
Bispecific Antibody Format and the Organization of Immunological Synapses in T Cell-Redirecting Strategies for Cancer Immunotherapy
 ,
,  ,
,
Abstract
:1. Introduction
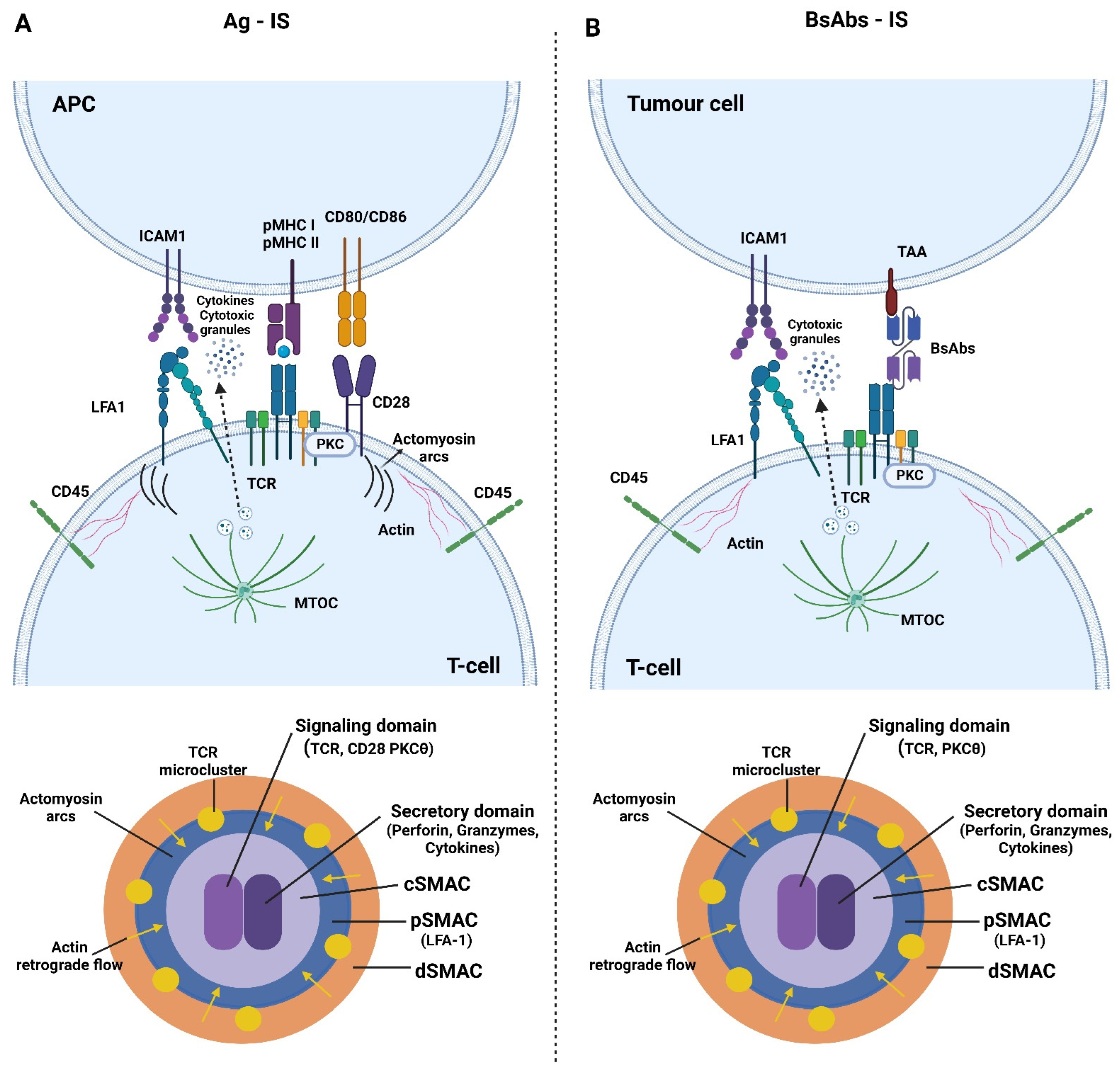
2. Immune Synapse Formation and Pathology
3. T Cell Is Organization and Effector Function Induced by bsAb
4. Format of bsAb and Immunological Synapse Organization
4.1. TAA-Binding
4.2. bsAbs Size and Spatial Requirements
5. bsAbs versus mAbs
6. bsAbs versus CARs
7. Improving Therapies Based on bsAbs
7.1. Direct Secretion of bsAbs to the Tumor Site
7.2. Combining Different bsAbs
8. Conclusions and Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021, 12, 626616. [Google Scholar] [CrossRef] [PubMed]
- Castaman, G.; Di Minno, G.; De Cristofaro, R.; Peyvandi, F. The Arrival of Gene Therapy for Patients with Hemophilia A. Int. J. Mol. Sci. 2022, 23, 228. [Google Scholar] [CrossRef] [PubMed]
- Rofo, F.; Meier, S.R.; Metzendorf, N.G.; Morrison, J.I.; Petrovic, A.; Syvanen, S.; Sehlin, D.; Hultqvist, G. A Brain-Targeting Bispecific-Multivalent Antibody Clears Soluble Amyloid-Beta Aggregates in Alzheimer’s Disease Mice. Neurotherapeutics 2022, 19, 1588–1602. [Google Scholar] [CrossRef] [PubMed]
- Blanco, B.; Dominguez-Alonso, C.; Alvarez-Vallina, L. Bispecific Immunomodulatory Antibodies for Cancer Immunotherapy. Clin. Cancer Res. 2021, 27, 5457–5464. [Google Scholar] [CrossRef] [PubMed]
- Nunez-Prado, N.; Compte, M.; Harwood, S.; Alvarez-Mendez, A.; Lykkemark, S.; Sanz, L.; Alvarez-Vallina, L. The coming of age of engineered multivalent antibodies. Drug Discov. Today 2015, 20, 588–594. [Google Scholar] [CrossRef]
- Muik, A.; Adams, H.C., 3rd; Gieseke, F.; Altintas, I.; Schoedel, K.B.; Blum, J.M.; Sanger, B.; Burm, S.M.; Stanganello, E.; Verzijl, D.; et al. DuoBody-CD40x4-1BB induces dendritic-cell maturation and enhances T-cell activation through conditional CD40 and 4-1BB agonist activity. J. Immunother. Cancer 2022, 10, e004322. [Google Scholar] [CrossRef]
- Jiang, C.; Zhang, L.; Xu, X.; Qi, M.; Zhang, J.; He, S.; Tian, Q.; Song, S. Engineering a Smart Agent for Enhanced Immunotherapy Effect by Simultaneously Blocking PD-L1 and CTLA-4. Adv. Sci. 2021, 8, e2102500. [Google Scholar] [CrossRef]
- La Motte-Mohs, R.; Shah, K.; Brown, J.G.; Smith, D.; Gorlatov, S.; Ciccarone, V.; Tamura, J.K.; Li, H.; Rillema, J.R.; Licea, M. Preclinical characterization of MGD013, a PD-1 x LAG-3 bispecific DART® molecule. Lung 2017, 28, 56. [Google Scholar]
- Moores, S.L.; Chiu, M.L.; Bushey, B.S.; Chevalier, K.; Luistro, L.; Dorn, K.; Brezski, R.J.; Haytko, P.; Kelly, T.; Wu, S.J.; et al. A Novel Bispecific Antibody Targeting EGFR and cMet Is Effective against EGFR Inhibitor-Resistant Lung Tumors. Cancer Res. 2016, 76, 3942–3953. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Kong, D.; Yao, Y.; Yang, L.; Yao, Q.; Zhu, Y.; Ding, Y.; Yang, F.; Gong, J.; Shen, L.; et al. Prediction of Human Pharmacokinetics and Clinical Effective Dose of SI-B001, an EGFR/HER3 Bi-specific Monoclonal Antibody. J. Pharm. Sci. 2020, 109, 3172–3180. [Google Scholar] [CrossRef]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef]
