
Oral Nanomedicines for siRNA Delivery to Treat Inflammatory Bowel Disease
 , and
, and
Abstract
:1. Introduction
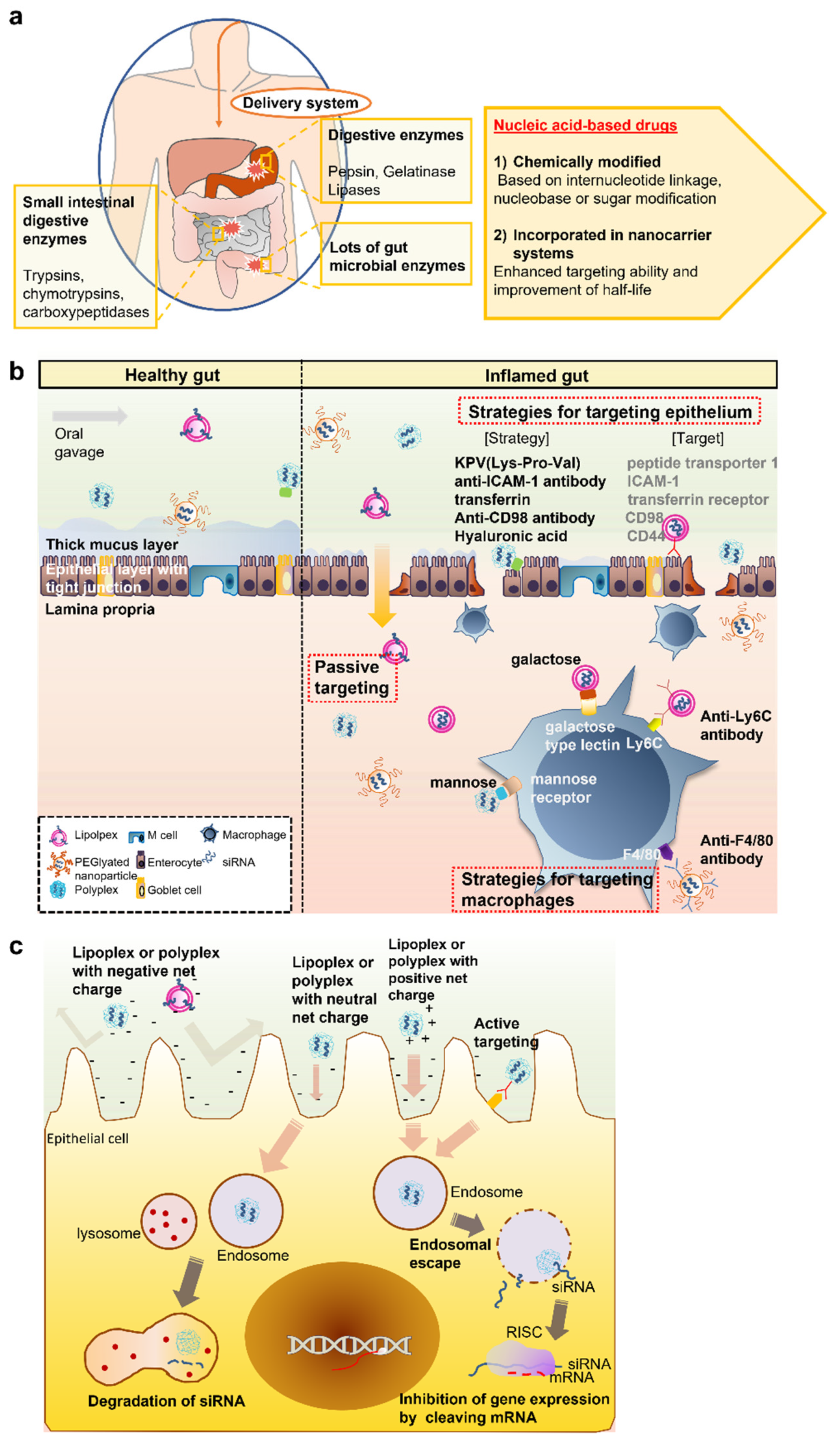
2. Physiological and Biological Barriers and Strategies for the Oral Delivery of siRNA Drugs
2.1. Biochemical Barriers
2.2. Intestinal Barrier: Mucus and the Intestinal Epithelium
2.3. Cellular Uptake Barrier
3. Oral siRNA Delivery for IBD Treatments
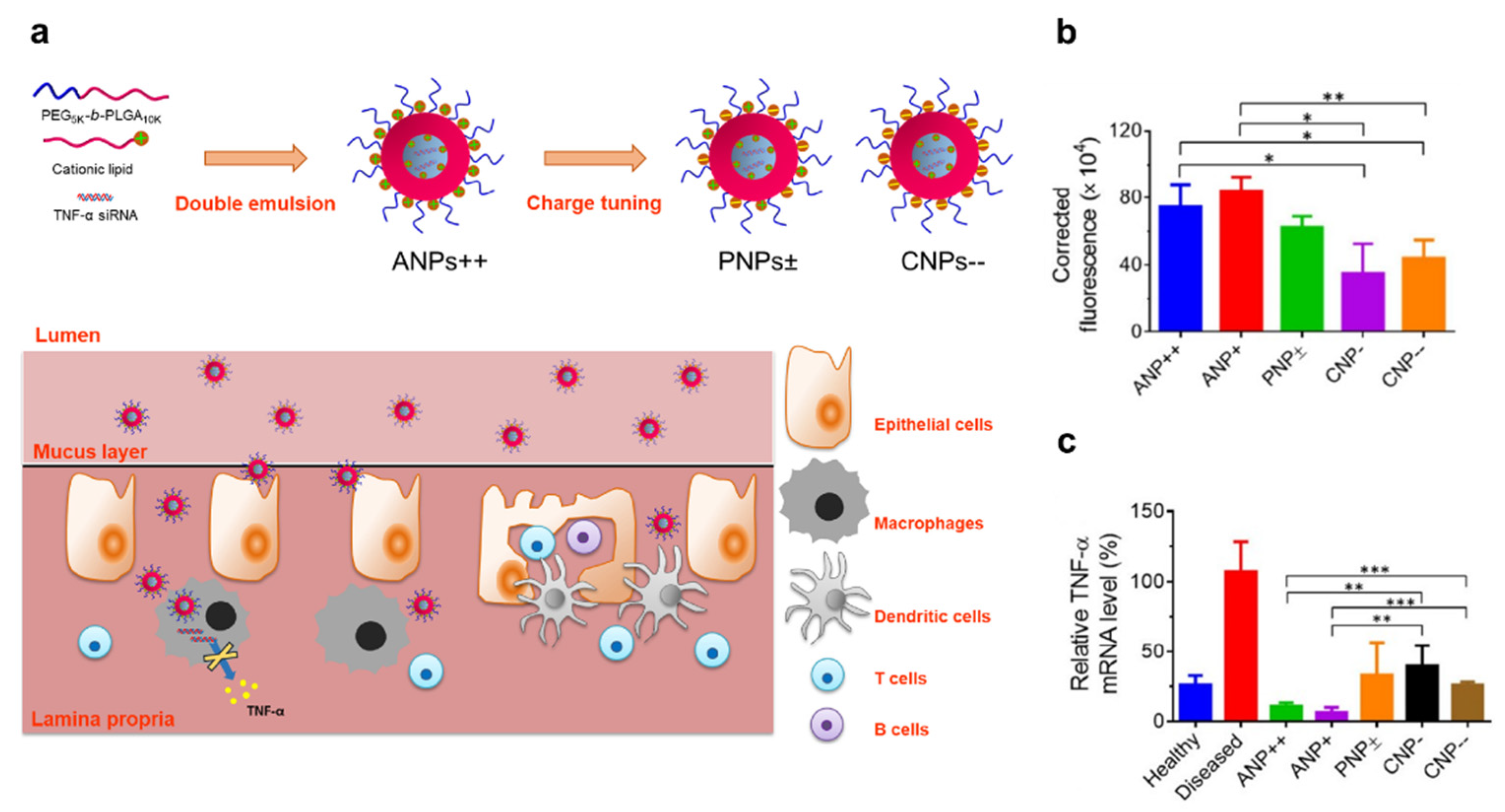
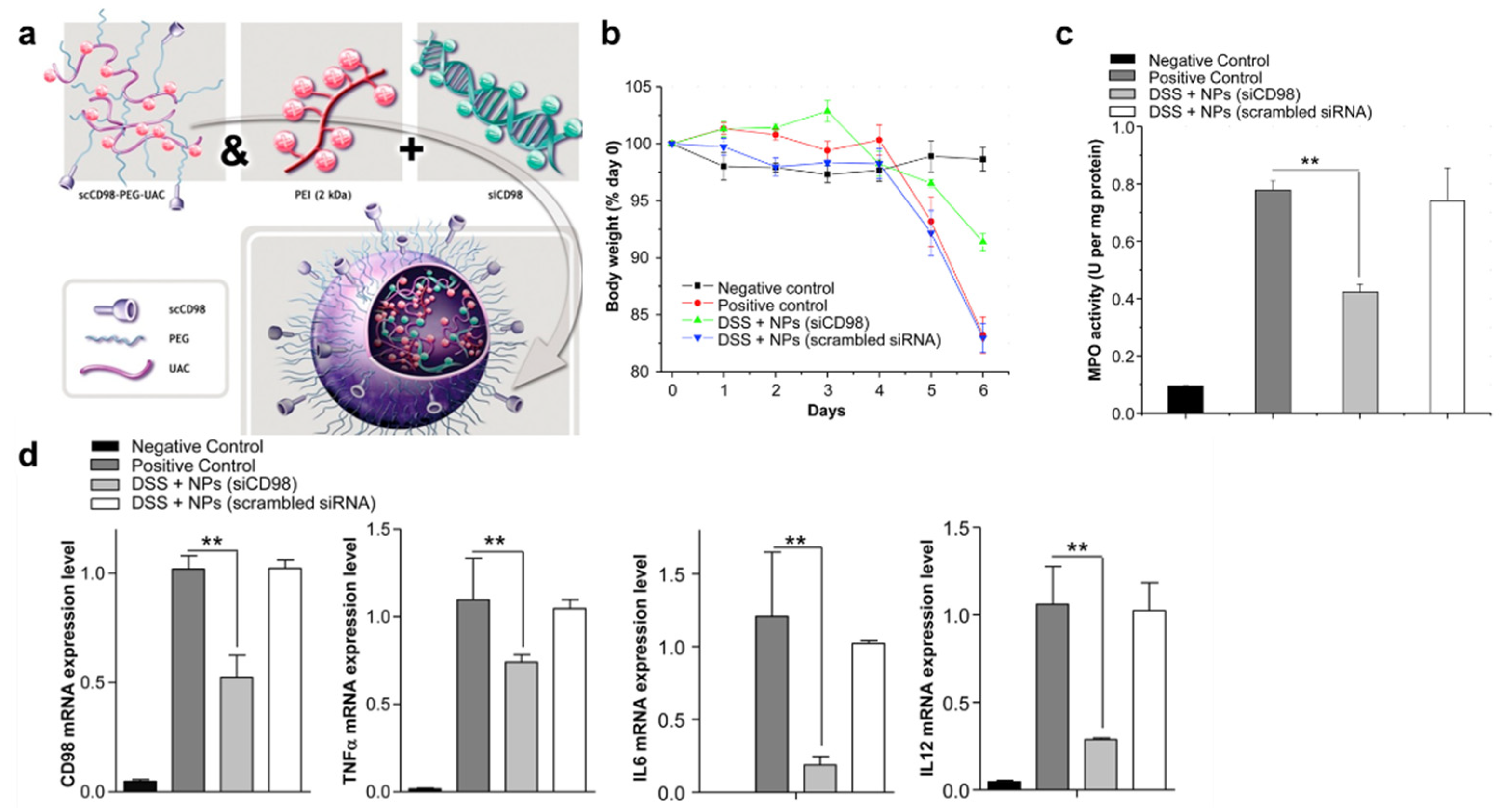
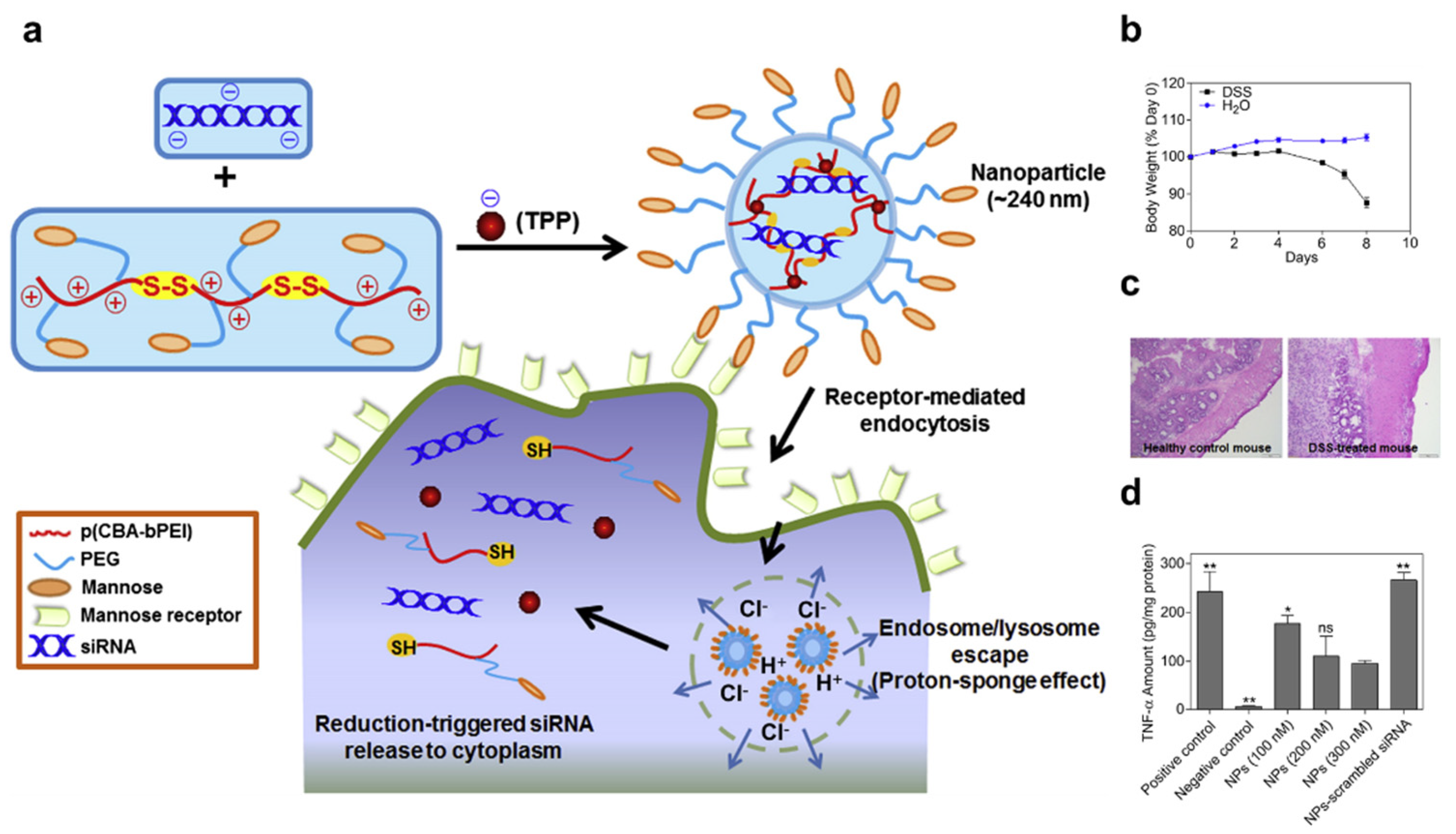
3.1. Polyplex or Lipoplex
3.2. Polyplex or Lipoplex Embedded in Hydrogels or Polymeric Materials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| siRNA Delivery Methods | Nanomaterials | siRNA | Target | References | |
|---|---|---|---|---|---|
| Lipo/polyplex formation | - | PEGylated poly/lipoplex | TNF-α siRNA | DSS-induced colitis (in vivo) | [89] |
| Polymeric micelles | Luciferase GL3 siRNA | Caco 2 cell (in vitro) | [90] | ||
| Ginger-derived lipid nanoparticle | CD98 siRNA | Colon-26 and RAW 264.7 cells (in vitro), CD98 mRNA expression in intestinal tract (in vivo) | [94] | ||
| With targeting ligands | Galactosylated trimethyl chitosan-cysteine nanoparticle | Map4k4 siRNA | Raw 264.7 cell (in vitro), DSS-induced colitis (in vivo) | [80] | |
| Mannosylated nanoparticle | TNF-α siRNA | Raw 264.7 cell (in vitro), DSS-induced colitis (ex vivo) | [98] | ||
| Lipo/polyplex embedded in hydrogels or polymeric materials | - | Type B gelatin nanoparticle in PCL microsphere | TNF-α siRNA | DSS-induced colitis (in vivo) | [100] |
| pH-responsive nanogel in trypsin-mediated degradable microgel | TNF-α siRNA | Raw 264.7 cell (in vitro) | [102] | ||
| 1,3-D-glucan-shell polyplex in PEI | Map4k4 siRNA | LPS-induced colitis (in vivo) | [103] | ||
| With targeting ligands | PEI polyplex in chitosan/alginate hydrogel with scCD98 antibody | CD98 siRNA | Colon-26 and RAW 264.7 cells (in vitro), DSS-induced colitis (in vivo) | [77] | |
| PEG-PLA nanoparticle in chitosan/alginate hydrogel with anti-F4/80 antibody | TNF-α siRNA | Raw 264.7 cell (in vitro), DSS-induced colitis (in vivo) | [81] | ||
4. Perspectives, Challenges, and Future Research Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANP | Aminated nanoparticle |
| CD | Crohn’s disease |
| CNP | Carboxylated nanoparticle |
| CRC | Colorectal cancer |
| DEAEMA | Diethylaminoethyl Methacrylate |
| DOPA | 1,2-dioleoyl-sn-glycero-3-phosphate |
| DOTA | 1,4,7,10-tetraazacyclododecane1,4,7,10-tetraacetic acid |
| DOTAP | 1,2-dioleoyl-3-trimethylammonium-propane |
| DSPC | Distearoylphosphatidylcholine |
| DSS | Dextran sodium sulfate |
| EDTA | Ethylenediamine tetraacetic acid |
| GAPDH | Glyceraldehyde-3-Phosphate Dehydrogenase |
| GDLN | Ginger-derived lipid nanoparticle |
| GeRP | Glucan-Encapsulated siRNA Particle |
| GI | Gastrointestinal |
| GIT | Gastrointestinal tract |
| HABN | Hyaluronic acid-bilirubin nanomedicine |
| hATTR | Hereditary transthyretin-mediated amyloidosis |
| IBD | Inflammatory bowel disease |
| ICAM-1 | Intercellular adhesion molecule-1 |
| LNP | Lipoid nanoparticles |
| LPS | Lipopolysaccharide |
| Ly6C | Lymphocyte antigen 6 complex |
| MAA | Methacrylic acid |
| MOA | Mode of action |
| MPO | Myeloperoxidase |
| NiMOS | Nanoparticles-in-microsphere oral system |
| NVP | N-Vinylpyrrolidone |
| PCL | poly(epsilon-caprolactone) |
| pDNA | Plasmid DNA |
| PEC | Polyelectrolyte complex |
| PEG | Polyethylene glycol |
| PEI | Polyethyleneimine |
| PLGA | Poly(lactic-co-glycolic acid) |
| PNP | Plain nanoparticle |
| RNAi | RNA interference |
