

Transethosomal Gel for the Topical Delivery of Celecoxib: Formulation and Estimation of Skin Cancer Progression

 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Formulation of CXB-Loaded Ethosomes (ES) and Transethsomes (TES)
2.3. Characterization of CXB-ES and CXB-TES
2.3.1. Vesicle Size and Surface Charge Measurements
2.3.2. Entrapment efficiency
2.3.3. Evaluation of CXB-Lipid Interaction Using the Differential Scanning Calorimetry (DSC)
2.3.4. Fourier Transform-Infra-Red Spectroscopy (FTIR)
2.3.5. Morphology
2.4. Preparation of CXB-Loaded Transethosomal (TES) Hydrogel
2.5. Evaluation of CXB-Loaded Transethosomal Hydrogel
Entrapment Efficiency (EE%)
2.6. In Vitro Drug Release Study
2.7. Ex Vivo Permeation Study
2.8. In Vitro Proliferation Study
2.9. Gene Expression Analysis by Quantitative RT-PCR
2.9.1. RNA Isolation and Reverse Transcription (RT) Reaction
2.9.2. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.10. DNA Damage in Normal and Cancer Skin Cell Lines Using the Comet Assay
2.11. DNA Fragmentation Assay
2.12. Statistical Analysis
3. Results and Discussion
3.1. Characterization of CXB-ES and CXB-TES
3.2. Entrapment Efficiency (EE%)
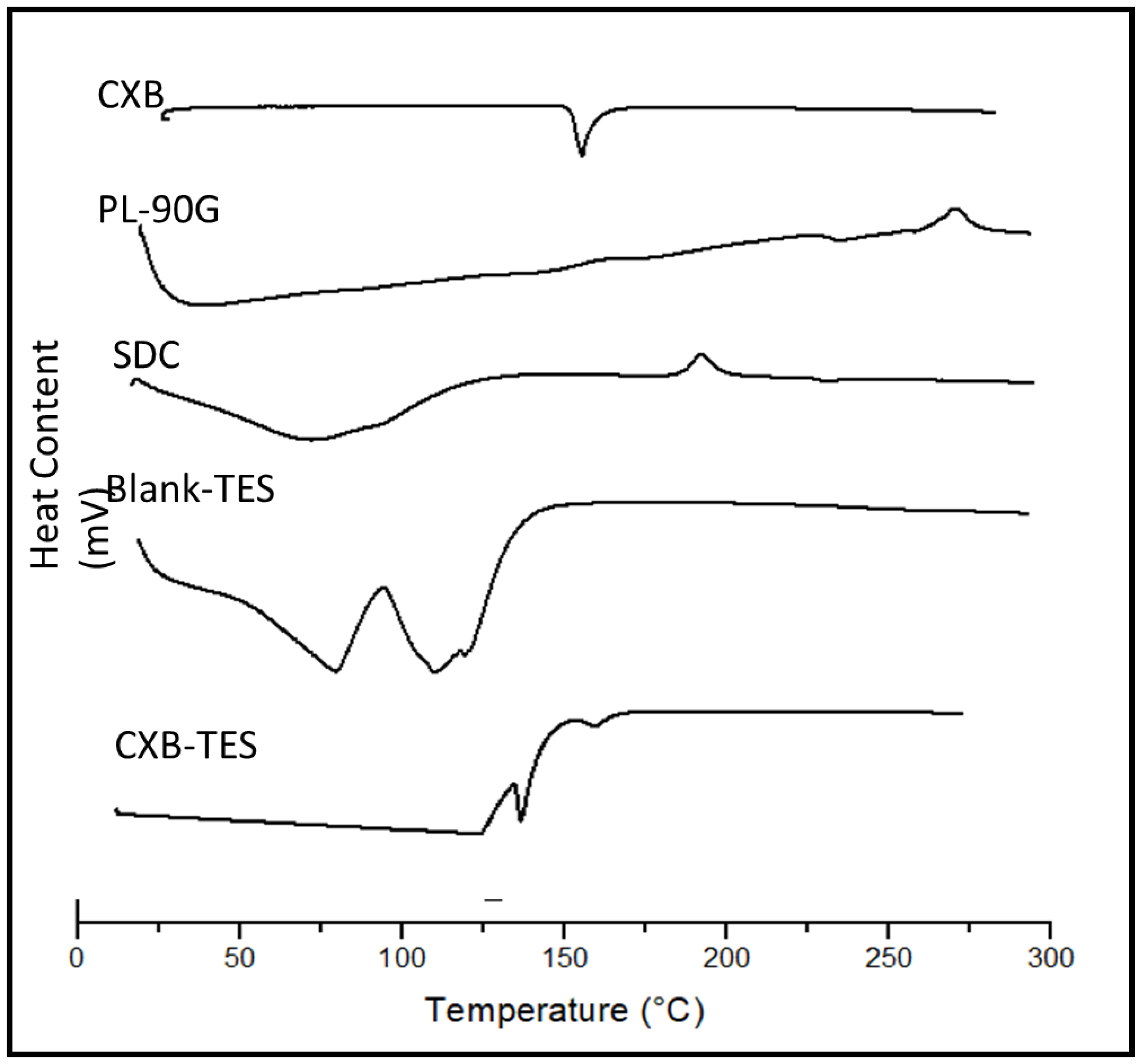
3.3. DSC
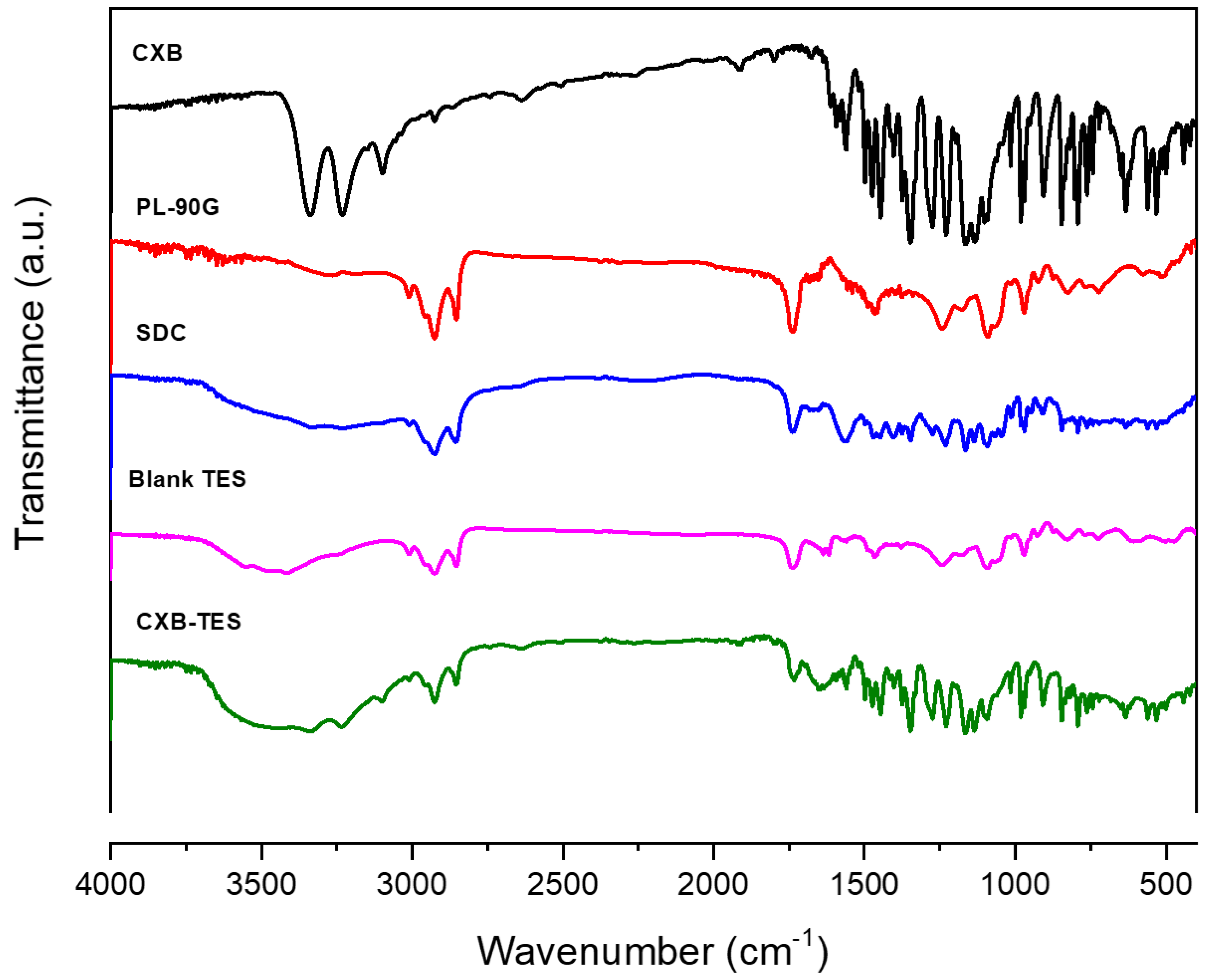
3.4. FT-IR
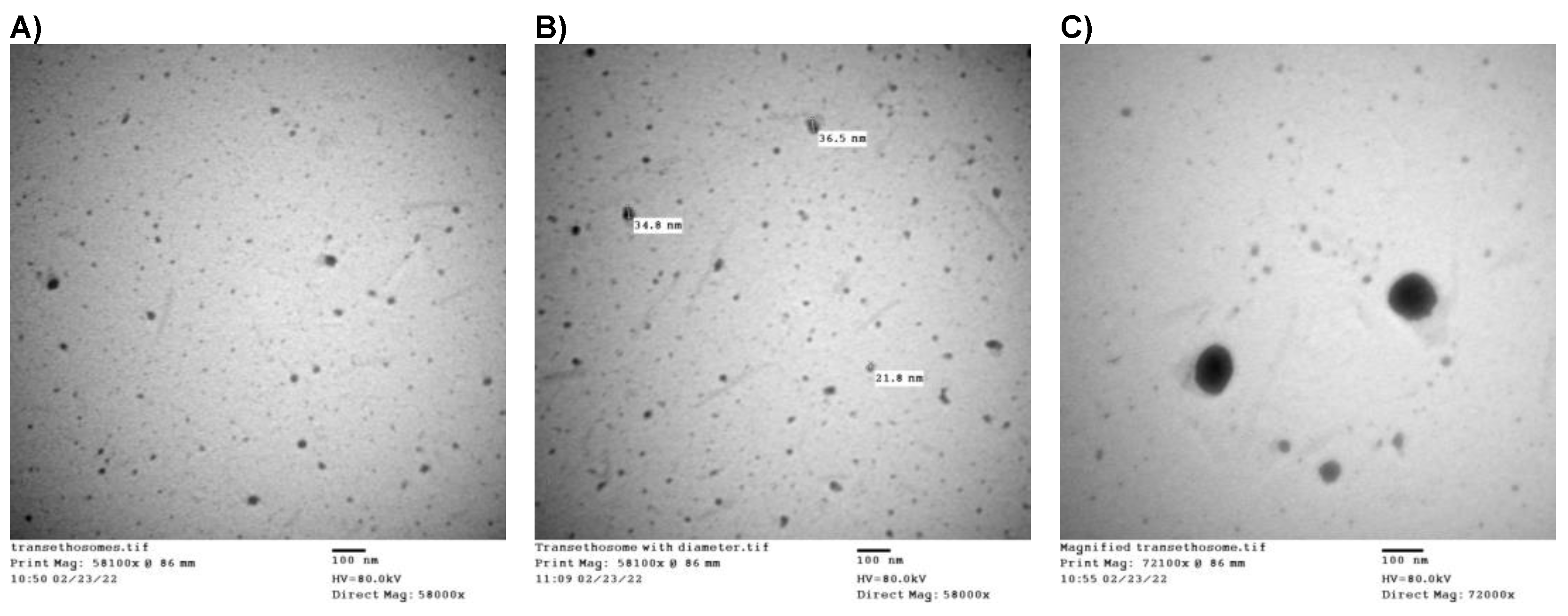
3.5. Morphological Analysis
3.6. Characterization of CXB-Transethosomal Hydrogel
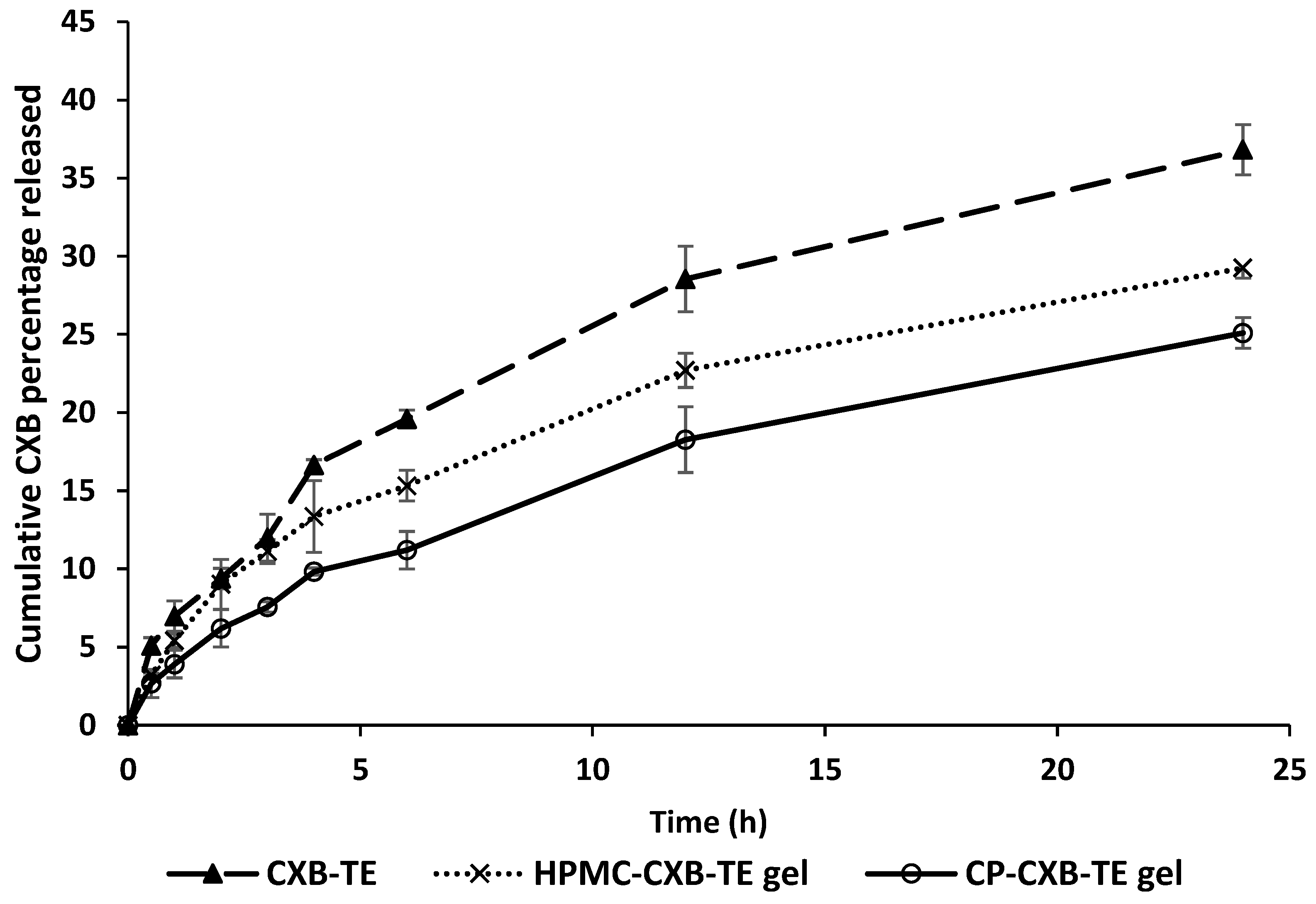
3.7. In Vitro Drug Release
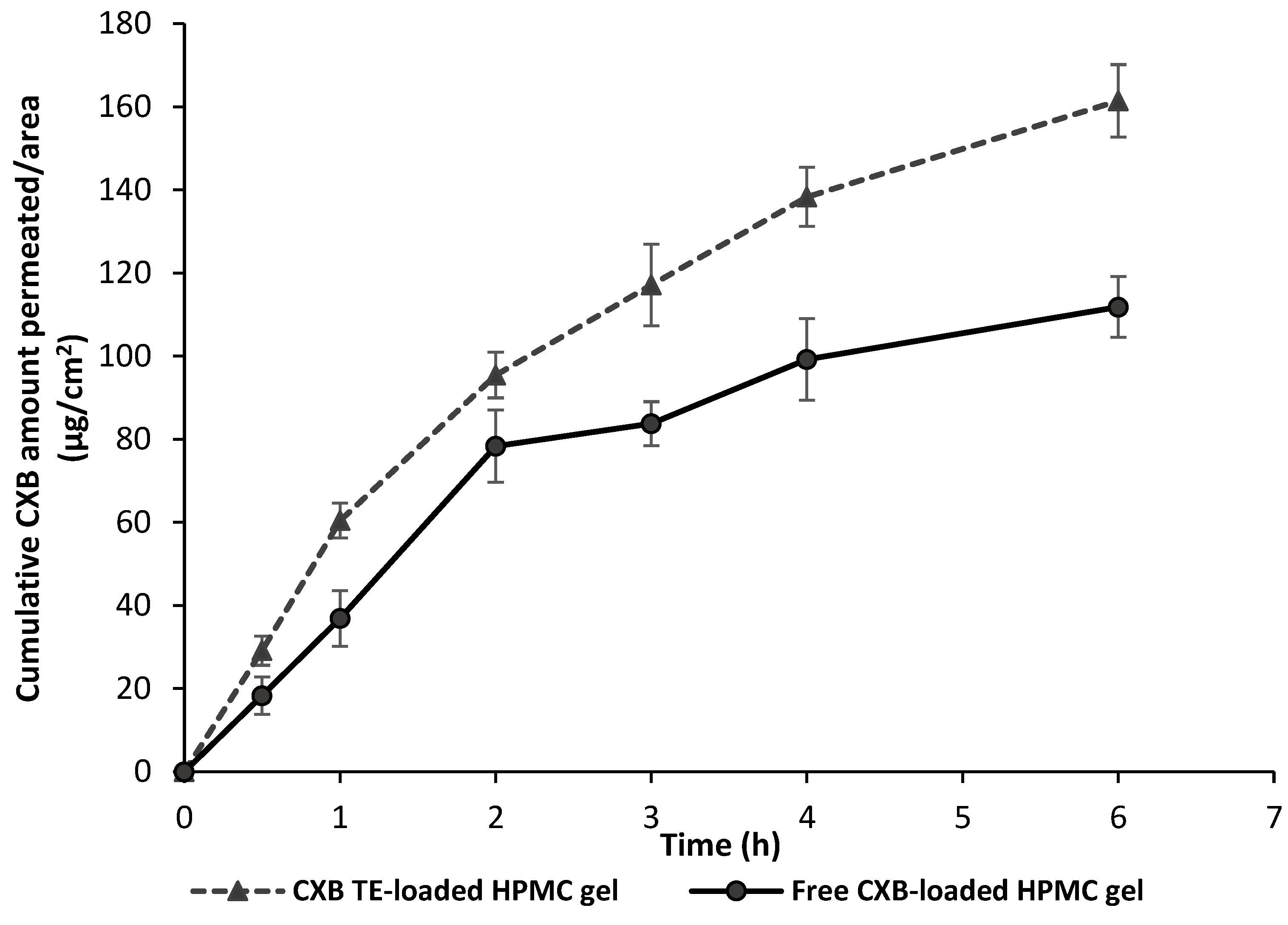
3.8. Ex Vivo Permeation Study
3.9. In Vitro Proliferation Study
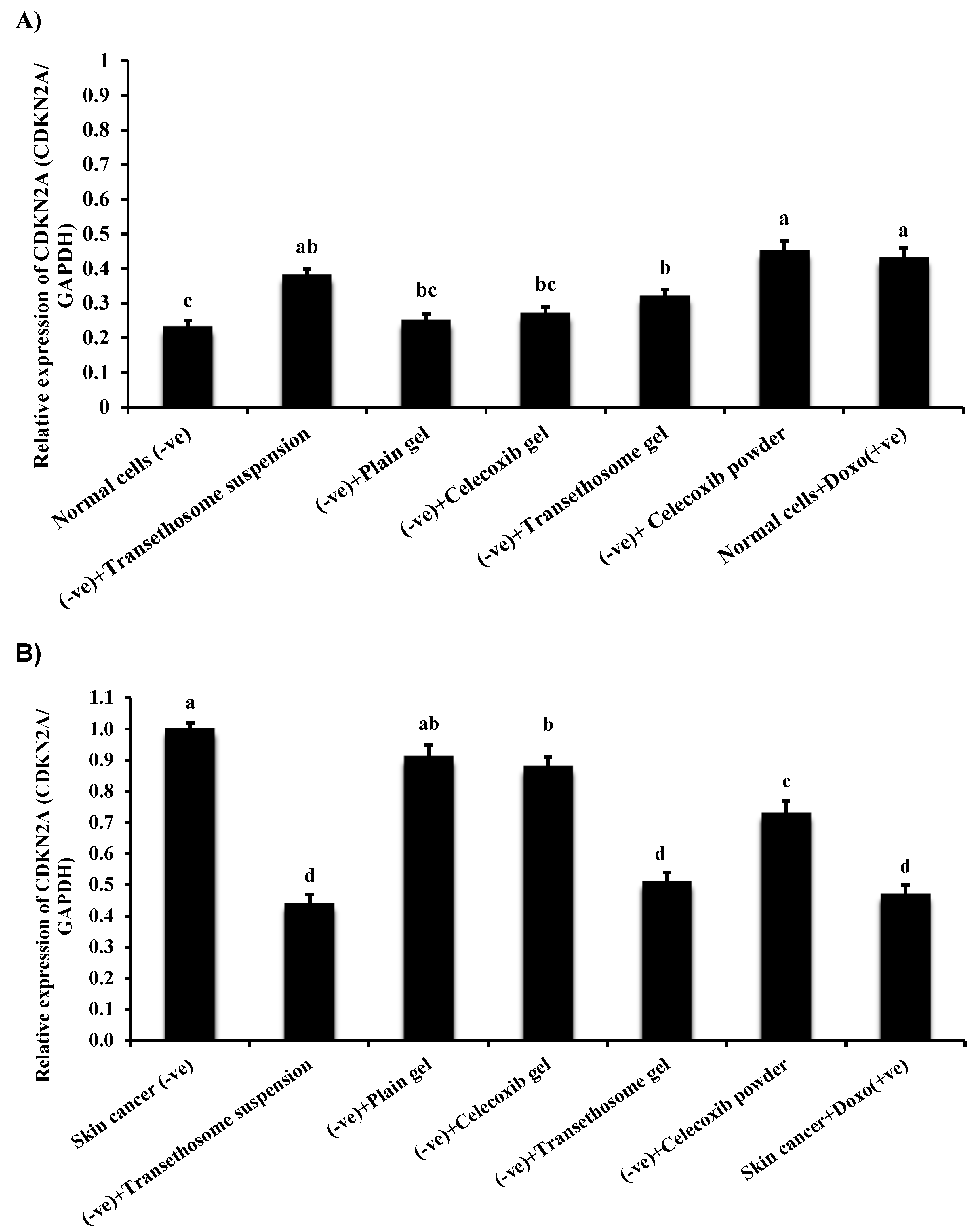
3.10. Gene Expression in Normal and Cancer Skin Cell Lines
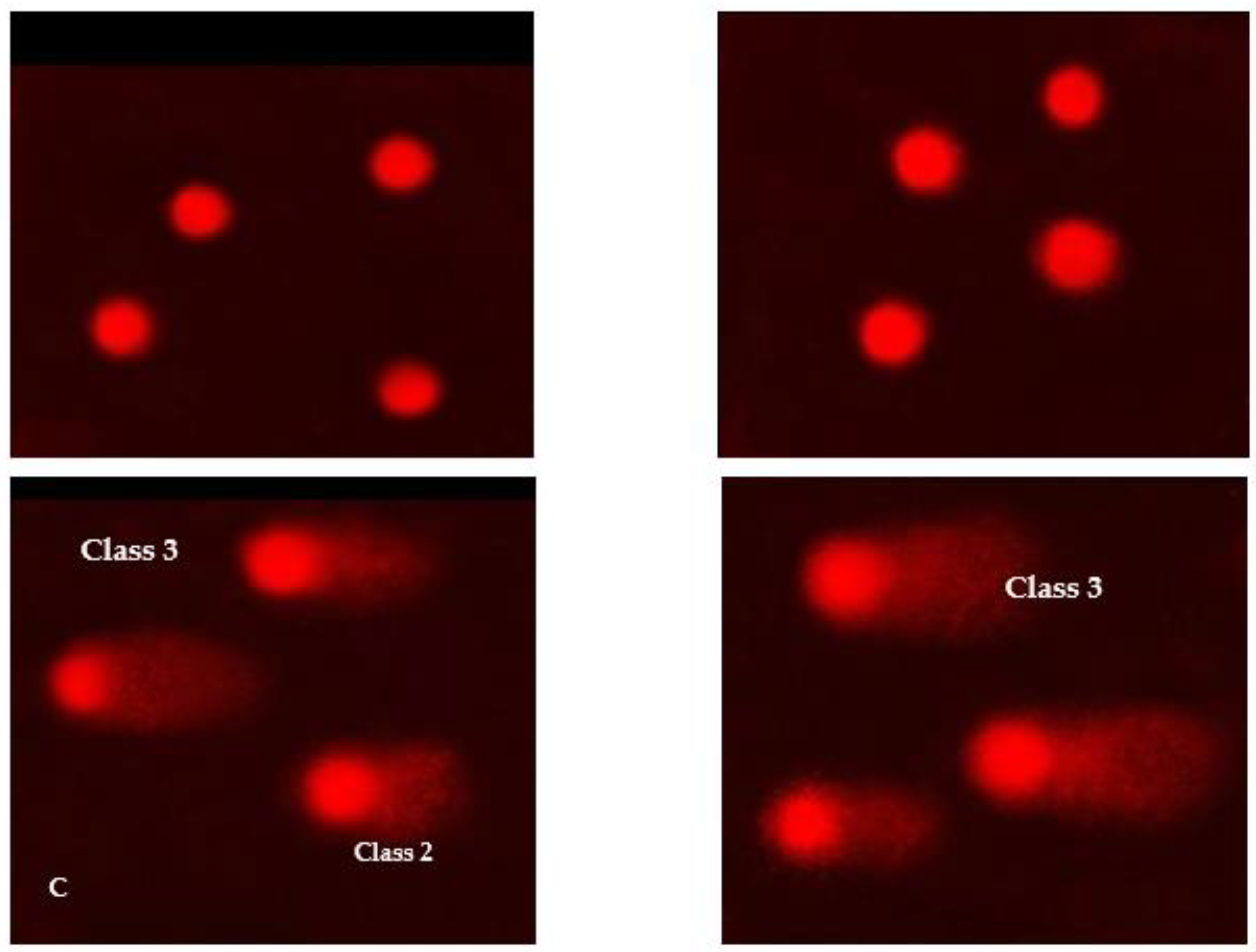
3.11. DNA Damage in Skin Cell Lines
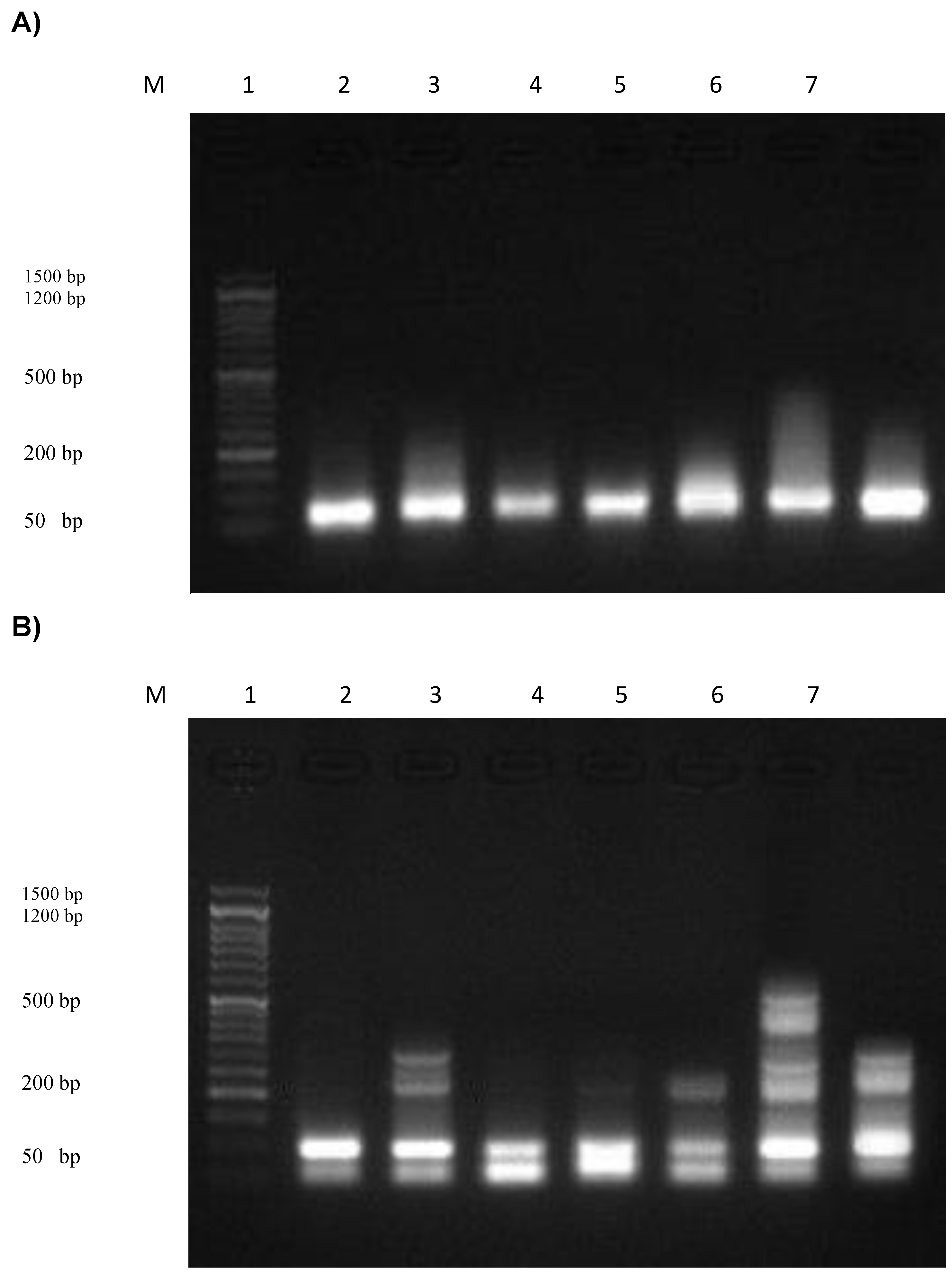
3.12. Assessment of the DNA Fragmentation in Normal and Cancer Skin Cell Lines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simões, M.F.; Sousa, J.S.; Pais, A.C. Skin cancer and new treatment perspectives: A review. Cancer Lett. 2015, 357, 8–42. [Google Scholar] [CrossRef]
- Gan, B.K.; Yong, C.Y.; Ho, K.L.; Omar, A.R.; Alitheen, N.B.; Tan, W.S. Targeted delivery of cell penetrating peptide virus-like nanoparticles to skin cancer cells. Sci. Rep. 2018, 8, 8499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.K.; Suh, H.-W.; Qureshi, M.; Lewis, J.M.; Yaqoob, S.; Moscato, Z.M.; Griff, S.; Lee, A.K.; Yin, E.S.; Saltzman, W.M. Nonsurgical treatment of skin cancer with local delivery of bioadhesive nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2020575118. [Google Scholar] [CrossRef]
- Tołoczko-Iwaniuk, N.; Dziemiańczyk-Pakieła, D.; Nowaszewska, B.K.; Celińska-Janowicz, K.; Miltyk, W. Celecoxib in cancer therapy and prevention–review. Curr. Drug Targets 2019, 20, 302–315. [Google Scholar] [CrossRef]
- Gao, L.; Wang, T.H.; Chen, C.P.; Xiang, J.J.; Zhao, X.-B.; Gui, R.-Y.; Liao, X.-H. Targeting COX-2 potently inhibits proliferation of cancer cells in vivo but not in vitro in cutaneous squamous cell carcinoma. Transl. Cancer Res. 2021, 10, 2219. [Google Scholar] [CrossRef]
- Fouad, E.A.; Yassin, A.E.B.; Alajami, H.N. Characterization of celecoxib-loaded solid lipid nanoparticles formulated with tristearin and softisan 100. Trop. J. Pharm. Res. 2015, 14, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Davies, N.M.; McLachlan, A.J.; Day, R.O.; Williams, K.M. Clinical pharmacokinetics and pharmacodynamics of celecoxib. Clin. Pharmacokinet. 2000, 38, 225–242. [Google Scholar] [CrossRef]
- Tindall, E. Celecoxib for the treatment of pain and inflammation: The preclinical and clinical results. J. Osteopath. Med. 1999, 99, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Sohrabi, M.; Soleimani, J.; Roshangar, L.; Vatansever, S.; Arbabi, F.; Khaki, A.A.; Abbasi, M.M.; Dustar, Y.; Javadzadeh, Y. The effect of dietary and topical celecoxib on 4-nitroquinoline-1-oxide-induced lingual epithelium alternations in rat. JPMA 2009, 59, 769–774. [Google Scholar]
