-7, caspase-9, cyclin B-1 and cyclin D-1 involvement, insignificant effects on singular treatment |
| Niclosamide (NIC) (Wntsignalinginhibitor) andDOX. Efficacy screened in MDA-MB-231, SKBR3 and MCF-7 BC cells. NIC: (0–128) μM, DOX: (0–64) μM, monitored for 24 h | The combination resulted in synergistically enhanced cell death, both in sequential andconcurrent treatment regimes, feasible for all clinical BC subtypes. Suppressed Wnt/β-catenin signaling with Go/G1 cell cycle arrest (by NIC) and aggravated ROS (both NIC and DOX) alongside the native DOX toxicity, contributed to synergistic response for both treatment regimes | For MDA-MB-231 cells:Of the 56 concurrent combinatorial stoichiometries, 47 caused >50% cell death. All DOX extents with intermediate to high NIC caused >70% cytotoxicity. Of the 47 combinations, 33 were highly synergistic (CI < 0.5). For SKBR3 and MCF-7 cells: All NIC dosages with intermediate to high (SKBR3), and high (MCF-7) numbers of cells were effective. 28 combinations screened in SKBR3 cells, with 15 synergistic outcomes. 6 of 49 combinations in MCF-7 cells revealed a synergism with 19–36% cell death. |
| siRNA (siPlk1) and miRNA (miR-200c) simultaneous delivery via encapsulated indocyanine green (ICG) to enable stealth trafficking alongside surface conjugation of iRGD peptide MDA-MB-231 TNBC cells siRNA andmiRNA: 100–200 nM, ICG: 0.96–1.92 μg·mL−1 monitored until 48 h | 91% EE for ICG, variations in ζ-potential (34.3 mV for –NH2-functionalized MSNPs, which decreased to 21.6 mV on loading of RNA and ICG cargos that further decreased (due to phospholipids and PEGylated lipids) to −19 mV implied successful siRNA + miRNA loading and lipidic surface stabilization | The photodynamic effect of ICG generated ROS and disrupted the endolysosomal membranes, succeeded by the liberation of entrapped therapeutic RNAs. iRGD conjugation augmented the infiltration of encapsulated siRNA + miRNA cargos. MSNPs loaded with Plk1 (siRNA), 200c(miRNA) and ICG combined with light irradiation suppressed the tumor growth to maximum, reducing the lung metastasis by 40% compared to 100% of all other combinations |
| Combining PTX-loaded mixed micelles (as gating agents) with DOX-loaded MSNPs viapH-sensitive Schiff base | Argine-glycine-aspartic acid (RGD) peptide was used as a targeting agent, with 81% PTX and 65% DOX release on 72 h monitoring | Though apoptotic induction was screened exact quantitative aspects could not be ascertained due to recent online availability. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rani, R.; Malik, P.; Dhania, S.; Mukherjee, T.K. Recent Advances in Mesoporous Silica Nanoparticle-Mediated Drug Delivery for Breast Cancer Treatment. Pharmaceutics 2023, 15, 227. https://doi.org/10.3390/pharmaceutics15010227
Rani R, Malik P, Dhania S, Mukherjee TK. Recent Advances in Mesoporous Silica Nanoparticle-Mediated Drug Delivery for Breast Cancer Treatment. Pharmaceutics. 2023; 15(1):227. https://doi.org/10.3390/pharmaceutics15010227
Chicago/Turabian StyleRani, Ruma, Parth Malik, Sunena Dhania, and Tapan Kumar Mukherjee. 2023. "Recent Advances in Mesoporous Silica Nanoparticle-Mediated Drug Delivery for Breast Cancer Treatment" Pharmaceutics 15, no. 1: 227. https://doi.org/10.3390/pharmaceutics15010227