
Scaffold Repurposing Reveals New Nanomolar Phosphodiesterase Type 5 (PDE5) Inhibitors Based on Pyridopyrazinone Scaffold: Investigation of In Vitro and In Silico Properties
 , , ,
, , ,  , ,
, ,  and
and
Abstract
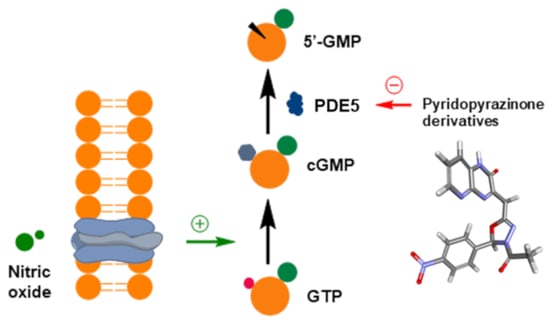
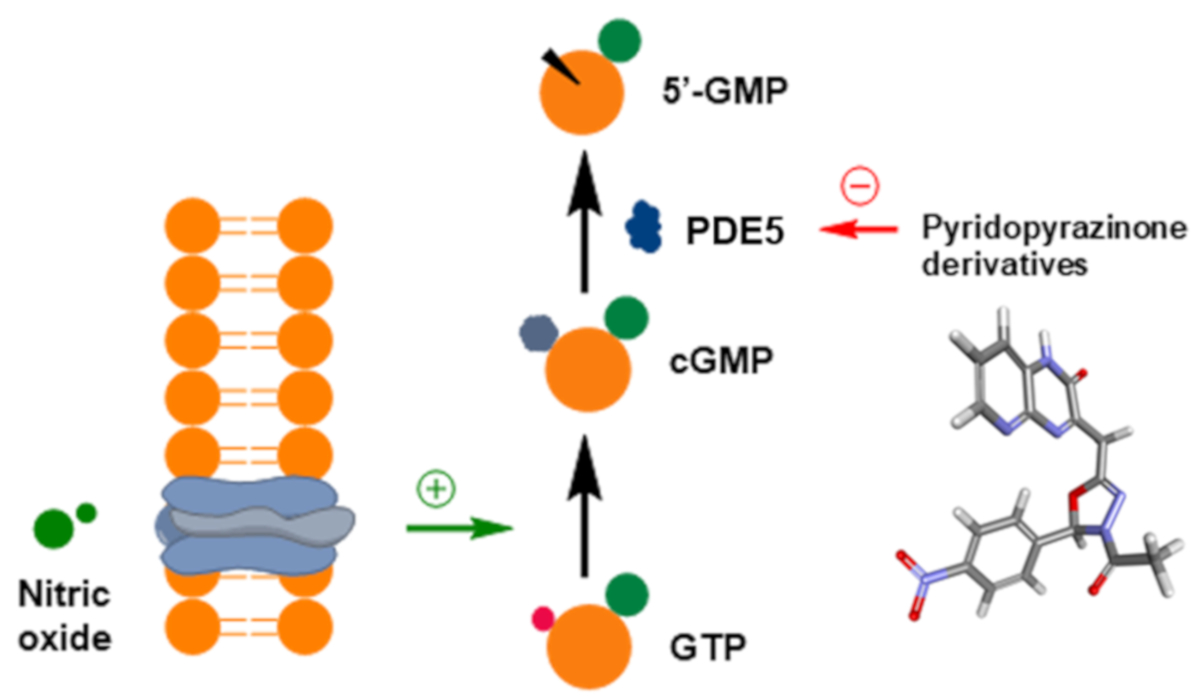
:
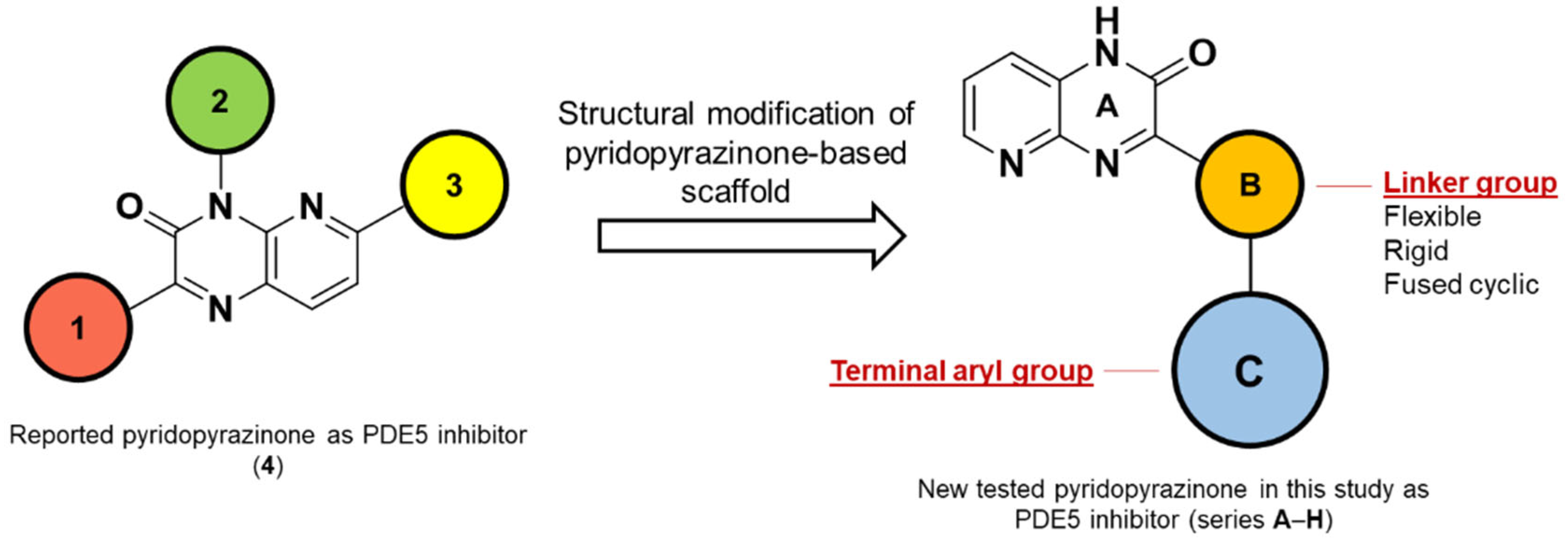
1. Introduction
2. Materials and Methods
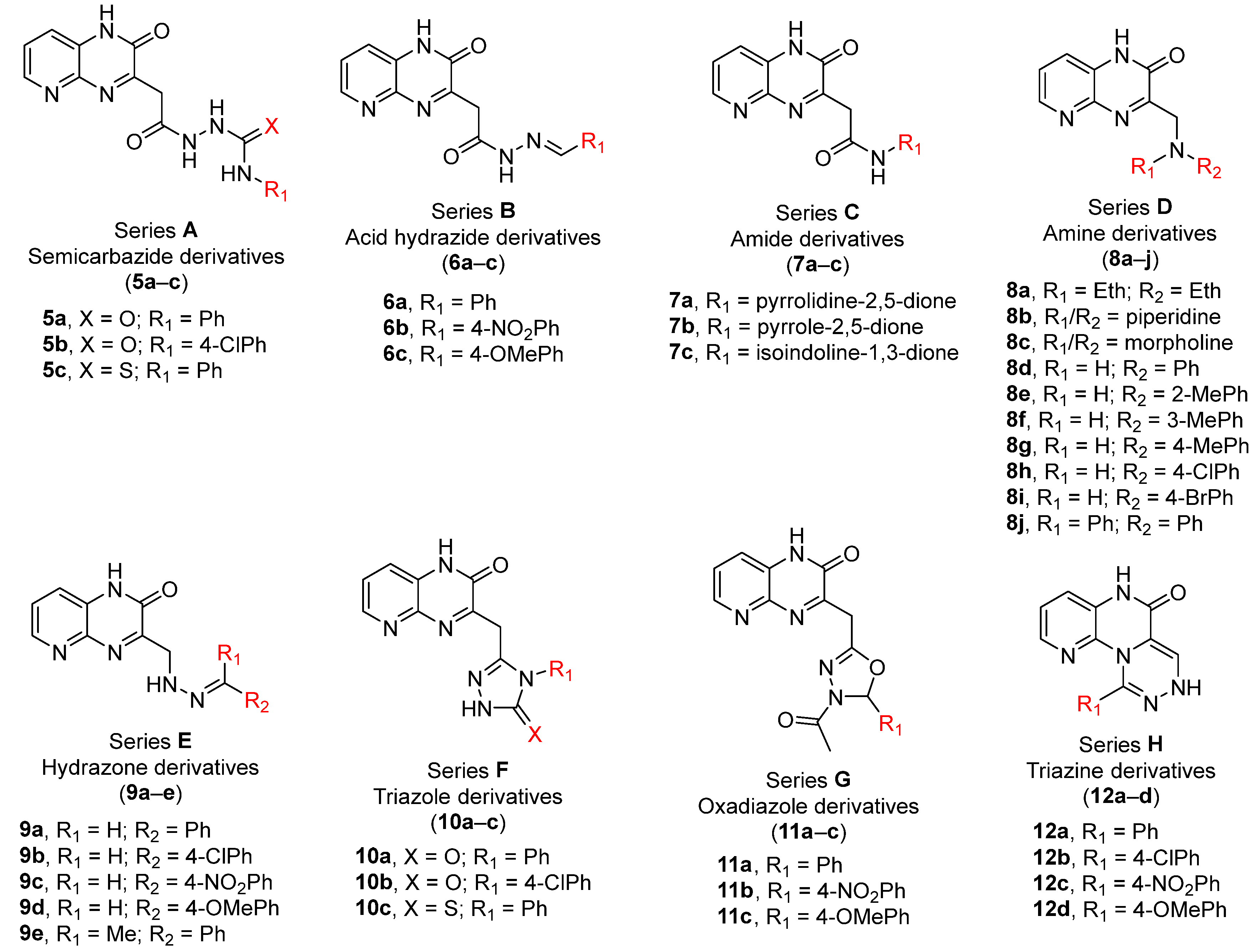
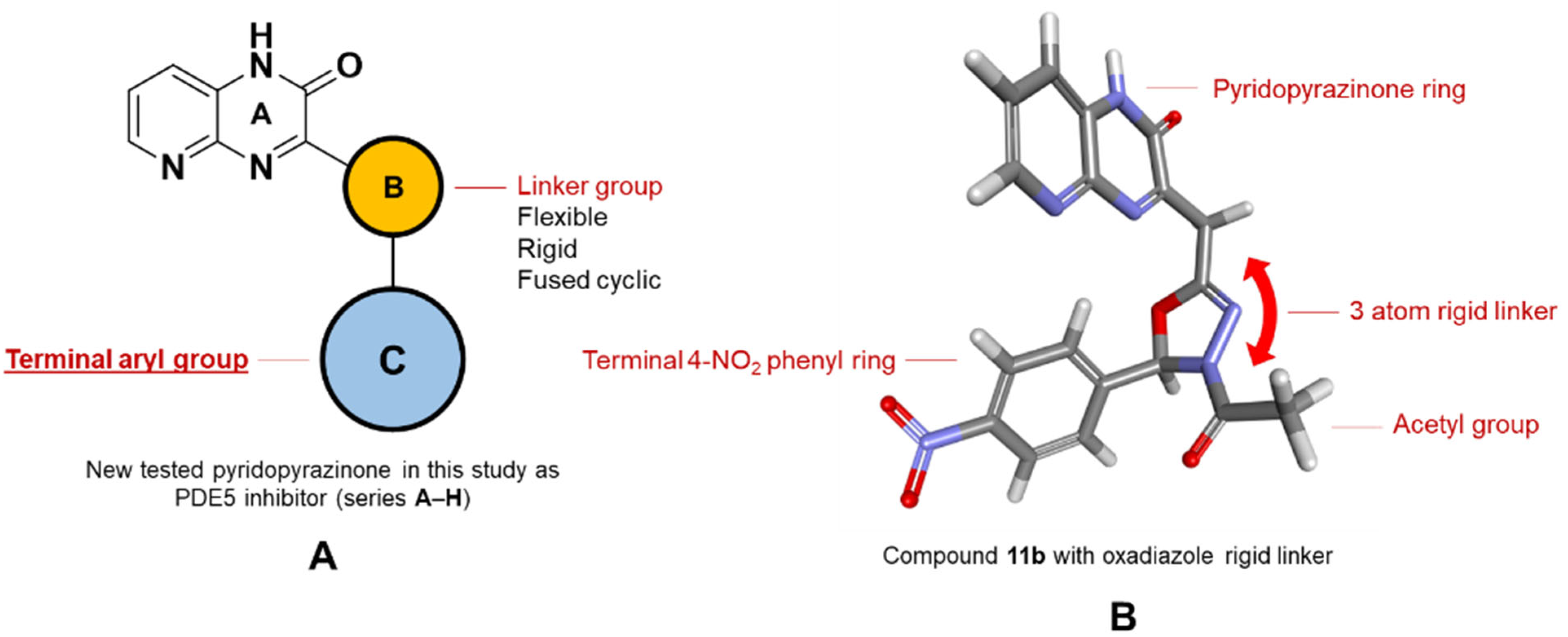
2.1. Chemistry
2.2. In-Silico Screening
2.2.1. 2D-QSAR Analysis
Assembling the Database for the Training Set and Test Set
Calculating the 2D-Descriptors
Pruning the Proposed 2D-Descriptors
Fitting the Experimental IC50 Values
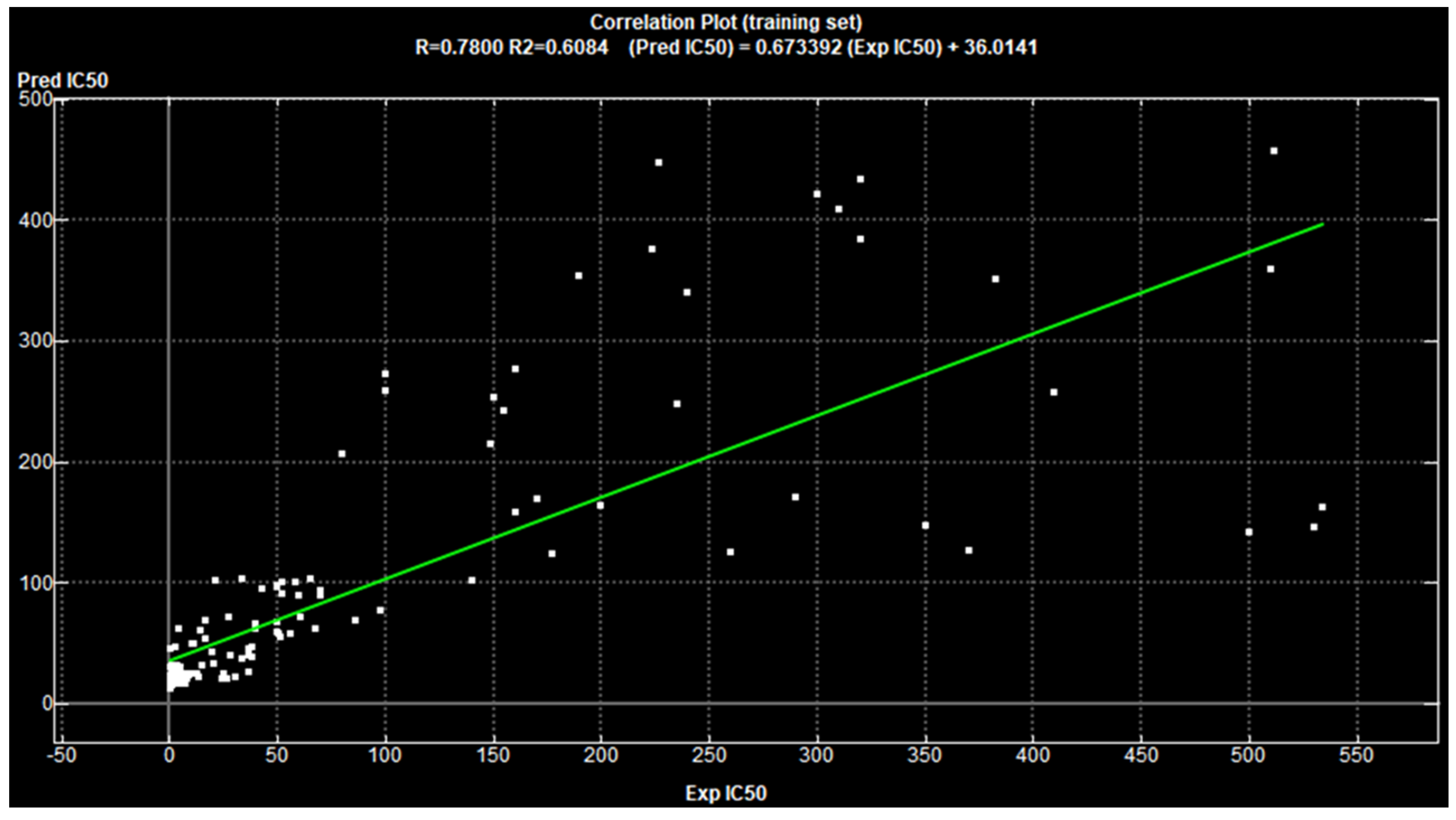
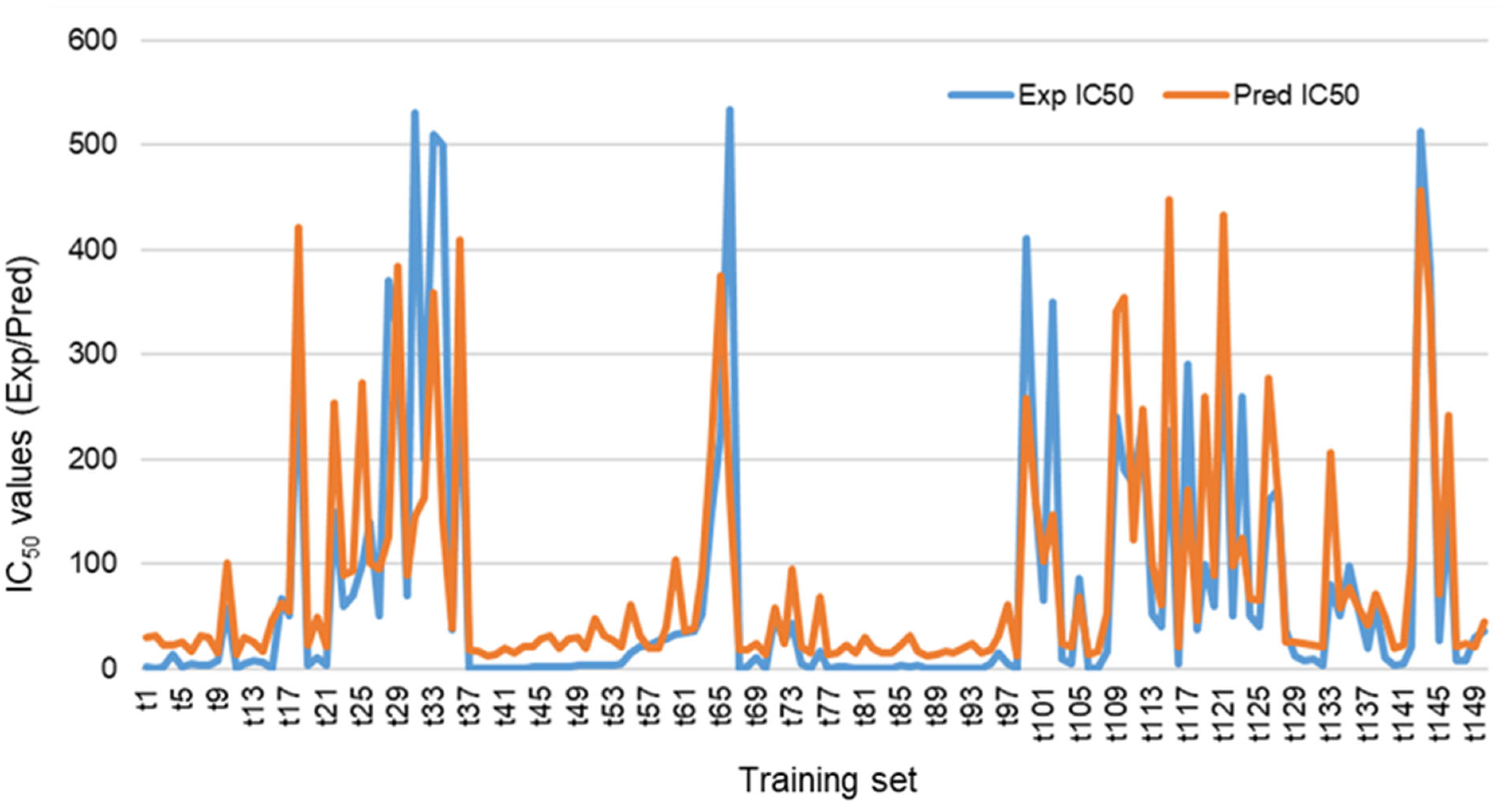
Cross Validating the Designed Model
Estimating the Predicted IC50 of the Test Set
2.2.2. Molecular Docking
2.3. In Vitro Enzyme Assay
2.4. Molecular Dynamic (MD) Simulation
3. Results and Discussion
3.1. In-Silico Screening
3.1.1. 2D-QSAR Analysis
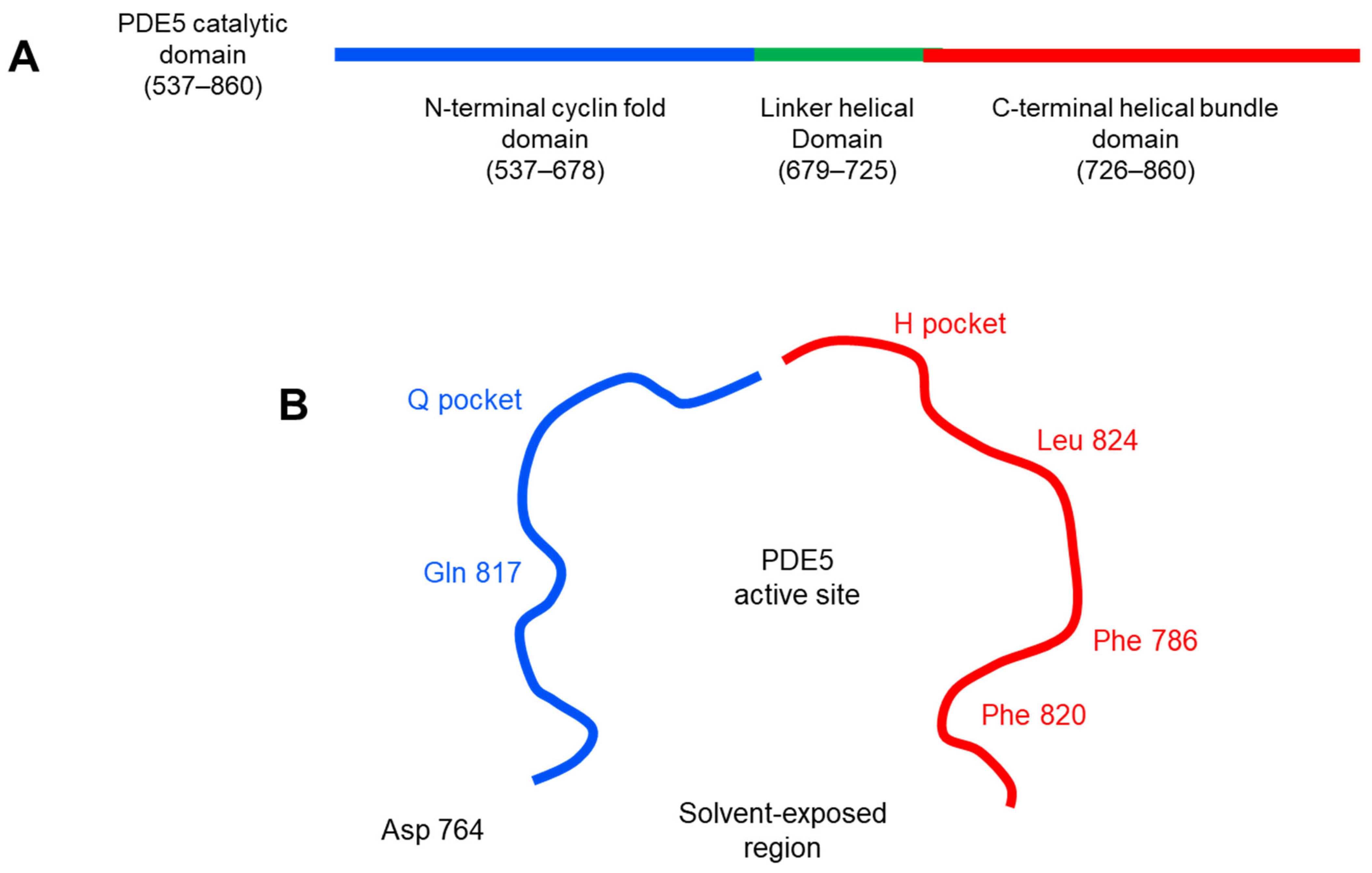
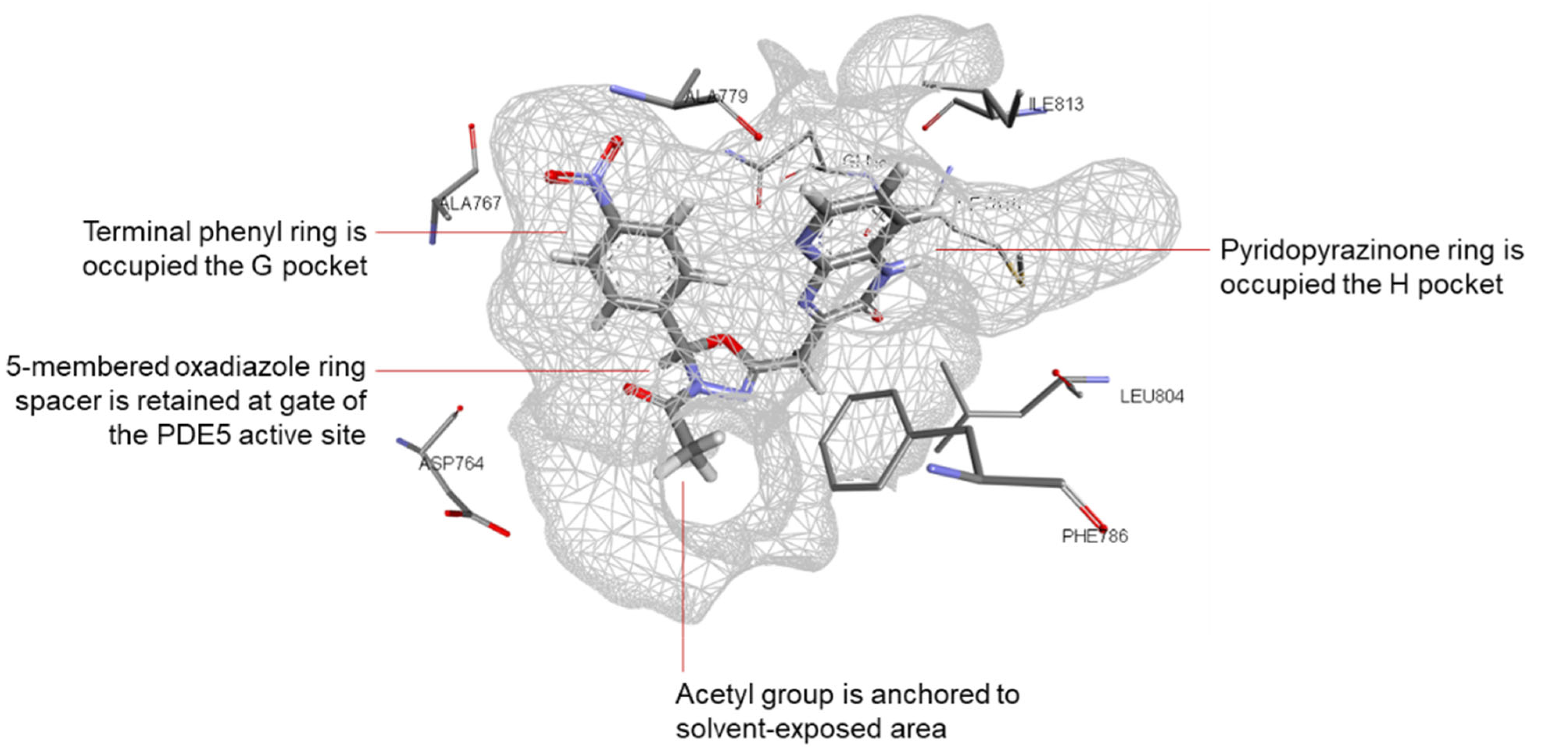
3.1.2. Molecular Docking
3.2. In Vitro Enzyme Assay
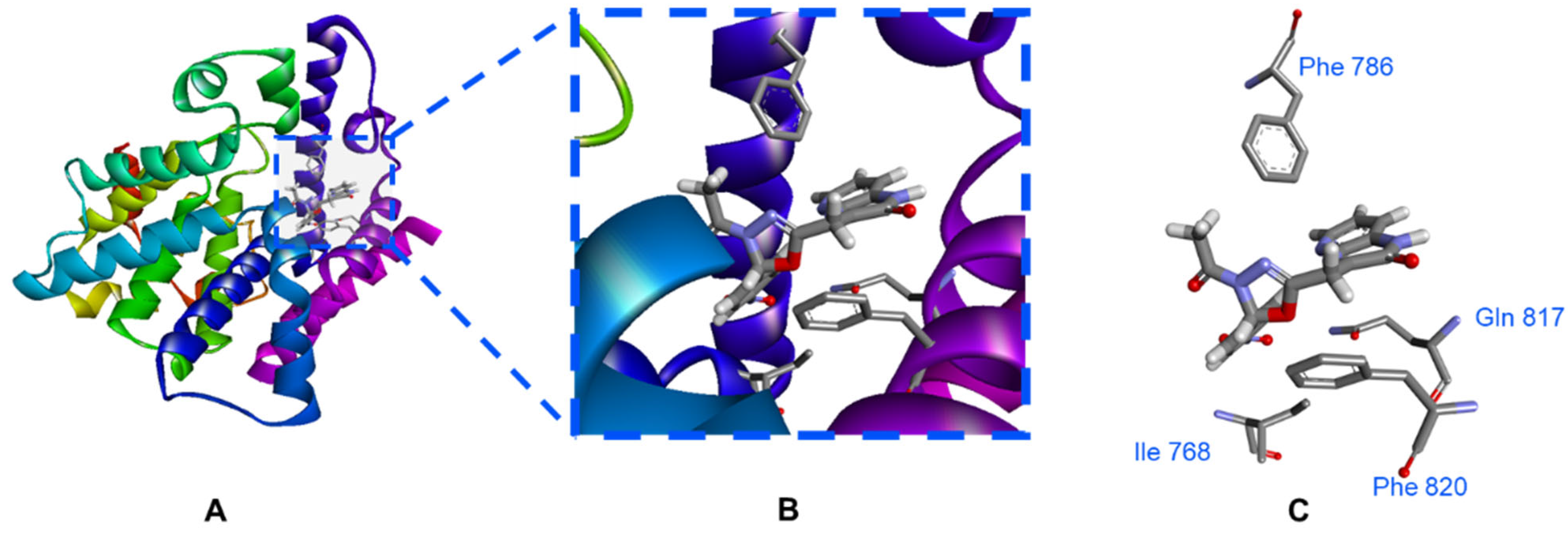
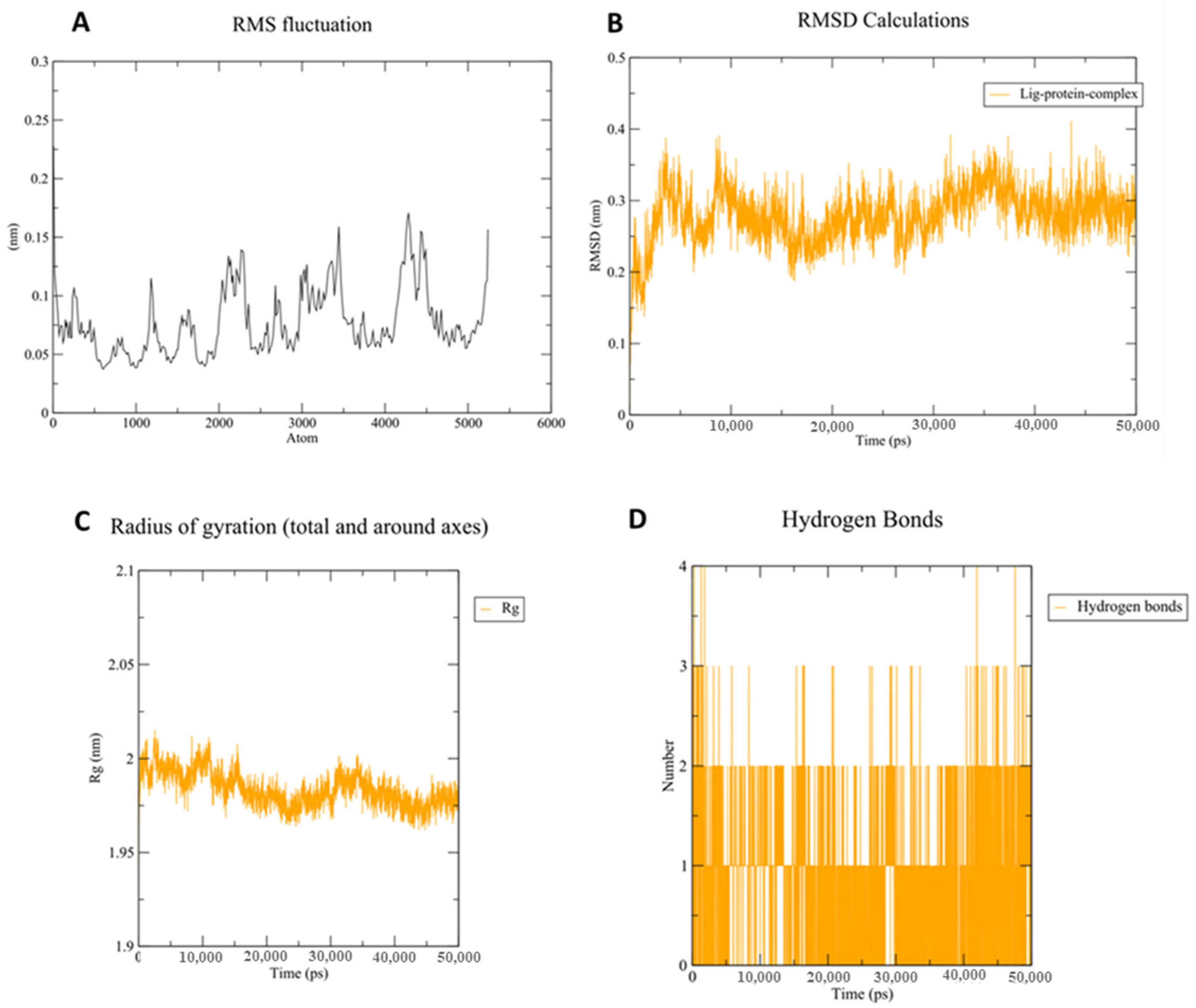
3.3. Molecular Dynamic (MD) Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Owen, D.R.; Walker, J.K.; Jacobsen, E.J.; Freskos, J.N.; Hughes, R.O.; Brown, D.L.; Bell, A.S.; Brown, D.G.; Phillips, C.; Mischke, B.V. Identification, synthesis and SAR of amino substituted pyrido [3, 2b] pyrazinones as potent and selective PDE5 inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 4088–4091. [Google Scholar] [CrossRef] [PubMed]