- Mojeiko, G.; de Brito, M.; Salata, G.C.; Lopes, L.B. Combination of microneedles and microemulsions to increase celecoxib topical delivery for potential application in chemoprevention of breast cancer. Int. J. Pharm. 2019, 560, 365–376. [Google Scholar] [CrossRef]
- Han, Y.; Chen, P.; Zhang, Y.; Lu, W.; Ding, W.; Luo, Y.; Wen, S.; Xu, R.; Liu, P.; Huang, P. Synergy between auranofin and celecoxib against colon cancer in vitro and in vivo through a novel redox-mediated mechanism. Cancers 2019, 11, 931. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.T.; Roth, M.D.; Fishbein, M.C.; Aberle, D.R.; Zhang, Z.F.; Rao, J.Y.; Tashkin, D.P.; Goodglick, L.; Holmes, E.C.; Cameron, R.B.; et al. Lung cancer chemoprevention with celecoxib in former smokers. Cancer Prev. Res. 2011, 4, 984–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabichi, A.L.; Lee, J.J.; Grossman, H.B.; Liu, S.; Richmond, E.; Czerniak, B.A.; De la Cerda, J.; Eagle, C.; Viner, J.L.; Palmer, J.L.; et al. A randomized controlled trial of celecoxib to prevent recurrence of nonmuscle-invasive bladder cancer. Cancer Prev. Res. 2011, 4, 1580–1589. [Google Scholar] [CrossRef] [Green Version]
- Paulson, S.K.; Vaughn, M.B.; Jessen, S.M.; Lawal, Y.; Gresk, C.J.; Yan, B.; Maziasz, T.J.; Cook, C.S.; Karim, A. Pharmacokinetics of celecoxib after oral administration in dogs and humans: Effect of food and site of absorption. J. Pharmacol. Exp. Ther. 2001, 297, 638–645. [Google Scholar]
- Auda, S.H.; Fathalla, D.; Fetih, G.; El-Badry, M.; Shakeel, F. Niosomes as transdermal drug delivery system for celecoxib: In vitro and in vivo studies. Polym. Bull. 2016, 73, 1229–1245. [Google Scholar] [CrossRef]
- Homayouni, A.; Sadeghi, F.; Varshosaz, J.; Garekani, H.A.; Nokhodchi, A. Comparing various techniques to produce micro/nanoparticles for enhancing the dissolution of celecoxib containing PVP. Eur. J. Pharm. Biopharm. 2014, 88, 261–274. [Google Scholar] [CrossRef]
- Salem, H.F.; Kharshoum, R.M.; Sayed, O.M.; Abdel Hakim, L.F. Formulation design and optimization of novel soft glycerosomes for enhanced topical delivery of celecoxib and cupferron by Box–Behnken statistical design. Drug Dev. Ind. Pharm. 2018, 44, 1871–1884. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.; Pfeffer, M.A.; Wittes, J.; Fowler, R.; Finn, P.; Anderson, W.F.; Zauber, A.; Hawk, E.; Bertagnolli, M. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N. Engl. J. Med. 2005, 352, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Ascenso, A.; Raposo, S.; Batista, C.; Cardoso, P.; Mendes, T.; Praça, F.G.; Bentley, M.V.L.B.; Simões, S. Development, characterization, and skin delivery studies of related ultradeformable vesicles: Transfersomes, ethosomes, and transethosomes. Int. J. Nanomed. 2015, 10, 5837. [Google Scholar] [CrossRef] [Green Version]
- Tawfeek, H.M.; Abdellatif, A.A.; Abdel-Aleem, J.A.; Hassan, Y.A.; Fathalla, D. Transfersomal gel nanocarriers for enhancement the permeation of lornoxicam. J. Drug Deliv. Sci. Technol. 2020, 56, 101540. [Google Scholar] [CrossRef]
- Abdellatif, A.A.; Tawfeek, H.M. Transfersomal nanoparticles for enhanced transdermal delivery of clindamycin. Aaps Pharmscitech 2016, 17, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Singh, H.; Bimbrawh, S.; Kumar Singh, S.; Gulati, M.; Vaidya, Y.; Kaur, P. Ethosomes and transfersomes: Principles, perspectives and practices. Curr. Drug Deliv. 2017, 14, 613–633. [Google Scholar] [CrossRef] [PubMed]
- Bragagni, M.; Mennini, N.; Maestrelli, F.; Cirri, M.; Mura, P. Comparative study of liposomes, transfersomes and ethosomes as carriers for improving topical delivery of celecoxib. Drug Deliv. 2012, 19, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Cevc, G.; Blume, G. New, highly efficient formulation of diclofenac for the topical, transdermal administration in ultradeformable drug carriers, Transfersomes. Biochim. Biophys. Acta (BBA)-Biomembr. 2001, 1514, 191–205. [Google Scholar] [CrossRef] [Green Version]
