
Pharmacokinetics and Pharmacodynamics (PK/PD) of Corallopyronin A against Methicillin-Resistant Staphylococcus aureus
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
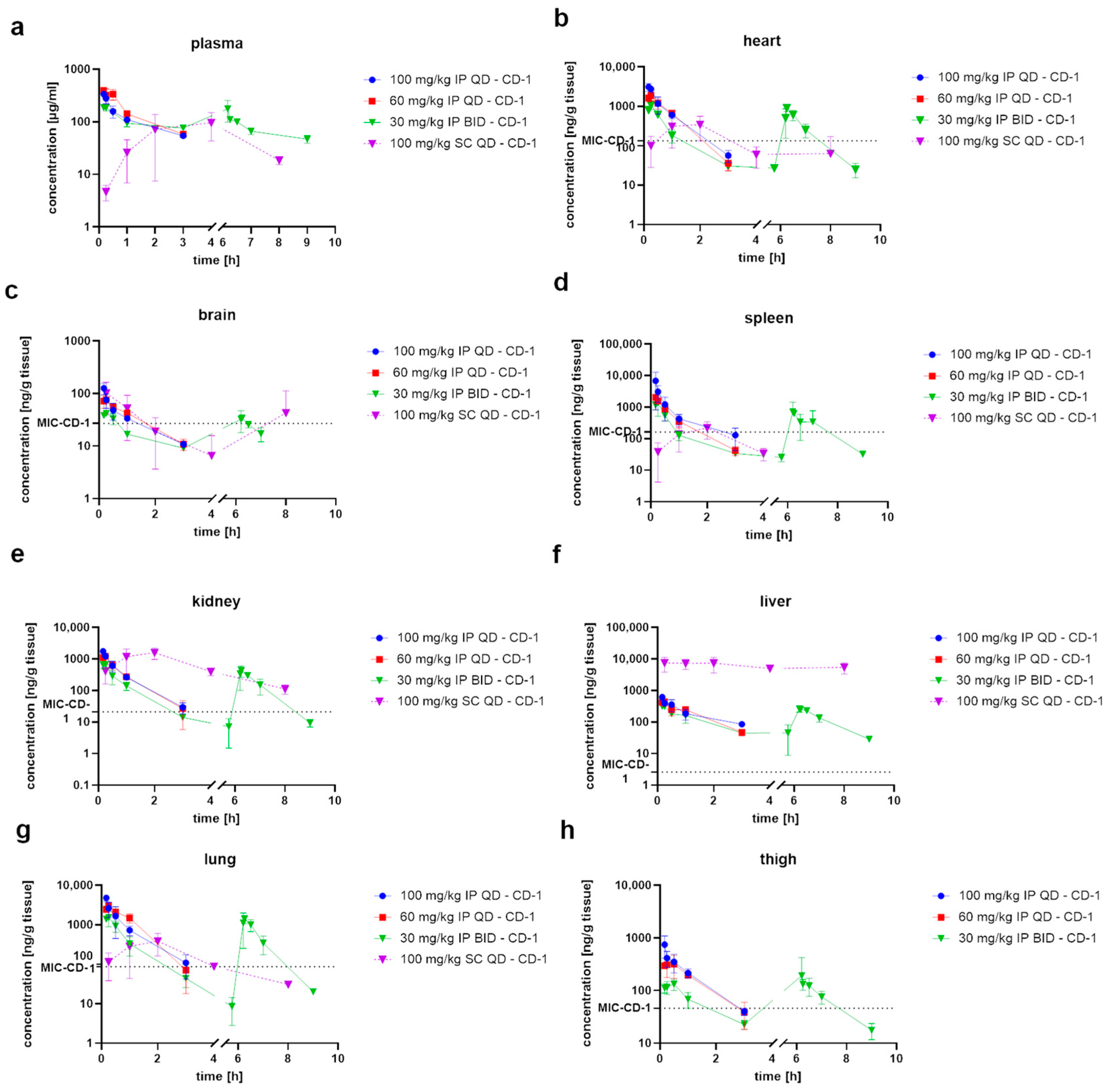
2.2. Pharmacokinetic Studies
2.3. Bioanalytics of PK and PD Studies
2.3.1. Bioanalytics of Plasma Samples
2.3.2. Bioanalytics of Organ Samples (PK and PD Samples)
2.4. Preparation of the Inoculum for Infection with S. aureus ATCC 33591
2.5. Neutropenic Thigh Infection Model with MRSA
2.6. Neutropenic Lung Infection Model with MRSA
2.7. Animals
2.8. Statistical Analysis
3. Results
3.1. Pharmacokinetic Evaluation of CorA
3.2. CorA Is Effective in the Neutropenic Thigh Infection Model
3.3. CorA Reduces Bacterial Burden in the Neutropenic Lung Infection Model
4. Discussion
4.1. Biodistribution Studies Form the Basis for Design of Efficacy Studies
4.2. CorA Shows Favorable Biodistribution after PO Administration Paving the Way for Additional Applications
4.3. In Vivo Proof of Concept of CorA as a Basis for Further Development
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014; ISBN 978-92-4-156474-8. [Google Scholar]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Miethke, M.; Pieroni, M.; Weber, T.; Brönstrup, M.; Hammann, P.; Halby, L.; Arimondo, P.B.; Glaser, P.; Aigle, B.; Bode, H.B.; et al. Towards the Sustainable Discovery and Development of New Antibiotics. Nat. Rev. Chem. 2021, 5, 726–749. [Google Scholar] [CrossRef] [PubMed]
- Irschik, H.; Jansen, R.; Höfle, G.; Gerth, K.; Reichenbach, H. The Corallopyronins, New Inhibitors of Bacterial RNA Synthesis from Myxobacteria. J. Antibiot. 1985, 38, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Krome, A.K.; Becker, T.; Kehraus, S.; Schiefer, A.; Gütschow, M.; Chaverra-Muñoz, L.; Hüttel, S.; Jansen, R.; Stadler, M.; Ehrens, A.; et al. Corallopyronin A: Antimicrobial Discovery to Preclinical Development. Nat. Prod. Rep. 2022, 39, 1705–1720. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, J.; Das, K.; Ismail, S.; Koppstein, D.; Jang, M.; Hudson, B.; Sarafianos, S.; Tuske, S.; Patel, J.; Jansen, R.; et al. The RNA Polymerase “Switch Region” Is a Target for Inhibitors. Cell 2008, 135, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Talaue, M.; Liu, S.; Degen, D.; Ebright, R.Y.; Sineva, E.; Chakraborty, A.; Druzhinin, S.Y.; Chatterjee, S.; Mukhopadhyay, J.; et al. New Target for Inhibition of Bacterial RNA Polymerase: ‘Switch Region’. Curr. Opin. Microbiol. 2011, 14, 532–543. [Google Scholar] [CrossRef] [Green Version]