- Nogami, A.; Sasaki, K. Therapeutic Advances in Immunotherapies for Hematological Malignancies. Int. J. Mol. Sci. 2022, 23, 11526. [Google Scholar] [CrossRef]
- Gokbuget, N.; Dombret, H.; Bonifacio, M.; Reichle, A.; Graux, C.; Faul, C.; Diedrich, H.; Topp, M.S.; Bruggemann, M.; Horst, H.A.; et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 2018, 131, 1522–1531. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.; Stein, A.; Gokbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foa, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Przepiorka, D.; Ko, C.W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, A.M.; Sainz-Pastor, N.; Bonet, J.; Oliva, B.; Alvarez-Vallina, L. Multivalent antibodies: When design surpasses evolution. Trends Biotechnol. 2010, 28, 355–362. [Google Scholar] [CrossRef]
- Sanchez-Martin, D.; Sorensen, M.D.; Lykkemark, S.; Sanz, L.; Kristensen, P.; Ruoslahti, E.; Alvarez-Vallina, L. Selection strategies for anticancer antibody discovery: Searching off the beaten path. Trends Biotechnol. 2015, 33, 292–301. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Camino, V.; Harwood, S.L.; Alvarez-Mendez, A.; Alvarez-Vallina, L. Efficacy and toxicity management of CAR-T-cell immunotherapy: A matter of responsiveness control or tumour-specificity? Biochem. Soc. Trans. 2016, 44, 406–411. [Google Scholar] [CrossRef]
- Garber, K. Bispecific antibodies rise again. Nat. Rev. Drug Discov. 2014, 13, 799–801. [Google Scholar] [CrossRef]
- Ruf, P.; Lindhofer, H. Induction of a long-lasting antitumor immunity by a trifunctional bispecific antibody. Blood 2001, 98, 2526–2534. [Google Scholar] [CrossRef] [Green Version]
- Bossi, G.; Buisson, S.; Oates, J.; Jakobsen, B.K.; Hassan, N.J. ImmTAC-redirected tumour cell killing induces and potentiates antigen cross-presentation by dendritic cells. Cancer Immunol. Immunother. 2014, 63, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Hua, G.; Carlson, D.; Starr, J.R. Tebentafusp-tebn: A Novel Bispecific T-Cell Engager for Metastatic Uveal Melanoma. J. Adv. Pract. Oncol. 2022, 13, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Bacac, M.; Fauti, T.; Sam, J.; Colombetti, S.; Weinzierl, T.; Ouaret, D.; Bodmer, W.; Lehmann, S.; Hofer, T.; Hosse, R.J.; et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin. Cancer Res. 2016, 22, 3286–3297. [Google Scholar] [CrossRef] [Green Version]
- Gandullo-Sanchez, L.; Ocana, A.; Pandiella, A. HER3 in cancer: From the bench to the bedside. J. Exp. Clin. Cancer Res. 2022, 41, 310. [Google Scholar] [CrossRef]
- Bartsch, R.; Bergen, E. ASCO 2018: Highlights in HER2-positive metastatic breast cancer. Memo 2018, 11, 280–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, R.; Pepper, C.; Brennan, P.; Nagorsen, D.; Man, S.; Fegan, C. Blinatumomab induces autologous T-cell killing of chronic lymphocytic leukemia cells. Haematologica 2013, 98, 1930–1938. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.L.; Ellerman, D.; Mathieu, M.; Hristopoulos, M.; Chen, X.; Li, Y.; Yan, X.; Clark, R.; Reyes, A.; Stefanich, E.; et al. Anti-CD20/CD3 T cell-dependent bispecific antibody for the treatment of B cell malignancies. Sci. Transl. Med. 2015, 7, 287ra270. [Google Scholar] [CrossRef]