- Elsayed, M.M.; Abdallah, O.; Naggar, V.; Khalafallah, N. Deformable liposomes and ethosomes as carriers for skin delivery of ketotifen. Die Pharm.-Int. J. Pharm. Sci. 2007, 62, 133–137. [Google Scholar]
- Rattanapak, T.; Young, K.; Rades, T.; Hook, S. Comparative study of liposomes, transfersomes, ethosomes and cubosomes for transcutaneous immunisation: Characterisation and in vitro skin penetration. J. Pharm. Pharmacol. 2012, 64, 1560–1569. [Google Scholar] [CrossRef]
- Faisal, W.; Soliman, G.M.; Hamdan, A.M. Enhanced skin deposition and delivery of voriconazole using ethosomal preparations. J. Liposome Res. 2018, 28, 14–21. [Google Scholar] [CrossRef]
- Fathalla, D.; Youssef, E.M.; Soliman, G.M. Liposomal and ethosomal gels for the topical delivery of anthralin: Preparation, comparative evaluation and clinical assessment in psoriatic patients. Pharmaceutics 2020, 12, 446. [Google Scholar] [CrossRef]
- Fathalla, D.; Soliman, G.; Fouad, E. Development and in vitro/in vivo evaluation of liposomal gels for the sustained ocular delivery of latanoprost. J Clin Exp Ophthalmol 2015, 6, 2. [Google Scholar]
- Dholakia, M.; Thakkar, V.; Patel, N.; Gandhi, T. Development and characterisation of thermo reversible mucoadhesive moxifloxacin hydrochloride in situ ophthalmic gel. J. Pharm. Bioallied Sci. 2012, 4 (Suppl. S1), S42. [Google Scholar] [CrossRef]
- Allam, A.; Elsabahy, M.; El Badry, M.; Eleraky, N.E. Betaxolol-loaded niosomes integrated within pH-sensitive in situ forming gel for management of glaucoma. Int. J. Pharm. 2021, 598, 120380. [Google Scholar] [CrossRef] [PubMed]
- Mircioiu, C.; Voicu, V.; Anuta, V.; Tudose, A.; Celia, C.; Paolino, D.; Fresta, M.; Sandulovici, R.; Mircioiu, I. Mathematical modeling of release kinetics from supramolecular drug delivery systems. Pharmaceutics 2019, 11, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Hixson, A.; Crowell, J. Dependence of reaction velocity upon surface and agitation. Ind. Eng. Chem. 1931, 23, 923–931. [Google Scholar] [CrossRef]
- Baker, R. Controlled release: Mechanisms and rates. Control. Release Biol. Act. Agents. 1974, 15. [Google Scholar]
- Jain, S.; Tiwary, A.K.; Sapra, B.; Jain, N.K. Formulation and evaluation of ethosomes for transdermal delivery of lamivudine. Aaps Pharmscitech 2007, 8, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Garber, J.C.; Wayne Barbee, R.; Bielitzki, J.T.; Clayton, L.A.; Donovan, J.C.; Hendriksen, C.; Kohn, D.F.; Lipman, N.S.; Locke, P.A.; Melcher, J. Committee for the Update of the Guide for the Care and Use of Laboratory Animals. In Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academy of Sciences: Washington, DC, USA, 2011. [Google Scholar]
- Sloan, K.B.; Beall, H.D.; Weimar, W.R.; Villanueva, R. The effect of receptor phase composition on the permeability of hairless mouse skin in diffusion cell experiments. Int. J. Pharm. 1991, 73, 97–104. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Hamed, M.A.; Aboul Naser, A.F.; Aboutabl, M.E.; Osman, A.F.; Hassan, E.E.; Aziz, W.M.; Khalil, W.K.; Farghaly, A.A.; El-Hagrassi, A.M. Bioactive compounds and therapeutic role of Brassica oleracea L. seeds in rheumatoid arthritis rats via regulating inflammatory signalling pathways and antagonizing interleukin-1 receptor action. Biomarkers 2021, 26, 788–807. [Google Scholar] [CrossRef]
- Salem, N.A.; Wahba, M.A.; Eisa, W.H.; El-Shamarka, M.; Khalil, W. Silver oxide nanoparticles alleviate indomethacin-induced gastric injury: A novel antiulcer agent. Inflammopharmacology 2018, 26, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Hamza, A.H.; Abdulfattah, H.M.; Mahmoud, R.H.; Khalil, W.K.; Ahmed, H.H. Current concepts in pathophysiology and management of hepatocellular carcinoma. Acta Biochim. Pol. 2015, 62. [Google Scholar] [CrossRef] [PubMed]