| ROS | Reactive oxygen species |
| scCD98 | Single-chain CD98 |
| SEDDS | Self-emulsifying drug delivery system |
| siRNA | Small interfering RNA |
| TKN | Thioketal nanoparticle |
| UC | Ulcerative colitis |
References
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastro. Hepat. 2015, 12, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.C.; Chong, C.A.; Chong, R.Y. National estimates of the burden of inflammatory bowel disease among racial and ethnic groups in the United States. J. Crohns Colitis 2014, 8, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastro. Hepat. 2015, 12, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kamada, N.; Moon, J.J. Oral nanomedicine for modulating immunity, intestinal barrier functions, and gut microbiome. Adv. Drug Deliver Rev. 2021, 179, 114021. [Google Scholar] [CrossRef]
- Lee, Y.; Sugihara, K.; Gillilland, M.G.; Jon, S.; Kamada, N.; Moon, J.J. Hyaluronic acid-bilirubin nanomedicine for targeted modulation of dysregulated intestinal barrier, microbiome and immune responses in colitis. Nat. Mater. 2020, 19, 118–126. [Google Scholar] [CrossRef]
- Cunliffe, R.N.; Scott, B.B. Review article: Monitoring for drug side-effects in inflammatory bowel disease. Aliment. Pharm. Ther. 2002, 16, 647–662. [Google Scholar] [CrossRef]
- Mason, M.; Siegel, C.A. Do Inflammatory Bowel Disease Therapies Cause Cancer? Inflamm. Bowel. Dis. 2013, 19, 1306–1321. [Google Scholar] [CrossRef]
- Kurosawa, M.; Nakagawa, S.; Mizuashi, M.; Sasaki, Y.; Kawamura, M.; Saito, M.; Aiba, S. A comparison of the efficacy, relapse rate and side effects among three modalities of systemic corticosteroid therapy for alopecia areata. Dermatology 2006, 212, 361–365. [Google Scholar] [CrossRef]
- Atreya, R.; Neurath, M.F.; Siegmund, B. Personalizing Treatment in IBD: Hype or Reality in 2020? Can We Predict Response to Anti-TNF? Front. Med.-Lausanne 2020, 7, 517. [Google Scholar] [CrossRef]
- Roy, K.; Mao, H.Q.; Huang, S.K.; Leong, K.W. Oral gene delivery with chitosan-DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat. Med. 1999, 5, 387–391. [Google Scholar] [CrossRef]
- Kriegel, C.; Amiji, M.M. Dual TNF-alpha/Cyclin D1 Gene Silencing With an Oral Polymeric Microparticle System as a Novel Strategy for the Treatment of Inflammatory Bowel Disease. Clin. Transl. Gastroenterol. 2011, 2, e2. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, X.; Fang, Z.; Zhou, H. Knockdown of KLK12 inhibits viability and induces apoptosis in human colorectal cancer HT-29 cell line. Int. J. Mol. Med. 2019, 44, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.R.; Liu, C.Q.; Feng, B.S.; Liu, Z.J. Dysregulation of mucosal immune response in pathogenesis of inflammatory bowel disease. World J. Gastroentero. 2014, 20, 3255–3264. [Google Scholar] [CrossRef] [PubMed]
- ten Hove, T.; van Montfrans, C.; Peppelenbosch, M.P.; van Deventer, S.J. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn’s disease. Gut 2002, 50, 206–211. [Google Scholar] [CrossRef]
- Flamant, M.; Paul, S.; Roblin, X. Golimumab for the treatment of ulcerative colitis. Expert Opin. Biol. Ther. 2017, 17, 879–886. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Hanauer, S.B.; Rutgeerts, P.; Fedorak, R.N.; Lukas, M.; MacIntosh, D.G.; Panaccione, R.; Wolf, D.; Kent, J.D.; Bittle, B.; et al. Adalimumab for maintenance treatment of Crohn’s disease: Results of the CLASSIC II trial. Gut 2007, 56, 1232–1239. [Google Scholar] [CrossRef]
- Moss, A.C.; Farrell, R.J. Infliximab for induction and maintenance therapy for ulcerative colitis. Gastroenterology 2006, 131, 1649–1651, discussion 1651. [Google Scholar] [CrossRef]