- Kirsch, S.H.; Haeckl, F.P.J.; Müller, R. Beyond the Approved: Target Sites and Inhibitors of Bacterial RNA Polymerase from Bacteria and Fungi. Nat. Prod. Rep. 2022, 39, 1226–1263. [Google Scholar] [CrossRef]
- Belogurov, G.A.; Vassylyeva, M.N.; Sevostyanova, A.; Appleman, J.R.; Xiang, A.X.; Lira, R.; Webber, S.E.; Klyuyev, S.; Nudler, E.; Artsimovitch, I.; et al. Transcription Inactivation through Local Refolding of the RNA Polymerase Structure. Nature 2009, 457, 332–335. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.L.; Balthazar, J.T.; Esposito, D.L.A.; Ayala, J.C.; Schiefer, A.; Pfarr, K.; Hoerauf, A.; Alt, S.; Hesterkamp, T.; Grosse, M.; et al. Potent In Vitro and Ex Vivo Anti-Gonococcal Activity of the RpoB Inhibitor Corallopyronin A. mSphere 2022, 7, e00362-22. [Google Scholar] [CrossRef]
- Shima, K.; Ledig, S.; Loeper, N.; Schiefer, A.; Pfarr, K.; Hoerauf, A.; Graspeuntner, S.; Rupp, J. Effective Inhibition of Rifampicin-Resistant Chlamydia Trachomatis by the Novel DNA-Dependent RNA Polymerase Inhibitor Corallopyronin A. Int. J. Antimicrob. Agents 2018, 52, 523–524. [Google Scholar] [CrossRef] [PubMed]
- Loeper, N.; Graspeuntner, S.; Ledig, S.; Kaufhold, I.; Hoellen, F.; Schiefer, A.; Henrichfreise, B.; Pfarr, K.; Hoerauf, A.; Shima, K.; et al. Elaborations on Corallopyronin A as a Novel Treatment Strategy Against Genital Chlamydial Infections. Front. Microbiol. 2019, 10, 943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kock, F.; Hauptmann, M.; Osterloh, A.; Schäberle, T.F.; Poppert, S.; Frickmann, H.; Menzel, K.-D.; Peschel, G.; Pfarr, K.; Schiefer, A.; et al. Orientia Tsutsugamushi Is Highly Susceptible to the RNA Polymerase Switch Region Inhibitor Corallopyronin A In Vitro and In Vivo. Antimicrob. Agents Chemother. 2018, 62, e01732-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariner, K.; McPhillie, M.; Trowbridge, R.; Smith, C.; O’Neill, A.J.; Fishwick, C.W.G.; Chopra, I. Activity of and Development of Resistance to Corallopyronin A, an Inhibitor of RNA Polymerase. Antimicrob. Agents Chemother. 2011, 55, 2413–2416. [Google Scholar] [CrossRef] [Green Version]
- Balansky, J.; Pfarr, K.; Szekat, C.; Kehraus, S.; Aden, T.; Grosse, M.; Jansen, R.; Hesterkamp, T.; Schiefer, A.; König, G.M.; et al. The RNA Polymerase Inhibitor Corallopyronin A Has a Lower Frequency of Resistance Than Rifampicin in Staphylococcus aureus. Antibiotics 2022, 11, 920. [Google Scholar] [CrossRef]