- Beavo, J.A. Cyclic nucleotide phosphodiesterases: Functional implications of multiple isoforms. Physiol. Rev. 1995, 75, 725–748. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J.D.; Francis, S.H. Cyclic GMP phosphodiesterase-5: Target of sildenafil. J. Biol. Chem. 1999, 274, 13729–13732. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.O.; Walker, J.K.; Cubbage, J.W.; Fobian, Y.M.; Rogier, D.J.; Heasley, S.E.; Blevis-Bal, R.M.; Benson, A.G.; Owen, D.R.; Jacobsen, E.J. Investigation of aminopyridiopyrazinones as PDE5 inhibitors: Evaluation of modifications to the central ring system. Bioorg. Med. Chem. Lett. 2009, 19, 4092–4096. [Google Scholar] [CrossRef]
- Bollenbach, M.; Lugnier, C.; Kremer, M.; Salvat, E.; Megat, S.; Bihel, F.; Bourguignon, J.-J.; Barrot, M.; Schmitt, M. Design and synthesis of 3-aminophthalazine derivatives and structural analogues as PDE5 inhibitors: Anti-allodynic effect against neuropathic pain in a mouse model. Eur. J. Med. Chem. 2019, 177, 269–290. [Google Scholar] [CrossRef]
- Kee, C.-L.; Ge, X.; Gilard, V.; Malet-Martino, M.; Low, M.-Y. A review of synthetic phosphodiesterase type 5 inhibitors (PDE5i) found as adulterants in dietary supplements. J. Pharmaceut. Biomed. 2018, 147, 250–277. [Google Scholar] [CrossRef]
- Wenzel, B.; Liu, J.; Dukic-Stefanovic, S.; Deuther-Conrad, W.; Teodoro, R.; Ludwig, F.-A.; Chezal, J.-M.; Moreau, E.; Brust, P.; Maisonial-Besset, A. Targeting cyclic nucleotide phosphodiesterase 5 (PDE5) in brain: Toward the development of a PET radioligand labeled with fluorine-18. Bioorg. Chem. 2019, 86, 346–362. [Google Scholar] [CrossRef]
- Korkmaz, S.; Radovits, T.; Barnucz, E.; Neugebauer, P.; Arif, R.; Hirschberg, K.; Loganathan, S.; Seidel, B.; Karck, M.; Szabó, G. Dose-dependent effects of a selective phosphodiesterase-5-inhibitor on endothelial dysfunction induced by peroxynitrite in rat aorta. Eur. J. Pharmacol. 2009, 615, 155–162. [Google Scholar] [CrossRef]