- Reinisch, W.; Sandborn, W.J.; Hommes, D.W.; D’Haens, G.; Hanauer, S.; Schreiber, S.; Panaccione, R.; Fedorak, R.N.; Tighe, M.B.; Huang, B.; et al. Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: Results of a randomised controlled trial. Gut 2011, 60, 780–787. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Marano, C.; Zhang, H.; Strauss, R.; Johanns, J.; Adedokun, O.J.; Guzzo, C.; Colombel, J.F.; Reinisch, W.; et al. Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology 2014, 146, 85–95, quiz e14–85. [Google Scholar] [CrossRef]
- Singh, S.; Heien, H.C.; Sangaralingham, L.R.; Schilz, S.R.; Kappelman, M.D.; Shah, N.D.; Loftus, E.V. Comparative effectiveness and safety of infliximab and adalimumab in patients with ulcerative colitis. Aliment. Pharmacol. Ther. 2016, 43, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- D’Haens, G.R.; Panaccione, R.; Higgins, P.D.; Vermeire, S.; Gassull, M.; Chowers, Y.; Hanauer, S.B.; Herfarth, H.; Hommes, D.W.; Kamm, M.; et al. The London Position Statement of the World Congress of Gastroenterology on Biological Therapy for IBD with the European Crohn’s and Colitis Organization: When to start, when to stop, which drug to choose, and how to predict response? Am. J. Gastroenterol. 2011, 106, 199–212; quiz 213. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, L.; Ron, Y.; Kivity, S.; Ben-Horin, S.; Israeli, E.; Fraser, G.M.; Dotan, I.; Chowers, Y.; Confino-Cohen, R.; Weiss, B. Infliximab-Related Infusion Reactions: Systematic Review. J. Crohns. Colitis. 2015, 9, 806–815. [Google Scholar] [CrossRef]
- Parakkal, D.; Sifuentes, H.; Semer, R.; Ehrenpreis, E.D. Hepatosplenic T-cell lymphoma in patients receiving TNF-alpha inhibitor therapy: Expanding the groups at risk. Eur. J. Gastroenterol. Hepatol. 2011, 23, 1150–1156. [Google Scholar] [CrossRef] [PubMed]
- Blair, S.A.; Kane, S.V.; Clayburgh, D.R.; Turner, J.R. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab. Invest. 2006, 86, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Marafini, I.; Monteleone, G. Inflammatory bowel disease: New therapies from antisense oligonucleotides. Ann. Med. 2018, 50, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Tieu, T.; Wei, Y.K.; Cifuentes-Rius, A.; Voelcker, N.H. Overcoming Barriers: Clinical Translation of siRNA Nanomedicines. Adv. Ther. Ger. 2021, 4, 2100108. [Google Scholar] [CrossRef]
- Sant, S.; Tao, S.L.; Fisher, O.Z.; Xu, Q.; Peppas, N.A.; Khademhosseini, A. Microfabrication technologies for oral drug delivery. Adv. Drug Deliv. Rev. 2012, 64, 496–507. [Google Scholar] [CrossRef]
- Sastry, S.V.; Nyshadham, J.R.; Fix, J.A. Recent technological advances in oral drug delivery-a review. Pharm. Sci. Technol. Today 2000, 3, 138–145. [Google Scholar] [CrossRef]
- Forbes, D.C.; Peppas, N.A. Oral delivery of small RNA and DNA. J. Control. Release 2012, 162, 438–445. [Google Scholar] [CrossRef]
- Akhtar, S. Oral delivery of siRNA and antisense oligonucleotides. J. Drug Target. 2009, 17, 491–495. [Google Scholar] [CrossRef]
- O’Neill, M.J.; Bourre, L.; Melgar, S.; O’Driscoll, C.M. Intestinal delivery of non-viral gene therapeutics: Physiological barriers and preclinical models. Drug Discov. Today 2011, 16, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Pereira de Sousa, I.; Bernkop-Schnurch, A. Pre-systemic metabolism of orally administered drugs and strategies to overcome it. J. Control. Release 2014, 192, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Neal-Kluever, A.; Fisher, J.; Grylack, L.; Kakiuchi-Kiyota, S.; Halpern, W. Physiology of the Neonatal Gastrointestinal System Relevant to the Disposition of Orally Administered Medications. Drug Metab. Dispos. 2019, 47, 296–313. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Leblond, C.P. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian Theory of the origin of the four epithelial cell types. Am. J. Anat. 1974, 141, 537–561. [Google Scholar] [CrossRef] [PubMed]