- Schiefer, A.; Schmitz, A.; Schäberle, T.F.; Specht, S.; Lämmer, C.; Johnston, K.L.; Vassylyev, D.G.; König, G.M.; Hoerauf, A.; Pfarr, K. Corallopyronin A Specifically Targets and Depletes Essential Obligate Wolbachia Endobacteria From Filarial Nematodes In Vivo. J. Infect. Dis. 2012, 206, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Schäberle, T.F.; Schiefer, A.; Schmitz, A.; König, G.M.; Hoerauf, A.; Pfarr, K. Corallopyronin A—A Promising Antibiotic for Treatment of Filariasis. Int. J. Med. Microbiol. 2014, 304, 72–78. [Google Scholar] [CrossRef]
- Schäberle, T.F.; Schmitz, A.; Zocher, G.; Schiefer, A.; Kehraus, S.; Neu, E.; Roth, M.; Vassylyev, D.G.; Stehle, T.; Bierbaum, G.; et al. Insights into Structure–Activity Relationships of Bacterial RNA Polymerase Inhibiting Corallopyronin Derivatives. J. Nat. Prod. 2015, 78, 2505–2509. [Google Scholar] [CrossRef]
- Schiefer, A.; Hübner, M.P.; Krome, A.; Lämmer, C.; Ehrens, A.; Aden, T.; Koschel, M.; Neufeld, H.; Chaverra-Muñoz, L.; Jansen, R.; et al. Corallopyronin A for Short-Course Anti-Wolbachial, Macrofilaricidal Treatment of Filarial Infections. PLoS Negl. Trop. Dis. 2020, 14, e0008930. [Google Scholar] [CrossRef]
- Pogorevc, D.; Panter, F.; Schillinger, C.; Jansen, R.; Wenzel, S.C.; Müller, R. Production Optimization and Biosynthesis Revision of Corallopyronin A, a Potent Anti-Filarial Antibiotic. Metab. Eng. 2019, 55, 201–211. [Google Scholar] [CrossRef]
- Ehrens, A.; Schiefer, A.; Krome, A.; Becker, T.; Rox, K.; Neufeld, H.; Aden, T.; Wagner, K.; Müller, R.; Grosse, M.; et al. Pharmacology and Early ADMET Data of Corallopyronin A, a Natural Product with Macrofilaricidal Anti-Wolbachial Activity in Filarial Nematodes. Front. Trop. Dis. 2022, 3, 983107. [Google Scholar] [CrossRef]
- Krome, A.K.; Becker, T.; Kehraus, S.; Schiefer, A.; Steinebach, C.; Aden, T.; Frohberger, S.J.; López Mármol, Á.; Kapote, D.; Jansen, R.; et al. Solubility and Stability Enhanced Oral Formulations for the Anti-Infective Corallopyronin A. Pharmaceutics 2020, 12, 1105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An Add-in Program for Pharmacokinetic and Pharmacodynamic Data Analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- Rox, K. Influence of Tramadol on Bacterial Burden in the Standard Neutropenic Thigh Infection Model. Sci. Rep. 2022, 12, 19606. [Google Scholar] [CrossRef] [PubMed]
- Lawther, B.K.; Kumar, S.; Krovvidi, H. Blood–Brain Barrier. Contin. Educ. Anaest. Crit. Care Pain 2011, 11, 128–132. [Google Scholar] [CrossRef]
- Gleeson, M.P. Generation of a Set of Simple, Interpretable ADMET Rules of Thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef]