- Broske, A.E.; Korfi, K.; Belousov, A.; Wilson, S.; Ooi, C.H.; Bolen, C.R.; Canamero, M.; Alcaide, E.G.; James, I.; Piccione, E.C.; et al. Pharmacodynamics and molecular correlates of response to glofitamab in relapsed/refractory non-Hodgkin lymphoma. Blood Adv. 2022, 6, 1025–1037. [Google Scholar] [CrossRef]
- Cremasco, F.; Menietti, E.; Speziale, D.; Sam, J.; Sammicheli, S.; Richard, M.; Varol, A.; Klein, C.; Umana, P.; Bacac, M.; et al. Cross-linking of T cell to B cell lymphoma by the T cell bispecific antibody CD20-TCB induces IFNgamma/CXCL10-dependent peripheral T cell recruitment in humanized murine model. PLoS ONE 2021, 16, e0241091. [Google Scholar] [CrossRef]
- Pillarisetti, K.; Powers, G.; Luistro, L.; Babich, A.; Baldwin, E.; Li, Y.; Zhang, X.; Mendonca, M.; Majewski, N.; Nanjunda, R.; et al. Teclistamab is an active T cell-redirecting bispecific antibody against B-cell maturation antigen for multiple myeloma. Blood Adv. 2020, 4, 4538–4549. [Google Scholar] [CrossRef]
- Al-Hussaini, M.; Rettig, M.P.; Ritchey, J.K.; Karpova, D.; Uy, G.L.; Eissenberg, L.G.; Gao, F.; Eades, W.C.; Bonvini, E.; Chichili, G.R.; et al. Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood 2016, 127, 122–131. [Google Scholar] [CrossRef]
- Alcover, A.; Di Bartolo, V.; Roda-Navarro, P. Editorial: Molecular Dynamics at the Immunological Synapse. Front. Immunol. 2016, 7, 632. [Google Scholar] [CrossRef] [Green Version]
- Monks, C.R.; Kupfer, H.; Tamir, I.; Barlow, A.; Kupfer, A. Selective modulation of protein kinase C-theta during T-cell activation. Nature 1997, 385, 83–86. [Google Scholar] [CrossRef]
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The immunological synapse: A molecular machine controlling T cell activation. Science 1999, 285, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Monks, C.R.; Freiberg, B.A.; Kupfer, H.; Sciaky, N.; Kupfer, A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 1998, 395, 82–86. [Google Scholar] [CrossRef]
- Lee, K.H.; Holdorf, A.D.; Dustin, M.L.; Chan, A.C.; Allen, P.M.; Shaw, A.S. T cell receptor signaling precedes immunological synapse formation. Science 2002, 295, 1539–1542. [Google Scholar] [CrossRef] [Green Version]
- Varma, R.; Campi, G.; Yokosuka, T.; Saito, T.; Dustin, M.L. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity 2006, 25, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Yokosuka, T.; Sakata-Sogawa, K.; Kobayashi, W.; Hiroshima, M.; Hashimoto-Tane, A.; Tokunaga, M.; Dustin, M.L.; Saito, T. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol. 2005, 6, 1253–1262. [Google Scholar] [CrossRef]