- Lugnier, C.; Schoeffter, P.; le Bec, A.; Strouthou, E.; Stoclet, J. Selective inhibition of cyclic nucleotide phosphodiesterases of human, bovine and rat aorta. Biochem. Pharmacol. 1986, 35, 1743–1751. [Google Scholar] [CrossRef]
- Francis, S.H.; Lincoln, T.; Corbin, J. Characterization of a novel cGMP binding protein from rat lung. J. Biol. Chem. 1980, 255, 620–626. [Google Scholar] [CrossRef]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Brit. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef]
- Galiè, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef]
- Wang, C.; Ashton, T.D.; Gustafson, A.; Bland, N.D.; Ochiana, S.O.; Campbell, R.K.; Pollastri, M.P. Synthesis and evaluation of human phosphodiesterases (PDE) 5 inhibitor analogs as trypanosomal PDE inhibitors. Part 1. Sildenafil analogs. Bioorg. Med. Chem. Lett. 2012, 22, 2579–2581. [Google Scholar] [CrossRef]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Therapeut. 2006, 109, 366–398. [Google Scholar] [CrossRef]
- Peixoto, C.A.; Nunes, A.K.S.; Garcia-Osta, A. Phosphodiesterase-5 inhibitors: Action on the signaling pathways of neuroinflammation, neurodegeneration, and cognition. Mediat. Inflamm. 2015, 2015, 940207. [Google Scholar] [CrossRef]
- Ochiana, S.O.; Gustafson, A.; Bland, N.D.; Wang, C.; Russo, M.J.; Campbell, R.K.; Pollastri, M.P. Synthesis and evaluation of human phosphodiesterases (PDE) 5 inhibitor analogs as trypanosomal PDE inhibitors. Part 2. Tadalafil analogs. Bioorg. Med. Chem. Lett. 2012, 22, 2582–2584. [Google Scholar] [CrossRef]