- Elhinnawi, M.A.; Mohareb, R.M.; Rady, H.M.; Khalil, W.K.; Abd Elhalim, M.M.; Elmegeed, G.A. Novel pregnenolone derivatives modulate apoptosis via Bcl-2 family genes in hepatocellular carcinoma in vitro. J. Steroid Biochem. Mol. Biol. 2018, 183, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Krähn, G.; Leiter, U.; Udart, M.; Kaskel, P.; Peter, R. UVB-induced Decrease of p16/CDKN2A Expression in Skin Cancer Patients. Pigment Cell Res. 2001, 14, 201–205. [Google Scholar] [CrossRef]
- Pacifico, A.; Goldberg, L.H.; Peris, K.; Chimenti, S.; Leone, G.; Ananthaswamy, H. Loss of CDKN2A and p14ARF expression occurs frequently in human nonmelanoma skin cancers. Br. J. Dermatol. 2008, 158, 291–297. [Google Scholar] [CrossRef]
- Patel, S.; Wilkinson, C.J.; Sviderskaya, E.V. Loss of both CDKN2A and CDKN2B allows for centrosome overduplication in melanoma. J. Investig. Dermatol. 2020, 140, 1837–1846. [Google Scholar] [CrossRef]
- Olive, P.L.; Banáth, J.P.; Durand, R.E. Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the “comet” assay. Radiat. Res. 2012, 178, AV35–AV42. [Google Scholar] [CrossRef]
- Collins, A.; Dušinská, M.; Franklin, M.; Somorovská, M.; Petrovská, H.; Duthie, S.; Fillion, L.; Panayiotidis, M.; Rašlová, K.; Vaughan, N. Comet assay in human biomonitoring studies: Reliability, validation, and applications. Environ. Mol. Mutagen. 1997, 30, 139–146. [Google Scholar] [CrossRef]
- Yawata, A.; Adachi, M.; Okuda, H.; Naishiro, Y.; Takamura, T.; Hareyama, M.; Takayama, S.; Reed, J.C.; Imai, K. Prolonged cell survival enhances peritoneal dissemination of gastric cancer cells. Oncogene 1998, 16, 2681–2686. [Google Scholar] [CrossRef] [Green Version]
- Abdulbaqi, I.M.; Darwis, Y.; Khan, N.A.K.; Abou Assi, R.; Khan, A.A. Ethosomal nanocarriers: The impact of constituents and formulation techniques on ethosomal properties, in vivo studies, and clinical trials. Int. J. Nanomed. 2016, 11, 2279. [Google Scholar] [CrossRef] [Green Version]
- El Zaafarany, G.M.; Awad, G.A.; Holayel, S.M.; Mortada, N.D. Role of edge activators and surface charge in developing ultradeformable vesicles with enhanced skin delivery. Int. J. Pharm. 2010, 397, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Albash, R.; Abdelbary, A.A.; Refai, H.; El-Nabarawi, M.A. Use of transethosomes for enhancing the transdermal delivery of olmesartan medoxomil: In vitro, ex vivo, and in vivo evaluation. Int. J. Nanomed. 2019, 14, 1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboud, H.M.; Ali, A.A.; El-Menshawe, S.F.; Elbary, A.A. Nanotransfersomes of carvedilol for intranasal delivery: Formulation, characterization and in vivo evaluation. Drug Deliv. 2016, 23, 2471–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunieda, H.; Ohyama, K.-I. Three-phase behavior and HLB numbers of bile salts and lecithin in a water-oil system. J. Colloid Interface Sci. 1990, 136, 432–439. [Google Scholar] [CrossRef]
- Samuel, G.; Nazim, U.; Sharma, A.; Manuel, V.; Elnaggar, M.G.; Taye, A.; Nasr, N.E.H.; Hofni, A.; Hakiem, A.F.A. Selective Targeting of the Novel CK-10 Nanoparticles to the MDA-MB-231 Breast Cancer Cells. J. Pharm. Sci. 2022, 111, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, M.; Sharma, V.; Pathak, K. Nanovesicles for transdermal delivery of felodipine: Development, characterization, and pharmacokinetics. Int. J. Pharm. Investig. 2014, 4, 119. [Google Scholar]
- Khalil, R.M.; Abdelbary, G.A.; Basha, M.; Awad, G.E.; El-Hashemy, H.A. Enhancement of lomefloxacin Hcl ocular efficacy via niosomal encapsulation: In vitro characterization and in vivo evaluation. J. Liposome Res. 2017, 27, 312–323. [Google Scholar] [CrossRef]
- Mbah, C.C.; Builders, P.F.; Attama, A.A. Nanovesicular carriers as alternative drug delivery systems: Ethosomes in focus. Expert Opin. Drug Deliv. 2014, 11, 45–59. [Google Scholar] [CrossRef]
- Ascenso, A.; Cruz, M.; Euletério, C.; Carvalho, F.A.; Santos, N.C.; Marques, H.C.; Simoes, S. Novel tretinoin formulations: A drug-in-cyclodextrin-in-liposome approach. J. Liposome Res. 2013, 23, 211–219. [Google Scholar] [CrossRef]