- Murugesan, S.; Hong, J.; Yi, J.; Li, D.; Beach, J.R.; Shao, L.; Meinhardt, J.; Madison, G.; Wu, X.; Betzig, E.; et al. Formin-generated actomyosin arcs propel T cell receptor microcluster movement at the immune synapse. J. Cell Biol. 2016, 215, 383–399. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.; Wu, X.S.; Crites, T.; Hammer, J.A., 3rd. Actin retrograde flow and actomyosin II arc contraction drive receptor cluster dynamics at the immunological synapse in Jurkat T cells. Mol. Biol. Cell 2012, 23, 834–852. [Google Scholar] [CrossRef]
- Babich, A.; Li, S.; O’Connor, R.S.; Milone, M.C.; Freedman, B.D.; Burkhardt, J.K. F-actin polymerization and retrograde flow drive sustained PLCgamma1 signaling during T cell activation. J. Cell Biol. 2012, 197, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Kobayashi, W.; Sakata-Sogawa, K.; Takamatsu, M.; Hashimoto-Tane, A.; Dustin, M.L.; Tokunaga, M.; Saito, T. Spatiotemporal regulation of T cell costimulation by TCR-CD28 microclusters and protein kinase C theta translocation. Immunity 2008, 29, 589–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, V.; Nal, B.; Dujeancourt, A.; Thoulouze, M.-I.; Galli, T.; Roux, P.; Dautry-Varsat, A.; Alcover, A. Activation-Induced Polarized Recycling Targets T Cell Antigen Receptors to the Immunological Synapse: Involvement of SNARE Complexes. Immunity 2004, 20, 577–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Cofreces, N.B.; Robles-Valero, J.; Cabrero, J.R.; Mittelbrunn, M.; Gordon-Alonso, M.; Sung, C.H.; Alarcon, B.; Vazquez, J.; Sanchez-Madrid, F. MTOC translocation modulates IS formation and controls sustained T cell signaling. J. Cell Biol. 2008, 182, 951–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huse, M.; Lillemeier, B.F.; Kuhns, M.S.; Chen, D.S.; Davis, M.M. T cells use two directionally distinct pathways for cytokine secretion. Nat. Immunol. 2006, 7, 247–255. [Google Scholar] [CrossRef]
- Stinchcombe, J.C.; Bossi, G.; Booth, S.; Griffiths, G.M. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity 2001, 15, 751–761. [Google Scholar] [CrossRef] [Green Version]
- Mittelbrunn, M.; Gutierrez-Vazquez, C.; Villarroya-Beltri, C.; Gonzalez, S.; Sanchez-Cabo, F.; Gonzalez, M.A.; Bernad, A.; Sanchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef] [Green Version]
- Choudhuri, K.; Llodra, J.; Roth, E.W.; Tsai, J.; Gordo, S.; Wucherpfennig, K.W.; Kam, L.C.; Stokes, D.L.; Dustin, M.L. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature 2014, 507, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Messina, L.; Rodriguez-Galan, A.; de Yebenes, V.G.; Gutierrez-Vazquez, C.; Tenreiro, S.; Seabra, M.C.; Ramiro, A.R.; Sanchez-Madrid, F. Transfer of extracellular vesicle-microRNA controls germinal center reaction and antibody production. EMBO Rep. 2020, 21, e48925. [Google Scholar] [CrossRef]
- Saliba, D.G.; Cespedes-Donoso, P.F.; Balint, S.; Compeer, E.B.; Korobchevskaya, K.; Valvo, S.; Mayya, V.; Kvalvaag, A.; Peng, Y.; Dong, T.; et al. Composition and structure of synaptic ectosomes exporting antigen receptor linked to functional CD40 ligand from helper T cells. eLife 2019, 8, e47528. [Google Scholar] [CrossRef]