- Ensign, L.M.; Cone, R.; Hanes, J. Oral drug delivery with polymeric nanoparticles: The gastrointestinal mucus barriers. Adv. Drug Deliv. Rev. 2012, 64, 557–570. [Google Scholar] [CrossRef]
- Dressman, J.B.; Berardi, R.R.; Dermentzoglou, L.C.; Russell, T.L.; Schmaltz, S.P.; Barnett, J.L.; Jarvenpaa, K.M. Upper gastrointestinal (GI) pH in young, healthy men and women. Pharm. Res. 1990, 7, 756–761. [Google Scholar] [CrossRef]
- Rouge, N.; Buri, P.; Doelker, E. Drug absorption sites in the gastrointestinal tract and dosage forms for site-specific delivery. Int. J. Pharm. 1996, 136, 117–139. [Google Scholar] [CrossRef]
- Zhang, Y.; Thanou, M.; Vllasaliu, D. Exploiting disease-induced changes for targeted oral delivery of biologics and nanomedicines in inflammatory bowel disease. Eur. J. Pharm. Biopharm. 2020, 155, 128–138. [Google Scholar] [CrossRef]
- Cui, M.; Zhang, M.; Liu, K. Colon-targeted drug delivery of polysaccharide-based nanocarriers for synergistic treatment of inflammatory bowel disease: A review. Carbohydr. Polym. 2021, 272, 118530. [Google Scholar] [CrossRef]
- Nunes, R.; Neves, J.D.; Sarmento, B. Nanoparticles for the regulation of intestinal inflammation: Opportunities and challenges. Nanomedicine 2019, 14, 2631–2644. [Google Scholar] [CrossRef]
- Hua, S.; Marks, E.; Schneider, J.J.; Keely, S. Advances in oral nano-delivery systems for colon targeted drug delivery in inflammatory bowel disease: Selective targeting to diseased versus healthy tissue. Nanomed. Nanotechnol. 2015, 11, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- Malayandi, R.; Kondamudi, P.K.; Ruby, P.K.; Aggarwal, D. Biopharmaceutical considerations and characterizations in development of colon targeted dosage forms for inflammatory bowel disease. Drug Deliv. Transl. Res. 2014, 4, 187–202. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, C.M.; Bernkop-Schnurch, A.; Friedl, J.D.; Preat, V.; Jannin, V. Oral delivery of non-viral nucleic acid-based therapeutics—do we have the guts for this? Eur. J. Pharm. Sci. 2019, 133, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.D.; Whitehead, K.A.; Mitragotri, S. Materials for oral delivery of proteins and peptides. Nat. Rev. Mater. 2020, 5, 127–148. [Google Scholar] [CrossRef]
- Duran-Lobato, M.; Niu, Z.; Alonso, M.J. Oral Delivery of Biologics for Precision Medicine. Adv. Mater. 2020, 32, e1901935. [Google Scholar] [CrossRef]
- Deleavey, G.F.; Damha, M.J. Designing Chemically Modified Oligonucleotides for Targeted Gene Silencing. Chem. Biol. 2012, 19, 937–954. [Google Scholar] [CrossRef]
- Loretz, B.; Foger, F.; Werle, M.; Bernkop-Schnurch, A. Oral gene delivery: Strategies to improve stability of pDNA towards intestinal digestion. J. Drug Target. 2006, 14, 311–319. [Google Scholar] [CrossRef]
- Lechanteur, A.; Sanna, V.; Duchemin, A.; Evrard, B.; Mottet, D.; Piel, G. Cationic Liposomes Carrying siRNA: Impact of Lipid Composition on Physicochemical Properties, Cytotoxicity and Endosomal Escape. Nanomaterials 2018, 8, 270. [Google Scholar] [CrossRef]
- Hauptstein, S.; Prufert, F.; Bernkop-Schnurch, A. Self-nanoemulsifying drug delivery systems as novel approach for pDNA drug delivery. Int. J. Pharm. 2015, 487, 25–31. [Google Scholar] [CrossRef]