- Lee, S.J.; Konishi, Y.; Yu, D.T.; Miskowski, T.A.; Riviello, C.M.; Macina, O.T.; Frierson, M.R.; Kondo, K.; Sugitani, M. Discovery of potent cyclic GMP phosphodiesterase inhibitors. 2-Pyridyl-and 2-imidazolylquinazolines possessing cyclic GMP phosphodiesterase and thromboxane synthesis inhibitory activities. J. Med. Chem. 1995, 38, 3547–3557. [Google Scholar] [CrossRef]
- Chekol, R.; Gheysens, O.; Ahamed, M.; Cleynhens, J.; Pokreisz, P.; Vanhoof, G.; Janssens, S.; Verbruggen, A.; Bormans, G. Carbon-11 and Fluorine-18 Radiolabeled Pyridopyrazinone Derivatives for Positron Emission Tomography (PET) Imaging of Phosphodiesterase-5 (PDE5). J. Med. Chem. 2016, 60, 486–496. [Google Scholar] [CrossRef]
- Reddy, G.L.; Dar, M.I.; Hudwekar, A.D.; Mahajan, P.; Nargotra, A.; Baba, A.M.; Nandi, U.; Wazir, P.; Singh, G.; Vishwakarma, R.A. Design, Synthesis and Biological Evaluation of Pyrazolopyrimidinone Based Potent and Selective PDE5 Inhibitors for Treatment of Erectile Dysfunction. Bioorg. Chem. 2019, 89, 103022. [Google Scholar] [CrossRef]
- Terrett, N.K.; Bell, A.S.; Brown, D.; Ellis, P. Sildenafil (VIAGRATM), a potent and selective inhibitor of type 5 cGMP phosphodiesterase with utility for the treatment of male erectile dysfunction. Bioorg. Med. Chem. Lett. 1996, 6, 1819–1824. [Google Scholar] [CrossRef]
- Zheng, H.; Wu, Y.; Sun, B.; Cheng, C.; Qiao, Y.; Jiang, Y.; Zhao, S.; Xie, Z.; Tan, J.; Lou, H. Discovery of furyl/thienyl β-carboline derivatives as potent and selective PDE5 inhibitors with excellent vasorelaxant effect. Eur. J. Med. Chem. 2018, 158, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, K.; Inoue, T.; Nishizawa, T.; Numata, S.; Ishii, T. Process for Preparing Piperonal. US patent 4157333, 5 June 1979. [Google Scholar]