- Barcia, C., Jr.; Gomez, A.; Gallego-Sanchez, J.M.; Perez-Valles, A.; Castro, M.G.; Lowenstein, P.R.; Barcia, C., Sr.; Herrero, M.T. Infiltrating CTLs in human glioblastoma establish immunological synapses with tumorigenic cells. Am. J. Pathol. 2009, 175, 786–798. [Google Scholar] [CrossRef]
- Barcia, C.; Thomas, C.E.; Curtin, J.F.; King, G.D.; Wawrowsky, K.; Candolfi, M.; Xiong, W.D.; Liu, C.; Kroeger, K.; Boyer, O.; et al. In vivo mature immunological synapses forming SMACs mediate clearance of virally infected astrocytes from the brain. J. Exp. Med. 2006, 203, 2095–2107. [Google Scholar] [CrossRef] [Green Version]
- Henrickson, S.E.; Mempel, T.R.; Mazo, I.B.; Liu, B.; Artyomov, M.N.; Zheng, H.; Peixoto, A.; Flynn, M.; Senman, B.; Junt, T.; et al. In vivo imaging of T cell priming. Sci. Signal 2008, 1 Pt 2. [Google Scholar] [CrossRef] [Green Version]
- Schubert, D.A.; Gordo, S.; Sabatino, J.J., Jr.; Vardhana, S.; Gagnon, E.; Sethi, D.K.; Seth, N.P.; Choudhuri, K.; Reijonen, H.; Nepom, G.T.; et al. Self-reactive human CD4 T cell clones form unusual immunological synapses. J. Exp. Med. 2012, 209, 335–352. [Google Scholar] [CrossRef]
- Roda-Navarro, P.; Alvarez-Vallina, L. Understanding the Spatial Topology of Artificial Immunological Synapses Assembled in T Cell-Redirecting Strategies: A Major Issue in Cancer Immunotherapy. Front. Cell Dev. Biol. 2019, 7, 370. [Google Scholar] [CrossRef]
- Went, P.T.; Lugli, A.; Meier, S.; Bundi, M.; Mirlacher, M.; Sauter, G.; Dirnhofer, S. Frequent EpCam protein expression in human carcinomas. Hum. Pathol. 2004, 35, 122–128. [Google Scholar] [CrossRef]
- Marme, A.; Strauss, G.; Bastert, G.; Grischke, E.M.; Moldenhauer, G. Intraperitoneal bispecific antibody (HEA125xOKT3) therapy inhibits malignant ascites production in advanced ovarian carcinoma. Int. J. Cancer 2002, 101, 183–189. [Google Scholar] [CrossRef]
- Hammarstrom, S. The carcinoembryonic antigen (CEA) family: Structures, suggested functions and expression in normal and malignant tissues. Semin. Cancer Biol. 1999, 9, 67–81. [Google Scholar] [CrossRef]
- Elkins, K.; Zheng, B.; Go, M.; Slaga, D.; Du, C.; Scales, S.J.; Yu, S.F.; McBride, J.; de Tute, R.; Rawstron, A.; et al. FcRL5 as a target of antibody-drug conjugates for the treatment of multiple myeloma. Mol. Cancer Ther. 2012, 11, 2222–2232. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Stagg, N.J.; Johnston, J.; Harris, M.J.; Menzies, S.A.; DiCara, D.; Clark, V.; Hristopoulos, M.; Cook, R.; Slaga, D.; et al. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer Cell 2017, 31, 383–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, B.; Ramirez-Fernandez, A.; Bueno, C.; Argemi-Muntadas, L.; Fuentes, P.; Aguilar-Sopena, O.; Gutierrez-Aguera, F.; Zanetti, S.R.; Tapia-Galisteo, A.; Diez-Alonso, L.; et al. Overcoming CAR-Mediated CD19 Downmodulation and Leukemia Relapse with T Lymphocytes Secreting Anti-CD19 T-cell Engagers. Cancer Immunol. Res. 2022, 10, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Harwood, S.L.; Alvarez-Cienfuegos, A.; Nunez-Prado, N.; Compte, M.; Hernandez-Perez, S.; Merino, N.; Bonet, J.; Navarro, R.; Van Bergen En Henegouwen, P.M.P.; Lykkemark, S.; et al. ATTACK, a novel