- Johnson, T.W.; Gallego, R.A.; Edwards, M.P. Lipophilic Efficiency as an Important Metric in Drug Design. J. Med. Chem. 2018, 61, 6401–6420. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.; Krome, A.K.; Vahdati, S.; Schiefer, A.; Pfarr, K.; Ehrens, A.; Aden, T.; Grosse, M.; Jansen, R.; Alt, S.; et al. In Vitro–In Vivo Relationship in Mini-Scale—Enabling Formulations of Corallopyronin A. Pharmaceutics 2022, 14, 1657. [Google Scholar] [CrossRef]
- Zelmer, A.R.; Nelson, R.; Richter, K.; Atkins, G.J. Can Intracellular Staphylococcus aureus in Osteomyelitis Be Treated Using Current Antibiotics? A Systematic Review and Narrative Synthesis. Bone Res. 2022, 10, 53. [Google Scholar] [CrossRef]
- Mouton, J.W.; Theuretzbacher, U.; Craig, W.A.; Tulkens, P.M.; Derendorf, H.; Cars, O. Tissue Concentrations: Do We Ever Learn? J. Antimicrob. Chemother. 2007, 61, 235–237. [Google Scholar] [CrossRef]
- Theuretzbacher, U. Tissue Penetration of Antibacterial Agents: How Should This Be Incorporated into Pharmacodynamic Analyses? Curr. Opin. Pharmacol. 2007, 7, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.; Scaglione, F.; Derendorf, H. Class-Dependent Relevance of Tissue Distribution in the Interpretation of Anti-Infective Pharmacokinetic/Pharmacodynamic Indices. Int. J. Antimicrob. Agents 2010, 35, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, P.G.; Bhavnani, S.M.; Rubino, C.M.; Louie, A.; Gumbo, T.; Forrest, A.; Drusano, G.L. Pharmacokinetics-Pharmacodynamics of Antimicrobial Therapy: It’s Not Just for Mice Anymore. Clin. Infect. Dis. 2007, 44, 79–86. [Google Scholar] [CrossRef]
- Andes, D.R.; Lepak, A.J. In Vivo Infection Models in the Pre-Clinical Pharmacokinetic/Pharmacodynamic Evaluation of Antimicrobial Agents. Curr. Opin. Pharmacol. 2017, 36, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Bulik, C.C.; Bader, J.C.; Zhang, L.; Wart, S.A.V.; Rubino, C.M.; Bhavnani, S.M.; Sweeney, K.L.; Ambrose, P.G. PK–PD Compass: Bringing Infectious Diseases Pharmacometrics to the Patient’s Bedside. J. Pharmacokinet. Pharmacodyn. 2017, 44, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Rox, K.; Heyner, M.; Krull, J.; Harmrolfs, K.; Rinne, V.; Hokkanen, J.; Perez Vilaro, G.; Díez, J.; Müller, R.; Kröger, A.; et al. Physiologically Based Pharmacokinetic/Pharmacodynamic Model for the Treatment of Dengue Infections Applied to the Broad Spectrum Antiviral Soraphen A. ACS Pharmacol. Transl. Sci. 2021, 4, 1499–1513. [Google Scholar] [CrossRef] [PubMed]