- Junyaprasert, V.B.; Singhsa, P.; Suksiriworapong, J.; Chantasart, D. Physicochemical properties and skin permeation of Span 60/Tween 60 niosomes of ellagic acid. Int. J. Pharm. 2012, 423, 303–311. [Google Scholar] [CrossRef]
- Mokhtar, M.; Sammour, O.A.; Hammad, M.A.; Megrab, N.A. Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int. J. Pharm. 2008, 361, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Zhao, F.; Li, N.; Yang, Y. Studies on a high encapsulation of colchicine by a niosome system. Int. J. Pharm. 2002, 244, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Alajami, H.N.; Fouad, E.A.; Ashour, A.E.; Kumar, A.; Yassin, A.E.B. Celecoxib-Loaded Solid Lipid Nanoparticles for Colon Delivery: Formulation Optimization and In Vitro Assessment of Anti-Cancer Activity. Pharmaceutics 2022, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Ogawa, M.; Hironaka, K.; Ito, K.; Sunada, H. A nifedipine coground mixture with sodium deoxycholate. II. Dissolution characteristics and stability. Drug Dev. Ind. Pharm. 2001, 27, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Pandya, V.M.; Patel, D.J.; Patel, J.K.; Patel, R.P. Formulation, characterization, and optimization of fast-dissolve tablets containing celecoxib solid dispersion. Dissolution Technol. 2009, 16, 22–27. [Google Scholar] [CrossRef]
- Salama, A.; Badran, M.; Elmowafy, M.; Soliman, G.M. Spironolactone-loaded leciplexes as potential topical delivery systems for female acne: In vitro appraisal and ex vivo skin permeability studies. Pharmaceutics 2019, 12, 25. [Google Scholar] [CrossRef]
- Sun, S.; Liang, N.; Kawashima, Y.; Xia, D.; Cui, F. Hydrophobic ion pairing of an insulin-sodium deoxycholate complex for oral delivery of insulin. Int. J. Nanomed. 2011, 6, 3049. [Google Scholar]
- Eleraky, N.E.; Omar, M.M.; Mahmoud, H.A.; Abou-Taleb, H.A. Nanostructured lipid carriers to mediate brain delivery of temazepam: Design and in vivo study. Pharmaceutics 2020, 12, 451. [Google Scholar] [CrossRef]
- Badria, F.; Mazyed, E. Formulation of Nanospanlastics as a Promising Approach for Improving the Topical Delivery of a Natural Leukotriene Inhibitor (3-Acetyl-11-Keto-β-Boswellic Acid): Statistical Optimization, in vitro Characterization, and ex vivo Permeation Study. Drug Des. Dev. Ther. 2020, 14, 3697. [Google Scholar] [CrossRef]
- Ana, R.; Mendes, M.; Sousa, J.; Pais, A.; Falcão, A.; Fortuna, A.; Vitorino, C. Rethinking carbamazepine oral delivery using polymer-lipid hybrid nanoparticles. Int. J. Pharm. 2019, 554, 352–365. [Google Scholar] [CrossRef]
- Elnaggar, M.G.; Jiang, K.; Eldesouky, H.E.; Pei, Y.; Park, J.; Yuk, S.A.; Meng, F.; Dieterly, A.M.; Mohammad, H.T.; Hegazy, Y.A. Antibacterial nanotruffles for treatment of intracellular bacterial infection. Biomaterials 2020, 262, 120344. [Google Scholar] [CrossRef] [PubMed]
- Fissan, H.; Ristig, S.; Kaminski, H.; Asbach, C.; Epple, M. Comparison of different characterization methods for nanoparticle dispersions before and after aerosolization. Anal. Methods 2014, 6, 7324–7334. [Google Scholar] [CrossRef] [Green Version]
- Abdellatif, A.A.; Rasheed, Z.; Alhowail, A.H.; Alqasoumi, A.; Alsharidah, M.; Khan, R.A.; Aljohani, A.S.; Aldubayan, M.A.; Faisal, W. Silver citrate nanoparticles inhibit PMA-induced TNFα expression via deactivation of NF-κB activity in human cancer cell-lines, MCF-7. Int. J. Nanomed. 2020, 15, 8479. [Google Scholar] [CrossRef] [PubMed]
- Mekkawy, A.I.; Eleraky, N.E.; Soliman, G.M.; Elnaggar, M.G.; Elnaggar, M.G. Combinatorial Therapy of Letrozole-and Quercetin-Loaded Spanlastics for Enhanced Cytotoxicity against MCF-7 Breast Cancer Cells. Pharmaceutics 2022, 14, 1727. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, M.M.; Hussein, A.K.; Sarhan, H.A.; Mansour, H.F. Flurbiprofen-loaded niosomes-in-gel system improves the ocular bioavailability of flurbiprofen in the aqueous humor. Drug Dev. Ind. Pharm. 2017, 43, 902–910. [Google Scholar] [CrossRef]