bispecific T cell-recruiting antibody with trivalent EGFR binding and monovalent CD3 binding for cancer immunotherapy. Oncoimmunology 2017, 7, e1377874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez-Fernandez, A.; Aguilar-Sopena, O.; Diez-Alonso, L.; Segura-Tudela, A.; Dominguez-Alonso, C.; Roda-Navarro, P.; Alvarez-Vallina, L.; Blanco, B. Synapse topology and downmodulation events determine the functional outcome of anti-CD19 T cell-redirecting strategies. Oncoimmunology 2022, 11, 2054106. [Google Scholar] [CrossRef]
- Etxeberria, I.; Glez-Vaz, J.; Teijeira, A.; Melero, I. New emerging targets in cancer immunotherapy: CD137/4-1BB costimulatory axis. ESMO Open 2020, 4, e000733. [Google Scholar] [CrossRef]
- Piechutta, M.; Berghoff, A.S. New emerging targets in cancer immunotherapy: The role of Cluster of Differentiation 40 (CD40/TNFR5). ESMO Open 2019, 4, e000510. [Google Scholar] [CrossRef] [Green Version]
- Kamakura, D.; Asano, R.; Yasunaga, M. T Cell Bispecific Antibodies: An Antibody-Based Delivery System for Inducing Antitumor Immunity. Pharmaceuticals 2021, 14, 1172. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [Green Version]
- Molgaard, K.; Harwood, S.L.; Compte, M.; Merino, N.; Bonet, J.; Alvarez-Cienfuegos, A.; Mikkelsen, K.; Nunez-Prado, N.; Alvarez-Mendez, A.; Sanz, L.; et al. Bispecific light T-cell engagers for gene-based immunotherapy of epidermal growth factor receptor (EGFR)-positive malignancies. Cancer Immunol. Immunother. 2018, 67, 1251–1260. [Google Scholar] [CrossRef]
- Tapia-Galisteo, A.; Sanchez Rodriguez, I.; Aguilar-Sopena, O.; Harwood, S.L.; Narbona, J.; Ferreras Gutierrez, M.; Navarro, R.; Martin-Garcia, L.; Corbacho, C.; Compte, M.; et al. Trispecific T-cell engagers for dual tumor-targeting of colorectal cancer. Oncoimmunology 2022, 11, 2034355. [Google Scholar] [CrossRef]
- Cartwright, A.N.; Griggs, J.; Davis, D.M. The immune synapse clears and excludes molecules above a size threshold. Nat. Commun. 2014, 5, 5479. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, B.C.; Cheng, L. Characterization of a novel monoclonal antibody against human perforin using transfected cell lines. Immunology 1994, 81, 291–295. [Google Scholar] [PubMed]
- Cultrera, J.L.; Dalia, S.M. Diffuse large B-cell lymphoma: Current strategies and future directions. Cancer Control 2012, 19, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andric, Z.; Geater, S.; et al. Atezolizumab for First-Line Treatment of PD-L1-Selected Patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.L.; Zhu, H.X.; Deng, L.; Meng, X.Q.; Li, K.; Xu, W.; Zhao, L.; Liu, Y.Q.; Zhu, Z.P.; Huang, H.M. Bispecific antibody simultaneously targeting PD1 and HER2 inhibits tumor growth via direct tumor cell killing in combination with PD1/PDL1 blockade and HER2 inhibition. Acta Pharmacol. Sin. 2022, 43, 672–680. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Rui, W.; Zheng, H.; Huang, D.; Yu, F.; Zhang, Y.; Dong, J.; Zhao, X.; Lin, X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020, 50, 712–724. [Google Scholar] [CrossRef]