- van Os, W.; Zeitlinger, M. Predicting Antimicrobial Activity at the Target Site: Pharmacokinetic/Pharmacodynamic Indices versus Time-Kill Approaches. Antibiotics 2021, 10, 1485. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose Translation from Animal to Human Studies Revisited. FASEB J. 2007, 22, 659–661. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| 100 mg/kg IP | 60 mg/kg IP | 30 mg/kg IP | 100 mg/kg SC | |
|---|---|---|---|---|
| t1/2 [h] | 1.87 ± 0.8 | 1.10 ± 0.2 | 2.33 ± 0.3 | |
| Tmax [h] | 0.19 ± 0.0 | 0.19 ± 0.0 | 0.19 ± 0.0 | 3.33 ± 1.2 |
| Cmax [µg/mL] | 349.40 ± 12.4 | 416.03 ± 39.11 | 194.13 ± 15.6 | 107.1 ± 59.2 |
| AUC0-t [µg/Ml × h] | 339.53 ± 28.4 | 463.87 ± 18.8 | 303.88 ± 21.9 | 459.32 ± 238.2 |
| MRT [h] | 2.51 ± 1.0 | 1.55 ± 0.2 | 3.61 ± 0.6 | |
| Vz/F_obs [l/kg] | 0.54 ± 0.2 | 0.17 ± 0.0 | 0.18 ± 0.0 | |
| Cl/F_obs [ml/min/kg] | 3.43 ± 0.4 | 1.80 ± 0.0 | 0.89 ± 0.0 |
| Cmax [µg/g] | AUC [µg/g × h] | |||||||
|---|---|---|---|---|---|---|---|---|
| IP | SC | IP | SC | |||||
| Organ | 100 mg/kg | 60 mg/kg | 30 mg/kg | 100 mg/kg | 100 mg/kg | 60 mg/kg | 30 mg/kg | 100 mg/kg |
| Liver | 0.61 | 0.43 | 0.31 | 2.3 | 0.59 | 0.57 | 0.40 | 12.44 |
| Heart | 3.1 | 1.8 | 1.1 | 1.48 | 2.10 | 1.83 | 1.08 | 3.53 |
| Spleen | 4.9 | 2.1 | 1.4 | 0.6 | 2.55 | 1.34 | 0.77 | 1.4 |
| Brain | 0.1 | 0.07 | 0.04 | 0.15 | 0.10 | 0.10 | 0.05 | 0.24 |
| Lung | 4.8 | 3.1 | 1.5 | 1.27 | 2.67 | 3.56 | 1.2 | 3.85 |
| Kidney | 1.8 | 1.2 | 0.7 | 1.76 | 1.01 | 0.95 | 0.49 | 5.61 |
| Thigh | 0.7 | 0.3 | 0.1 | - | 0.60 | 0.49 | 0.19 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rox, K.; Becker, T.; Schiefer, A.; Grosse, M.; Ehrens, A.; Jansen, R.; Aden, T.; Kehraus, S.; König, G.M.; Krome, A.K.; et al. Pharmacokinetics and Pharmacodynamics (PK/PD) of Corallopyronin A against Methicillin-Resistant Staphylococcus aureus. Pharmaceutics 2023, 15, 131. https://doi.org/10.3390/pharmaceutics15010131
Rox K, Becker T, Schiefer A, Grosse M, Ehrens A, Jansen R, Aden T, Kehraus S, König GM, Krome AK, et al. Pharmacokinetics and Pharmacodynamics (PK/PD) of Corallopyronin A against Methicillin-Resistant Staphylococcus aureus. Pharmaceutics. 2023; 15(1):131. https://doi.org/10.3390/pharmaceutics15010131
Chicago/Turabian StyleRox, Katharina, Tim Becker, Andrea Schiefer, Miriam Grosse, Alexandra Ehrens, Rolf Jansen, Tilman Aden, Stefan Kehraus, Gabriele M. König, Anna K. Krome, and et al. 2023. "Pharmacokinetics and Pharmacodynamics (PK/PD) of Corallopyronin A against Methicillin-Resistant Staphylococcus aureus" Pharmaceutics 15, no. 1: 131. https://doi.org/10.3390/pharmaceutics15010131