- Haning, H.; Niewöhner, U.; Schenke, T.; Es-Sayed, M.; Schmidt, G.; Lampe, T.; Bischoff, E. Imidazo [5, 1-f], triazin-4 (3H)-ones, a new class of potent PDE 5 inhibitors. Bioorg. Med. Chem. Lett. 2002, 12, 865–868. [Google Scholar] [CrossRef]
- Zheng, H.; Li, L.; Sun, B.; Gao, Y.; Song, W.; Zhao, X.; Gao, Y.; Xie, Z.; Zhang, N.; Ji, J. Design and synthesis of furyl/thineyl pyrroloquinolones based on natural alkaloid perlolyrine, lead to the discovery of potent and selective PDE5 inhibitors. Eur. J. Med. Chem. 2018, 150, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Dzierba, C.D.; Sielecki, T.M.; Arvanitis, A.G.; Galka, A.; Johnson, T.L.; Takvorian, A.G.; Rafalski, M.; Kasireddy-Polam, P.; Vig, S.; Dasgupta, B. Synthesis and structure–activity relationships of pyrido [3, 2-b] pyrazin-3 (4H)-ones and pteridin-7 (8H)-ones as corticotropin-releasing factor-1 receptor antagonists. Bioorg. Med. Chem. Lett. 2012, 22, 4986–4989. [Google Scholar] [CrossRef]
- Hemley, C.; McCluskey, A.; Keller, P.A. Corticotropin releasing hormone-a GPCR drug target. Curr. Drug. Target. 2007, 8, 105–115. [Google Scholar] [CrossRef]
- Ising, M.; Holsboer, F. CRH₁ receptor antagonists for the treatment of depression and anxiety. Exp. Clin. Psychopharm. 2007, 15, 519. [Google Scholar] [CrossRef]
- Valdez, G.R. CRF receptors as a potential target in the development of novel pharmacotherapies for depression. Curr. Pharm. Design. 2009, 15, 1587–1594. [Google Scholar] [CrossRef]
- Zorrilla, E.P.; Koob, G.F. Progress in corticotropin-releasing factor-1 antagonist development. Drug. Disov. Today 2010, 15, 371–383. [Google Scholar] [CrossRef]