- Girod, S.; Zahm, J.M.; Plotkowski, C.; Beck, G.; Puchelle, E. Role of the physiochemical properties of mucus in the protection of the respiratory epithelium. Eur. Respir. J. 1992, 5, 477–487. [Google Scholar] [PubMed]
- Harbitz, O.; Jenssen, A.O.; Smidsrod, O. Lysozyme and lactoferrin in sputum from patients with chronic obstructive lung disease. Eur. J. Respir. Dis. 1984, 65, 512–520. [Google Scholar] [PubMed]
- Lieleg, O.; Ribbeck, K. Biological hydrogels as selective diffusion barriers. Trends. Cell Biol. 2011, 21, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Sanders, N.; Rudolph, C.; Braeckmans, K.; De Smedt, S.C.; Demeester, J. Extracellular barriers in respiratory gene therapy. Adv. Drug Deliv. Rev. 2009, 61, 115–127. [Google Scholar] [CrossRef]
- Thornton, D.J.; Sheehan, J.K. From mucins to mucus: Toward a more coherent understanding of this essential barrier. Proc. Am. Thorac. Soc. 2004, 1, 54–61. [Google Scholar] [CrossRef]
- Lamblin, G.; Lhermitte, M.; Klein, A.; Houdret, N.; Scharfman, A.; Ramphal, R.; Roussel, P. The carbohydrate diversity of human respiratory mucins: A protection of the underlying mucosa? Am. Rev. Respir. Dis. 1991, 144, S19–S24. [Google Scholar] [CrossRef]
- Thornton, D.J.; Rousseau, K.; McGuckin, M.A. Structure and function of the polymeric mucins in airways mucus. Annu. Rev. Physiol. 2008, 70, 459–486. [Google Scholar] [CrossRef]
- Cao, X.; Bansil, R.; Bhaskar, K.R.; Turner, B.S.; LaMont, J.T.; Niu, N.; Afdhal, N.H. pH-dependent conformational change of gastric mucin leads to sol-gel transition. Biophys. J. 1999, 76, 1250–1258. [Google Scholar] [CrossRef]
- Raynal, B.D.; Hardingham, T.E.; Sheehan, J.K.; Thornton, D.J. Calcium-dependent protein interactions in MUC5B provide reversible cross-links in salivary mucus. J. Biol. Chem. 2003, 278, 28703–28710. [Google Scholar] [CrossRef]
- de Sousa, I.P.; Steiner, C.; Schmutzler, M.; Wilcox, M.D.; Veldhuis, G.J.; Pearson, J.P.; Huck, C.W.; Salvenmoser, W.; Bernkop-Schnurch, A. Mucus permeating carriers: Formulation and characterization of highly densely charged nanoparticles. Eur. J. Pharm. Biopharm. 2015, 97, 273–279. [Google Scholar] [CrossRef]
- Ball, R.L.; Bajaj, P.; Whitehead, K.A. Oral delivery of siRNA lipid nanoparticles: Fate in the GI tract. Sci. Rep. 2018, 8, 2178. [Google Scholar] [CrossRef]
- Griesser, J.; Hetenyi, G.; Kadas, H.; Demarne, F.; Jannin, V.; Bernkop-Schnurch, A. Self-emulsifying peptide drug delivery systems: How to make them highly mucus permeating. Int. J. Pharm. 2018, 538, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.S.; Xu, Q.G.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radical Res. 2018, 52, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Gao, Z.; Chen, L.; Kang, L.; Huang, W.; Jin, M.; Wang, Q.; Bae, Y.H. Multifunctional oral delivery systems for enhanced bioavailability of therapeutic peptides/proteins. Acta. Pharm. Sin. B 2019, 9, 902–922. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Ho, S.B. Intestinal goblet cells and mucins in health and disease: Recent insights and progress. Curr. Gastroenterol. Rep. 2010, 12, 319–330. [Google Scholar] [CrossRef]
- Johansson, M.E.; Larsson, J.M.; Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 4659–4665. [Google Scholar] [CrossRef]
- Sellers, R.S.; Morton, D. The colon: From banal to brilliant. Toxicol. Pathol. 2014, 42, 67–81. [Google Scholar] [CrossRef]
- Nicoletti, C. Unsolved mysteries of intestinal M cells. Gut 2000, 47, 735–739. [Google Scholar] [CrossRef]