- Tomasik, J.; Jasinski, M.; Basak, G.W. Next generations of CAR-T cells—new therapeutic opportunities in hematology? Front. Immunol. 2022, 13, 1034707. [Google Scholar] [CrossRef]
- Dalal, P.J.; Patel, N.P.; Feinstein, M.J.; Akhter, N. Adverse Cardiac Effects of CAR T-Cell Therapy: Characteristics, Surveillance, Management, and Future Research Directions. Technol. Cancer Res. Treat. 2022, 21, 15330338221132927. [Google Scholar] [CrossRef]
- Mukherjee, M.; Mace, E.M.; Carisey, A.F.; Ahmed, N.; Orange, J.S. Quantitative Imaging Approaches to Study the CAR Immunological Synapse. Mol. Ther. 2017, 25, 1757–1768. [Google Scholar] [CrossRef]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, E2068–E2076. [Google Scholar] [CrossRef] [PubMed]
- Blanco, B.; Ramirez-Fernandez, A.; Alvarez-Vallina, L. Engineering Immune Cells for in vivo Secretion of Tumor-Specific T Cell-Redirecting Bispecific Antibodies. Front. Immunol. 2020, 11, 1792. [Google Scholar] [CrossRef] [PubMed]
- Qiao, G.; Kone, L.B.; Phillips, E.H.; Lee, S.S.; Brown, G.E.; Khetani, S.R.; Thakur, A.; Lum, L.G.; Prabhakar, B.S.; Maker, A.V. LIGHT enhanced bispecific antibody armed T-cells to treat immunotherapy resistant colon cancer. Oncogene 2022, 41, 2054–2068. [Google Scholar] [CrossRef]
- Hallek, M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef] [Green Version]
- Blanco, B.; Compte, M.; Lykkemark, S.; Sanz, L.; Alvarez-Vallina, L. T Cell-Redirecting Strategies to ‘STAb’ Tumors: Beyond CARs and Bispecific Antibodies. Trends Immunol. 2019, 40, 243–257. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [Green Version]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef]
- Skokos, D.; Waite, J.C.; Haber, L.; Crawford, A.; Hermann, A.; Ullman, E.; Slim, R.; Godin, S.; Ajithdoss, D.; Ye, X.; et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci. Transl. Med. 2020, 12, eaaw7888. [Google Scholar] [CrossRef]
- Cassioli, C.; Patrussi, L.; Valitutti, S.; Baldari, C.T. Learning from TCR Signaling and Immunological Synapse Assembly to Build New Chimeric Antigen Receptors (CARs). Int. J. Mol. Sci. 2022, 23, 14255. [Google Scholar] [CrossRef]
- Stamova, S.; Feldmann, A.; Cartellieri, M.; Arndt, C.; Koristka, S.; Apel, F.; Wehner, R.; Schmitz, M.; Bornhauser, M.; von Bonin, M.; et al. Generation of single-chain bispecific green fluorescent protein fusion antibodies for imaging of antibody-induced T cell synapses. Anal. Biochem. 2012, 423, 261–268. [Google Scholar] [CrossRef]


{kind=link}
| BsAbs Name | Format | Target Protein 1 (Gene; Cell) | Target Protein 2 (Gene; Cell) | Mechanism of Action | Type of Cancer | Phase (Identifier) | |
|---|---|---|---|---|---|---|---|
| Catumaxomab | Triomab | CD3 (CD3E; T cell) | EpCAM (EPCAM; cancer cell) | Recruitment and activation of T cells [20] | Malignant ascites | Solid tumors | Approved by EMA * |
| Tebentafusp | ImmTAC | PMEL peptide 280–288 (PMEL; cancer cell) | Recruitment and activation of T cells [21,22] | Unresectable or metastatic uveal melanoma | Approved by EMA and FDA | ||