- Mohanty, D.; Rani, M.J.; Haque, M.A.; Bakshi, V.; Jahangir, M.A.; Imam, S.S.; Gilani, S.J. Preparation and evaluation of transdermal naproxen niosomes: Formulation optimization to preclinical anti-inflammatory assessment on murine model. J. Liposome Res. 2020, 30, 377–387. [Google Scholar] [CrossRef]
- Kaul, S.; Jain, N.; Nagaich, U. Ultra deformable vesicles for boosting transdermal delivery of 2-arylpropionic acid class drug for management of musculoskeletal pain. J. Pharm. Investig. 2022, 52, 217–231. [Google Scholar] [CrossRef]
- Faria, M.J.; Machado, R.; Ribeiro, A.; Gonçalves, H.; Real Oliveira, M.E.C.; Viseu, T.; das Neves, J.; Lúcio, M. Rational development of liposomal hydrogels: A strategy for topical vaginal antiretroviral drug delivery in the context of HIV prevention. Pharmaceutics 2019, 11, 485. [Google Scholar] [CrossRef] [Green Version]
- Ricci, E.; Lunardi, L.O.; Nanclares, D.; Marchetti, J.M. Sustained release of lidocaine from Poloxamer 407 gels. Int. J. Pharm. 2005, 288, 235–244. [Google Scholar] [CrossRef]
- El-Badry, M.; Fetih, G. Preparation, charactarization and anti-inflammatory activity of celecoxib chitosan gel formulations. J. Drug Deliv. Sci. Technol. 2011, 21, 201–206. [Google Scholar] [CrossRef]
- Baig, M.S.; Ahad, A.; Aslam, M.; Imam, S.S.; Aqil, M.; Ali, A. Application of Box–Behnken design for preparation of levofloxacin-loaded stearic acid solid lipid nanoparticles for ocular delivery: Optimization, in vitro release, ocular tolerance, and antibacterial activity. Int. J. Biol. Macromol. 2016, 85, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Allam, A.A.; Eleraky, N.E.; Diab, N.H.; Elsabahy, M.; Mohamed, S.A.; Abdel-Ghaffar, H.S.; Hassan, N.A.; Shouman, S.A.; Omran, M.M.; Hassan, S.B. Development of Sedative Dexmedetomidine Sublingual In Situ Gels: In Vitro and In Vivo Evaluations. Pharmaceutics 2022, 14, 220. [Google Scholar] [CrossRef] [PubMed]
- Salem, H.F.; Kharshoum, R.M.; Abou-Taleb, H.A.; AbouTaleb, H.A.; AbouElhassan, K.M. Progesterone-loaded nanosized transethosomes for vaginal permeation enhancement: Formulation, statistical optimization, and clinical evaluation in anovulatory polycystic ovary syndrome. J. Liposome Res. 2019, 29, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Salem, H.F.; Nafady, M.M.; Kharshoum, R.M.; Abd el-Ghafar, O.A.; Farouk, H.O. Novel Enhanced Therapeutic Efficacy of Dapoxetine HCl by Nano-Vesicle Transdermal Gel for Treatment of Carrageenan-Induced Rat Paw Edema. AAPS PharmSciTech 2020, 21, 113. [Google Scholar] [CrossRef] [PubMed]
- Barry, B.W. Novel mechanisms and devices to enable successful transdermal drug delivery. Eur. J. Pharm. Sci. 2001, 14, 101–114. [Google Scholar] [CrossRef]
- Mishra, K.K.; Kaur, C.D.; Verma, S.; Sahu, A.K.; Dash, D.K.; Kashyap, P.; Mishra, S.P. Transethosomes and nanoethosomes: Recent approach on transdermal drug delivery system. Nanomedicine 2019, 2, 33–54. [Google Scholar]
- Shaji, J.; Bajaj, R. Transethosomes: A new prospect for enhanced transdermal delivery. Int. J. Pharm. Sci. Res. 2018, 9, 2681–2685. [Google Scholar]
- Ferrara, F.; Benedusi, M.; Sguizzato, M.; Cortesi, R.; Baldisserotto, A.; Buzzi, R.; Valacchi, G.; Esposito, E. Ethosomes and Transethosomes as Cutaneous Delivery Systems for Quercetin: A Preliminary Study on Melanoma Cells. Pharmaceutics 2022, 14, 1038. [Google Scholar] [CrossRef]
- Rosas, C.; Sinning, M.; Ferreira, A.; Fuenzalida, M.; Lemus, D. Celecoxib decreases growth and angiogenesis and promotes apoptosis in a tumor cell line resistant to chemotherapy. Biol. Res. 2014, 47, 27. [Google Scholar] [CrossRef]