- Fox, C.B.; Kim, J.; Le, L.V.; Nemeth, C.L.; Chirra, H.D.; Desai, T.A. Micro/nanofabricated platforms for oral drug delivery. J. Control. Release 2015, 219, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Citi, S. Intestinal barriers protect against disease. Science 2018, 359, 1097–1098. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Merlin, D. Nanoparticle-Mediated Drug Delivery Systems For The Treatment Of IBD: Current Perspectives. Int. J. Nanomed. 2019, 14, 8875–8889. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sun, M.H.; Wang, D.; Li, G.Y.; Huang, J.G.; Tan, S.W.; Bao, L.; Li, Q.; Li, G.; Si, L.Q. A PepT1 mediated medicinal nano-system for targeted delivery of cyclosporine A to alleviate acute severe ulcerative colitis. Biomater. Sci. 2019, 7, 4299–4309. [Google Scholar] [CrossRef] [PubMed]
- Dalmasso, G.; Charrier-Hisamuddin, L.; Nguyen, H.T.T.; Yan, Y.; Sitaraman, S.; Merlin, D. PepT1-mediated tripeptide KPV uptake reduces intestinal inflammation. Gastroenterology 2008, 134, 166–178. [Google Scholar] [CrossRef]
- Mane, V.; Muro, S. Biodistribution and endocytosis of ICAM-1-targeting antibodies versus nanocarriers in the gastrointestinal tract in mice. Int. J. Nanomed. 2012, 7, 4223–4237. [Google Scholar] [CrossRef]
- Harel, E.; Rubinstein, A.; Nissan, A.; Khazanov, E.; Milbauer, M.N.; Barenholz, Y.; Tirosh, B. Enhanced Transferrin Receptor Expression by Proinflammatory Cytokines in Enterocytes as a Means for Local Delivery of Drugs to Inflamed Gut Mucosa. PLoS ONE 2011, 6, e24202. [Google Scholar] [CrossRef]
- Xiao, B.; Laroui, H.; Viennois, E.; Ayyadurai, S.; Charania, M.A.; Zhang, Y.C.; Zhang, Z.; Baker, M.T.; Zhang, B.Y.; Gewirtz, A.T.; et al. Nanoparticles With Surface Antibody Against CD98 and Carrying CD98 Small Interfering RNA Reduce Colitis in Mice. Gastroenterology 2014, 146, 1289–1300. [Google Scholar] [CrossRef]
- Xiao, B.; Zhang, Z.; Viennois, E.; Kang, Y.J.; Zhang, M.Z.; Hang, M.K.; Chen, J.C.; Merlin, D. Combination Therapy for Ulcerative Colitis: Orally Targeted Nanoparticles Prevent Mucosal Damage and Relieve Inflammation. Theranostics 2016, 6, 2250–2266. [Google Scholar] [CrossRef]
- He, C.B.; Yue, H.M.; Xu, L.; Liu, Y.F.; Song, Y.D.; Tang, C.; Yin, C.H. siRNA release kinetics from polymeric nanoparticles correlate with RNAi efficiency and inflammation therapy via oral delivery. Acta. Biomater. 2020, 103, 213–222. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, C.; Yin, C.H. Galactosylated trimethyl chitosan-cysteine nanoparticles loaded with Map4k4 siRNA for targeting activated macrophages. Biomaterials 2013, 34, 3667–3677. [Google Scholar] [CrossRef]
- Laroui, H.; Viennois, E.; Xiao, B.; Canup, B.S.B.; Geem, D.; Denning, T.L.; Merlin, D. Fab’-bearing siRNA TNF alpha-loaded nanoparticles targeted to colonic macrophages offer an effective therapy for experimental colitis. J. Control. Release 2014, 186, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Veiga, N.; Goldsmith, M.; Granot, Y.; Rosenblum, D.; Dammes, N.; Kedmi, R.; Ramishetti, S.; Peer, D. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 2018, 9, 4493. [Google Scholar] [CrossRef] [PubMed]
- Shinn, J.; Kwon, N.; Lee, S.A.; Lee, Y. Smart pH-responsive nanomedicines for disease therapy. J. Pharm. Invest. 2022, 52, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Escriou, V.; Ciolina, C.; Helbling-Leclerc, A.; Wils, P.; Scherman, D. Cationic lipid-mediated gene transfer: Analysis of cellular uptake and nuclear import of plasmid DNA. Cell Biol. Toxicol. 1998, 14, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Poon, G.M.K.; Gariepy, J. Cell-surface proteoglycans as molecular portals for cationic peptide and polymer entry into cells. Biochem. Soc. T 2007, 35, 788–793. [Google Scholar] [CrossRef]