- Argyros, O.; Lougiakis, N.; Kouvari, E.; Papafotika, A.; Raptopoulou, C.P.; Psycharis, V.; Christoforidis, S.; Pouli, N.; Marakos, P.; Tamvakopoulos, C. Design and synthesis of novel 7-aminosubstituted pyrido [2, 3-b] pyrazines exhibiting anti-breast cancer activity. Eur. J. Med. Chem. 2017, 126, 954–968. [Google Scholar] [CrossRef]
- Gopal, A.K.; Kahl, B.S.; de Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Tolaney, S.M.; Tan, S.; Guo, H.; Barry, W.; van Allen, E.; Wagle, N.; Brock, J.; Larrabee, K.; Paweletz, C.; Ivanova, E. Phase II study of tivantinib (ARQ 197) in patients with metastatic triple-negative breast cancer. Investig. New Drug 2015, 33, 1108–1114. [Google Scholar] [CrossRef]
- Seipelt, I.; Claus, E.; Schuster, T.; Polymeropoulos, E.; Teifel, M.; Guenther, E. New generation of anilino-substituted pyridopyrazine-urea derivatives show highly selective PI3K-inhibition. Cancer Res. 2007, 67, 2379. [Google Scholar]
- Ammar, U.M.; Abdel-Maksoud, M.S.; Oh, C.-H. Recent advances of RAF (rapidly accelerated fibrosarcoma) inhibitors as anti-cancer agents. Eur. J. Med. Chem. 2018, 158, 144–166. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Abdel-Maksoud, M.S.; El-Din, M.M.G.; Shin, J.-S.; Lee, K.-T.; Yoo, K.H.; Oh, C.-H. Synthesis, in vitro antiproliferative and antiinflammatory activities, and kinase inhibitory effects of new 1, 3, 4-triarylpyrazole derivatives. Anti-Cancer Agent Med. Chem. 2017, 17, 75–84. [Google Scholar] [CrossRef]
- Abdel-Maksoud, M.S.; Ammar, U.M.; Oh, C.-H. Anticancer profile of newly synthesized BRAF inhibitors possess 5-(pyrimidin-4-yl) imidazo [2, 1-b] thiazole scaffold. Bioorg. Med. Chem. 2019, 27, 2041–2051. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Khan, M.A.; Tarazi, H.; Abdel-Maksoud, M.S.; El-Din, M.M.G.; Yoo, K.H.; Oh, C.-H. Design and synthesis of new RAF kinase-inhibiting antiproliferative quinoline derivatives. Part 2: Diarylurea derivatives. Eur. J. Med. Chem. 2017, 127, 413–423. [Google Scholar] [CrossRef]