- Perumal, V.; Banerjee, S.; Das, S.; Sen, R.K.; Mandal, M. Effect of liposomal celecoxib on proliferation of colon cancer cell and inhibition of DMBA-induced tumor in rat model. Cancer Nanotechnol. 2011, 2, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Whiteman, D.C.; Green, A.C. Melanoma and sun exposure: Where are we now? Int. J. Dermatol. 1999, 38, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Florell, S.R.; Boucher, K.M.; Garibotti, G.; Astle, J.; Kerber, R.; Mineau, G.; Wiggins, C.; Noyes, R.D.; Tsodikov, A.; Cannon-Albright, L.A. Population-based analysis of prognostic factors and survival in familial melanoma. J. Clin. Oncol. 2005, 23, 7168–7177. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.M.; Chan, M.; Harland, M.; Hayward, N.K.; Demenais, F.; Bishop, D.T.; Azizi, E.; Bergman, W.; Bianchi-Scarra, G.; Bruno, W. Features associated with germline CDKN2A mutations: A GenoMEL study of melanoma-prone families from three continents. J. Med. Genet. 2007, 44, 99–106. [Google Scholar] [CrossRef]
- Su, C.-C.; Wang, M.-J.; Chiu, T.-L. The anti-cancer efficacy of curcumin scrutinized through core signaling pathways in glioblastoma. Int. J. Mol. Med. 2010, 26, 217–224. [Google Scholar] [PubMed] [Green Version]
- Liu, W.; Chen, Y.; Wang, W.; Keng, P.; Finkelstein, J.; Hu, D.; Liang, L.; Guo, M.; Fenton, B.; Okunieff, P. Combination of radiation and celebrex (celecoxib) reduce mammary and lung tumor growth. Am. J. Clin. Oncol. 2003, 26, S103–S109. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.B.; Zhu, C.; Yong, S.K.; Gao, Q.; Wong, M.C. Enhanced sensitivity of celecoxib in human glioblastoma cells: Induction of DNA damage leading to p53-dependent G1 cell cycle arrest and autophagy. Mol. Cancer 2009, 8, 66. [Google Scholar] [CrossRef] [Green Version]
- Kalantar, M.; Rezaei, M.; Moghimipour, E.; Bavarsad, N.; Kalantari, H.; Varnaseri, G.; Forouzan, A. Evaluation of Apoptosis Induced by Celecoxib Loaded Liposomes in Isolated Rat Hepatocytes. Jundishapur J. Nat. Pharm. Prod. 2015, 10, e25421. [Google Scholar] [CrossRef]
- Pritchard, R.; Rodríguez-Enríquez, S.; Pacheco-Velázquez, S.C.; Bortnik, V.; Moreno-Sánchez, R.; Ralph, S. Celecoxib inhibits mitochondrial O2 consumption, promoting ROS dependent death of murine and human metastatic cancer cells via the apoptotic signalling pathway. Biochem. Pharmacol. 2018, 154, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Ansari, K.M.; Rundhaug, J.E.; Fischer, S.M. Multiple Signaling Pathways Are Responsible for Prostaglandin E2–Induced Murine Keratinocyte Proliferation. Mol. Cancer Res. 2008, 6, 1003–1016. [Google Scholar] [CrossRef]
- Rundhaug, J.E.; Pavone, A.; Kim, E.; Fischer, S.M. The effect of cyclooxygenase-2 overexpression on skin carcinogenesis is context dependent. Mol. Carcinog. Publ. Coop. Univ. Tex. MD Cancer Cent. 2007, 46, 981–992. [Google Scholar] [CrossRef]
- Tjiu, J.-W.; Chen, J.-S.; Shun, C.-T.; Lin, S.-J.; Liao, Y.-H.; Chu, C.-Y.; Tsai, T.-F.; Chiu, H.-C.; Dai, Y.-S.; Inoue, H. Tumor-associated macrophage-induced invasion and angiogenesis of human basal cell carcinoma cells by cyclooxygenase-2 induction. J. Investig. Dermatol. 2009, 129, 1016–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmets, C.A.; Viner, J.L.; Pentland, A.P.; Cantrell, W.; Lin, H.-Y.; Bailey, H.; Kang, S.; Linden, K.G.; Heffernan, M.; Duvic, M. Chemoprevention of nonmelanoma skin cancer with celecoxib: A randomized, double-blind, placebo-controlled trial. J. Natl. Cancer Inst. 2010, 102, 1835–1844. [Google Scholar] [CrossRef] [PubMed]
- Shureiqi, I.; Chen, D.; Lotan, R.; Yang, P.; Newman, R.A.; Fischer, S.M.; Lippman, S.M. 15-Lipoxygenase-1 mediates nonsteroidal anti-inflammatory drug-induced apoptosis independently of cyclooxygenase-2 in colon cancer cells. Cancer Res. 2000, 60, 6846–6850. [Google Scholar] [PubMed]