- Zhang, S.B.; Zhao, B.; Jiang, H.M.; Wang, B.; Ma, B.C. Cationic lipids and polymers mediated vectors for delivery of siRNA. J. Control. Release 2007, 123, 1–10. [Google Scholar] [CrossRef]
- Wang, T.; Upponi, J.R.; Torchilin, V.P. Design of multifunctional non-viral gene vectors to overcome physiological barriers: Dilemmas and strategies. Int. J. Pharm. 2012, 427, 3–20. [Google Scholar] [CrossRef]
- Bernkop-Schnurch, A. Strategies to overcome the polycation dilemma in drug delivery. Adv. Drug Deliv. Rev. 2018, 136, 62–72. [Google Scholar] [CrossRef]
- Iqbal, S.; Du, X.J.; Wang, J.L.; Li, H.J.; Yuan, Y.Y.; Wang, J. Surface charge tunable nanoparticles for TNF-alpha siRNA oral delivery for treating ulcerative colitis. Nano. Res. 2018, 11, 2872–2884. [Google Scholar] [CrossRef]
- Guo, J.F.; O’Mahony, A.M.; Cheng, W.P.; O’Driscoll, C.M. Amphiphilic polyallylamine based polymeric micelles for siRNA delivery to the gastrointestinal tract: In vitro investigations. Int. J. Pharm. 2013, 447, 150–157. [Google Scholar] [CrossRef]
- Dalby, B.; Cates, S.; Harris, A.; Ohki, E.C.; Tilkins, M.L.; Price, P.J.; Ciccarone, V.C. Advanced transfection with Lipofectamine 2000 reagent: Primary neurons, siRNA, and high-throughput applications. Methods 2004, 33, 95–103. [Google Scholar] [CrossRef]
- Ming, X.; Sato, K.; Juliano, R.L. Unconventional internalization mechanisms underlying functional delivery of antisense oligonucleotides via cationic lipoplexes and polyplexes. J. Control. Release 2011, 153, 83–92. [Google Scholar] [CrossRef]
- Lv, H.T.; Zhang, S.B.; Wang, B.; Cui, S.H.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef]
- Zhang, M.Z.; Wang, X.Y.; Han, M.K.; Collins, J.F.; Merlin, D. Oral administration of ginger-derived nanolipids loaded with siRNA as a novel approach for efficient siRNA drug delivery to treat ulcerative colitis. Nanomedicine 2017, 12, 1927–1943. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Zumbuehl, A.; Goldberg, M.; Leshchiner, E.S.; Busini, V.; Hossain, N.; Bacallado, S.A.; Nguyen, D.N.; Fuller, J.; Alvarez, R.; et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol. 2008, 26, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Wileman, T.E.; Lennartz, M.R.; Stahl, P.D. Identification of the macrophage mannose receptor as a 175-kDa membrane protein. Proc. Natl. Acad. Sci. USA 1986, 83, 2501–2505. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, S.J.; Saeland, E.; van Kooyk, Y. Sweet preferences of MGL: Carbohydrate specificity and function. Trends Immunol. 2008, 29, 83–90. [Google Scholar] [CrossRef]
- Xiao, B.; Laroui, H.; Ayyadurai, S.; Viennois, E.; Charania, M.A.; Zhang, Y.C.; Merlin, D. Mannosylated bioreducible nanoparticle-mediated macrophage-specific TNF-alpha RNA interference for IBD therapy. Biomaterials 2013, 34, 7471–7482. [Google Scholar] [CrossRef]
- Boegh, M.; Nielsen, H.M. Mucus as a Barrier to Drug Delivery-Understanding and Mimicking the Barrier Properties. Basic Clin. Pharm. 2015, 116, 179–186. [Google Scholar] [CrossRef]