| RO6958688 | CrossMab/KIH(IgG-like bsAbs) | CEA (CEACAM5; cancer cell) | Recruitment and activation of T cells [23] | CEA-positive tumors | Phase I (NCT02324257) | ||
| Amivantamab | Duobody | EGFR (EGFR; cancer cell) | METcMet (MET; cancer cell) | Blocking of dual signal pathways [9] | Non-small cell lung cancer (NSCLC) | Approved by EMA and FDA | |
| SI-B001 | IgG-(scFv)2 | HER3 (ERBB3; cancer cell) | Blocking of dual signal pathways [10,24] | NSCLC | Phase I (NCT04603287) | ||
| GEN1402 | Duobody | CD137 (TNFRSF9; T cell) | CD40 (CD40; APC) | Costimulating molecule engaging for efficient T cell activating signals [6] | NSCLC, Colorectal Cancer and Melanoma | Phase II (NCT04083599) | |
| Zanidatamab | Asymmetric | HER2 (ERBB2; cancer cell) | HER2 (ERBB2; cancer cell) | Blocking of dual signal pathways [25] | Gastro-oesophagealadenocarcinoma | Phase II (NCT04513665) | |
| Erfonrilimab | CRIB | CTLA-4 (CTLA4; T cell) | PD-L1 (CD274; cancer cell) | Blocking of immune checkpoints [7] | NSCLC and pancreatic ductal adenocarcinoma | Phase II (NCT03838848) | |
| Tebotelimab | DART | PD1 (CD80; T cell) | LAG3 (LAG3; cancer cell) | Blocking of immune checkpoints [8] | Gastric Cancer | Phase III (NCT04082364) | |
| Blinatumomab | BiTE | CD3 (CD3E; T cell) | CD19 (CD19; cancer cell) | Recruitment and activation of T cells [26] | Acute lymphoblastic leukaemia B | Haematological tumors | Approved by EMA and FDA |
| Mosunetuzumab | KIH (IgG1-like bsAb) | CD20 (CD20; cancer cell) | Recruitment and activation of T cells [27] | Relapsed or refractory follicular lymphoma | Approved by EMA | ||
| Glofitamab | CrossMab/KIH(IgG-like bsAbs) | Recruitment and activation of T cells [1,28,29] | Diffuse large B-cell lymphoma | Phase II/III (NCT03075696, NCT04408638) | |||
| Teclistamab | Duobody | BCMA (TNFRSF17; cancer cell) | Recruitment and activation of T cells [30] | Multiple myeloma | Approved by EMA | ||
| Flotetuzumab | DART | CD123 (IL3RA; cancer cell) | Recruitment and activation of T cells [31] | Acute myeloid leukaemia | Phase II (NCT03739606) * | ||
| TCEs | CAR-T Cells |
|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrasco-Padilla, C.; Hernaiz-Esteban, A.; Álvarez-Vallina, L.; Aguilar-Sopeña, O.; Roda-Navarro, P. Bispecific Antibody Format and the Organization of Immunological Synapses in T Cell-Redirecting Strategies for Cancer Immunotherapy. Pharmaceutics 2023, 15, 132. https://doi.org/10.3390/pharmaceutics15010132
Carrasco-Padilla C, Hernaiz-Esteban A, Álvarez-Vallina L, Aguilar-Sopeña O, Roda-Navarro P. Bispecific Antibody Format and the Organization of Immunological Synapses in T Cell-Redirecting Strategies for Cancer Immunotherapy. Pharmaceutics. 2023; 15(1):132. https://doi.org/10.3390/pharmaceutics15010132
Chicago/Turabian StyleCarrasco-Padilla, Carlos, Alicia Hernaiz-Esteban, Luis Álvarez-Vallina, Oscar Aguilar-Sopeña, and Pedro Roda-Navarro. 2023. "Bispecific Antibody Format and the Organization of Immunological Synapses in T Cell-Redirecting Strategies for Cancer Immunotherapy" Pharmaceutics 15, no. 1: 132. https://doi.org/10.3390/pharmaceutics15010132