- Vaish, V.; Sanyal, S.N. Role of Sulindac and Celecoxib in chemoprevention of colorectal cancer via intrinsic pathway of apoptosis: Exploring NHE-1, intracellular calcium homeostasis and Calpain 9. Biomed. Pharmacother. 2012, 66, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Piplani, H.; Rana, C.; Vaish, V.; Vaiphei, K.; Sanyal, S. Dolastatin, along with Celecoxib, stimulates apoptosis by a mechanism involving oxidative stress, membrane potential change and PI3-K/AKT pathway down regulation. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 5142–5156. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Walther, U.; Borchert, P.; Laufer, S.; Linnebacher, M.; Hinz, B. Induction but not inhibition of COX-2 confers human lung cancer cell apoptosis by celecoxib. J. Lipid Res. 2013, 54, 3116–3129. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yue, P.; Zhou, Z.; Khuri, F.R.; Sun, S.-Y. Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J. Natl. Cancer Inst. 2004, 96, 1769–1780. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula Number | Type of Nanovesicles | Type of EA | Amount of EA (mg) |
|---|---|---|---|
| F1 | Ethosome | NA | NA |
| F2 | Transethosome | Na deoxycholate | 10 |
| F3 | Transethosome | Na deoxycholate | 20 |
| F4 | Transethosome | Na deoxycholate | 30 |
| F5 | Transethosome | Tween 80 | 10 |
| F6 | Transethosome | Tween 80 | 20 |
| F7 | Transethosome | Tween 80 | 30 |
| F8 | Transethosome | Span 60 | 10 |
| F9 | Transethosome | Span 60 | 20 |
| F10 | Transethosome | Span 60 | 30 |
| Gene | Primer Sequence | GenBank (Accession No.) |
|---|---|---|
| CDKN2A | F: CAC CCC GCT TTC GTA GTT TT R: CCA ACA CAG TGA AAA GGC AGA | NM_058195.4 |
| GAPDH | F: CCA AGG AGT AAG ACC CCT GG R: TGG TTG AGC ACA GGG TAC TT | NM_001256799.3 |
| Formula Number | EE (%) | Particle Size (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|---|
| F1 | 59.9 ± 3.4 | 363.2 ± 21.6 | 0.9 ± 0.03 | −21.5 ± 0.02 |
| F2 | 86.2 ± 2.6 | 53.3 ± 14.2 | 0.7 ± 0.05 | −34.3 ± 0.58 |
| F3 | 87.9 ± 4.0 | 70.2 ± 8.8 | 0.7 ± 0.08 | −43.3 ± 0.57 |
| F4 | 88.8 ± 7.2 | 75.9 ± 11.4 | 0.4 ± 0.01 | −44.7 ± 1.52 |
| F5 | 80.4 ± 2.5 | 105.0 ± 2.3 | 0.3 ± 0.08 | −22.0 ± 1.00 |
| F6 | 78.5 ± 1.4 | 92.2 ± 2.3 | 0.9 ± 0.01 | −24.0 ± 0.01 |
| F7 | 77.9 ±1.5 | 85.0 ± 3.4 | 0.7 ± 0.11 | −18.6 ± 1.52 |
| F8 | 89.4 ± 1.9 | 143.6 ± 11.8 | 0.5 ± 0.03 | −20.7 ± 0.58 |
| F9 | 87.1 ± 2.0 | 138.5 ± 2.1 | 0.7 ± 0.07 | −19.3 ± 0.58 |
| F10 | 86.1 ± 4.5 | 95.7 ± 5.9 | 0.6 ± 0.04 | −23.3 ± 1.52 |
| Formulation | Papp × 10−2 (cm/h) | Jss (µg/cm2/h) | Drug Permeated after 6 h (µg/cm2) | ER a |
|---|---|---|---|---|
| CXB TES-loaded HPMC gel | 2.394 ± 0.102 * | 26.33 ± 1.9 * | 161.42 ± 1.17 * | 1.4 |
| Free CXB-loaded HPMC gel | 1.702 ± 0.2 | 18.72 ± 1.3 | 111.81 ± 3.31 | ---- |
| Formulations | % Mortality at 100 µg/mL (A431 Cell Line) | IC50 (µg/mL) | % Mortality at 100 µg/mL (Bj1 Cell Line) | IC50 (µg/mL) |
|---|---|---|---|---|
| CXB-TES dispersion | 100 | 1.4 | 54.3 | 82.7 |
| Blank HPMC gel | 10.3 | ------ | 22.5 | ----- |
| Free CXB gel 1% | 18.4 | ----- | 31.8 | ------ |
| CXB -TES HPMC gel 1% | 100 | 17.5 | 43.9 | 101.1 |
| CXB powder | 100 | 1.8 | 100 | 1.6 |
| Negative control | 1 | ---- | 2.3 | ----- |
| Positive control (doxorubicin) | 100 | 23.5 | 100 | 18.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdellatif, A.A.H.; Aldosari, B.N.; Al-Subaiyel, A.; Alhaddad, A.; Samman, W.A.; Eleraky, N.E.; Elnaggar, M.G.; Barakat, H.; Tawfeek, H.M. Transethosomal Gel for the Topical Delivery of Celecoxib: Formulation and Estimation of Skin Cancer Progression. Pharmaceutics 2023, 15, 22. https://doi.org/10.3390/pharmaceutics15010022
Abdellatif AAH, Aldosari BN, Al-Subaiyel A, Alhaddad A, Samman WA, Eleraky NE, Elnaggar MG, Barakat H, Tawfeek HM. Transethosomal Gel for the Topical Delivery of Celecoxib: Formulation and Estimation of Skin Cancer Progression. Pharmaceutics. 2023; 15(1):22. https://doi.org/10.3390/pharmaceutics15010022
Chicago/Turabian StyleAbdellatif, Ahmed A. H., Basmah Nasser Aldosari, Amal Al-Subaiyel, Aisha Alhaddad, Waad A. Samman, Nermin E. Eleraky, Marwa G. Elnaggar, Hassan Barakat, and Hesham M. Tawfeek. 2023. "Transethosomal Gel for the Topical Delivery of Celecoxib: Formulation and Estimation of Skin Cancer Progression" Pharmaceutics 15, no. 1: 22. https://doi.org/10.3390/pharmaceutics15010022