- Kriegel, C.; Amiji, M. Oral TNF-alpha gene silencing using a polymeric microsphere-based delivery system for the treatment of inflammatory bowel disease. J. Control. Release 2011, 150, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Bhavsar, M.D.; Amiji, M.M. Gastrointestinal distribution and in vivo gene transfection studies with nanoparticles-in-microsphere oral system (NiMOS). J. Control. Release 2007, 119, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Knipe, J.M.; Strong, L.E.; Peppas, N.A. Enzyme- and pH-Responsive Microencapsulated Nanogels for Oral Delivery of siRNA to Induce TNF-alpha Knockdown in the Intestine. Biomacromolecules 2016, 17, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Aouadi, M.; Tesz, G.J.; Nicoloro, S.M.; Wang, M.X.; Chouinard, M.; Soto, E.; Ostroff, G.R.; Czech, M.P. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature 2009, 458, 1180–1184. [Google Scholar] [CrossRef] [PubMed]
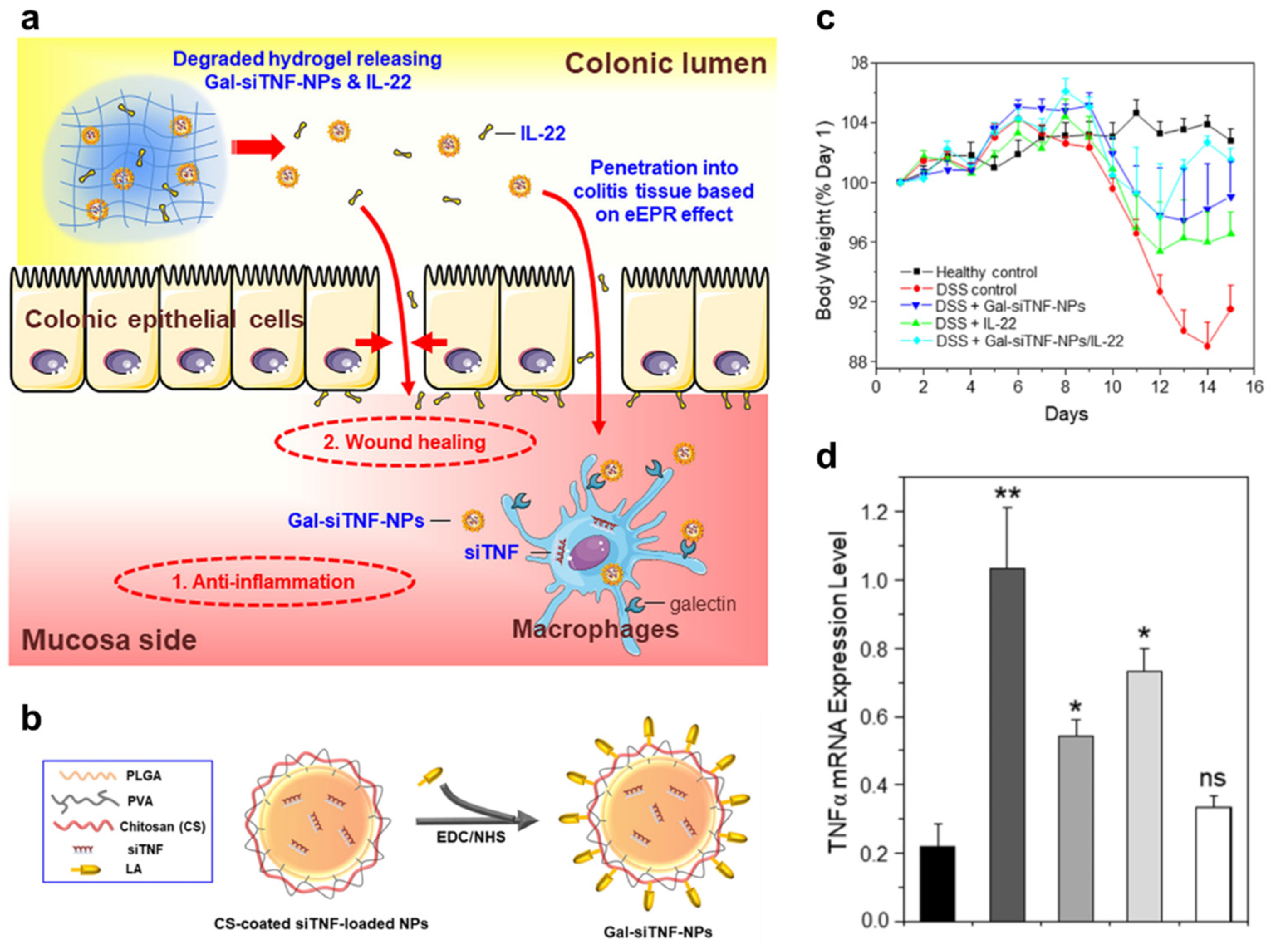
- Xiao, B.; Chen, Q.B.; Zhang, Z.; Wang, L.X.; Kang, Y.J.; Denning, T.; Merlin, D. TNF alpha gene silencing mediated by orally targeted nanoparticles combined with interleukin-22 for synergistic combination therapy of ulcerative colitis. J. Control. Release 2018, 287, 235–246. [Google Scholar] [CrossRef]
- Babbs, C.F. Oxygen radicals in ulcerative colitis. Free Radic. Biol. Med. 1992, 13, 169–181. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef]
- Simmonds, N.J.; Rampton, D.S. Inflammatory bowel disease--a radical view. Gut 1993, 34, 865–868. [Google Scholar] [CrossRef]
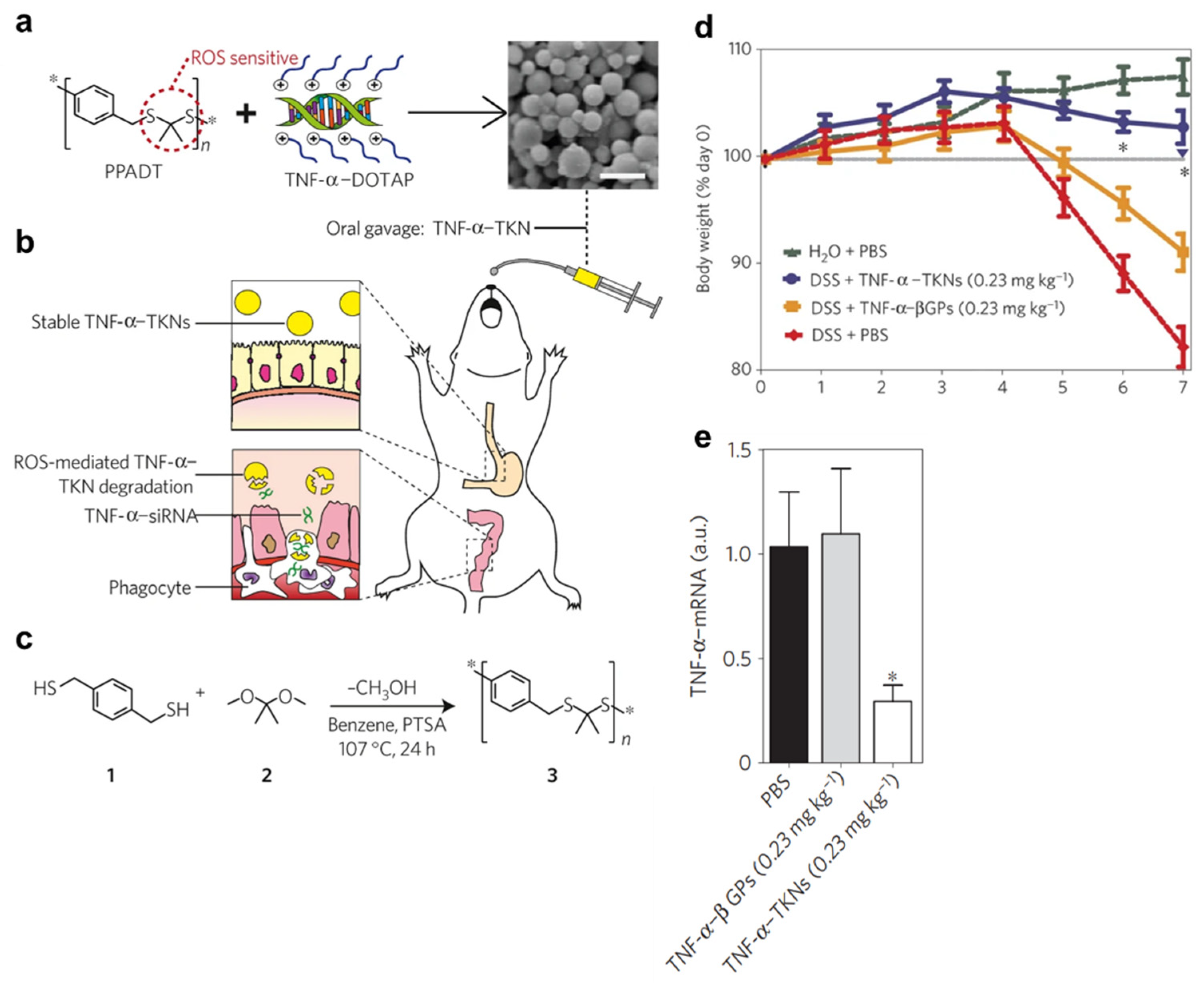
- Wilson, D.S.; Dalmasso, G.; Wang, L.X.; Sitaraman, S.V.; Merlin, D.; Murthy, N. Orally delivered thioketal nanoparticles loaded with TNF-alpha-siRNA target inflammation and inhibit gene expression in the intestines. Nat. Mater. 2010, 9, 923–928. [Google Scholar] [CrossRef]
- Zhang, X.; Goel, V.; Robbie, G.J. Pharmacokinetics of Patisiran, the First Approved RNA Interference Therapy in Patients With Hereditary Transthyretin-Mediated Amyloidosis. J. Clin. Pharm. 2020, 60, 573–585. [Google Scholar] [CrossRef]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 2012, 51, 8529–8533. [Google Scholar] [CrossRef]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiro, P.A.; Rees, D.C.; Stolzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Garrelfs, S.F.; Frishberg, Y.; Hulton, S.A.; Koren, M.J.; O’Riordan, W.D.; Cochat, P.; Deschenes, G.; Shasha-Lavsky, H.; Saland, J.M.; Van’t Hoff, W.G.; et al. Lumasiran, an RNAi Therapeutic for Primary Hyperoxaluria Type 1. N. Engl. J. Med. 2021, 384, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vazquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]







| siRNA Delivery Methods | Advantages | Drawbacks | |
|---|---|---|---|
| Chemical modification |
|
| |
| Co-administration of nuclease inhibitors |
|
| |
| Lipo-polyplex formation | - |
|
|
| With targeting ligands |
|
| |
| Lipo-polyplex embedded in hydrogels or polymeric materials | - |
|
|
| With targeting ligands |
|
| |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shinn, J.; Lee, J.; Lee, S.A.; Lee, S.J.; Choi, A.H.; Kim, J.S.; Kim, S.J.; Kim, H.J.; Lee, C.; Kim, Y.; et al. Oral Nanomedicines for siRNA Delivery to Treat Inflammatory Bowel Disease. Pharmaceutics 2022, 14, 1969. https://doi.org/10.3390/pharmaceutics14091969
Shinn J, Lee J, Lee SA, Lee SJ, Choi AH, Kim JS, Kim SJ, Kim HJ, Lee C, Kim Y, et al. Oral Nanomedicines for siRNA Delivery to Treat Inflammatory Bowel Disease. Pharmaceutics. 2022; 14(9):1969. https://doi.org/10.3390/pharmaceutics14091969
Chicago/Turabian StyleShinn, Jongyoon, Juyeon Lee, Seon Ah Lee, Seon Ju Lee, Ah Hyun Choi, Jung Seo Kim, Su Jin Kim, Hyo Jin Kim, Cherin Lee, Yejin Kim, and et al. 2022. "Oral Nanomedicines for siRNA Delivery to Treat Inflammatory Bowel Disease" Pharmaceutics 14, no. 9: 1969. https://doi.org/10.3390/pharmaceutics14091969