- Abdel-Maksoud, M.S.; Ammar, U.M.; El-Gamal, M.I.; El-Din, M.M.G.; Mersal, K.I.; Ali, E.M.; Yoo, K.H.; Lee, K.-T.; Oh, C.-H. Design, synthesis, and anticancer activity of imidazo [2, 1-b] oxazole-based RAF kinase inhibitors. Bioorg. Chem. 2019, 93, 103349. [Google Scholar] [CrossRef]
- Amin, K.M.; El-Badry, O.M.; Rahman, D.E.A.; Ammar, U.M.; Abdalla, M.M. Design, synthesis, anticancer evaluation and molecular docking of new V600EBRAF inhibitors derived from pyridopyrazinone. Eur. J. Chem. 2016, 7, 19–29. [Google Scholar] [CrossRef]
- Amin, K.; El-Badry, O.; Rahman, D.A.; Ammar, U. Synthesis and In Vitro Biological Evaluation of New Pyrido [2, 3-b] pyrazinone-Based Cytotoxic Agents and Molecular Docking as BRAF Inhibitors. ChemistrySelect 2019, 4, 8882–8885. [Google Scholar] [CrossRef]
- Molecular Operating Environment. Available online: https://www.chemcomp.com (accessed on 17 January 2021).
- Ammar, U.M.; Abdel-Maksoud, M.S.; Ali, E.M.H.; Mersal, K.I.; Yoo, K.H.; Oh, C.H. Structural optimization of imidazothiazole derivatives affords a new promising series as B-Raf V600E inhibitors; synthesis, in vitro assay and in silico screening. Bioorg. Chem. 2020, 100, 103967. [Google Scholar] [CrossRef]
- Huss, K.L.; Blonigen, P.E.; Campbell, R.M. Development of a Transcreener™ kinase assay for protein kinase A and demonstration of concordance of data with a filter-binding assay format. J. Biomol. Screen. 2007, 12, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Nada, H.; Lee, K.; Gotina, L.; Pae, A.N.; Elkamhawy, A. Identification of novel discoidin domain receptor 1 (DDR1) inhibitors using E-pharmacophore modeling, structure-based virtual screening, molecular dynamics simulation and MM-GBSA approaches. J. Comp. Bio. Med. 2022, 142, 105217. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Ammar, U.M.; Paik, S.; Abdellattif, M.H.; Elsherbeny, M.H.; Lee, K.; Roh, E.J. Scaffold repurposing of in-house small molecule candidates leads to discovery of first-in-class CDK-1/HER-2 dual inhibitors: In vitro and in silico screening. Molecules 2021, 17, 5324. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Ali, E.M.H.; Lee, K. New horizons in drug discovery of lymphocyte-specific protein tyrosine kinase (Lck) inhibitors: A decade review (2011–2021) focussing on structure–activity relationship (SAR) and docking insights. J. Enzyme Inhib. Med. Chem. 2021, 36, 1572–1600. [Google Scholar] [CrossRef]
- Nada, H.; Elkamhawy, A.; Lee, K. Structure activity relationship of key heterocyclic anti-angiogenic leads of promising potential in the fight against cancer. Molecules 2021, 26, 553. [Google Scholar] [CrossRef]
- Lee, K.; Nada, H.; Byun, H.J.; Lee, C.H.; Elkamhawy, A. Hit identification of a Novel quinazoline sulfonamide as a promising EphB3 inhibitor: Design, virtual combinatorial library, synthesis, biological evaluation, and docking simulation studies. Pharmaceuticals 2021, 14, 1247. [Google Scholar] [CrossRef]
- Huang, X.; Xu, P.; Cao, Y.; Liu, L.; Song, G.; Xu, L. Exploring the binding mechanisms of PDE5 with chromeno [2, 3-c] pyrrol-9 (2 H)-one by theoretical approaches. RSC Adv. 2018, 8, 30481–30490. [Google Scholar] [CrossRef]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein–ligand docking: Current status and future challenges. Proteins Struct. Funct. Bioinf. 2006, 65, 15–26. [Google Scholar] [CrossRef]












{kind=link}
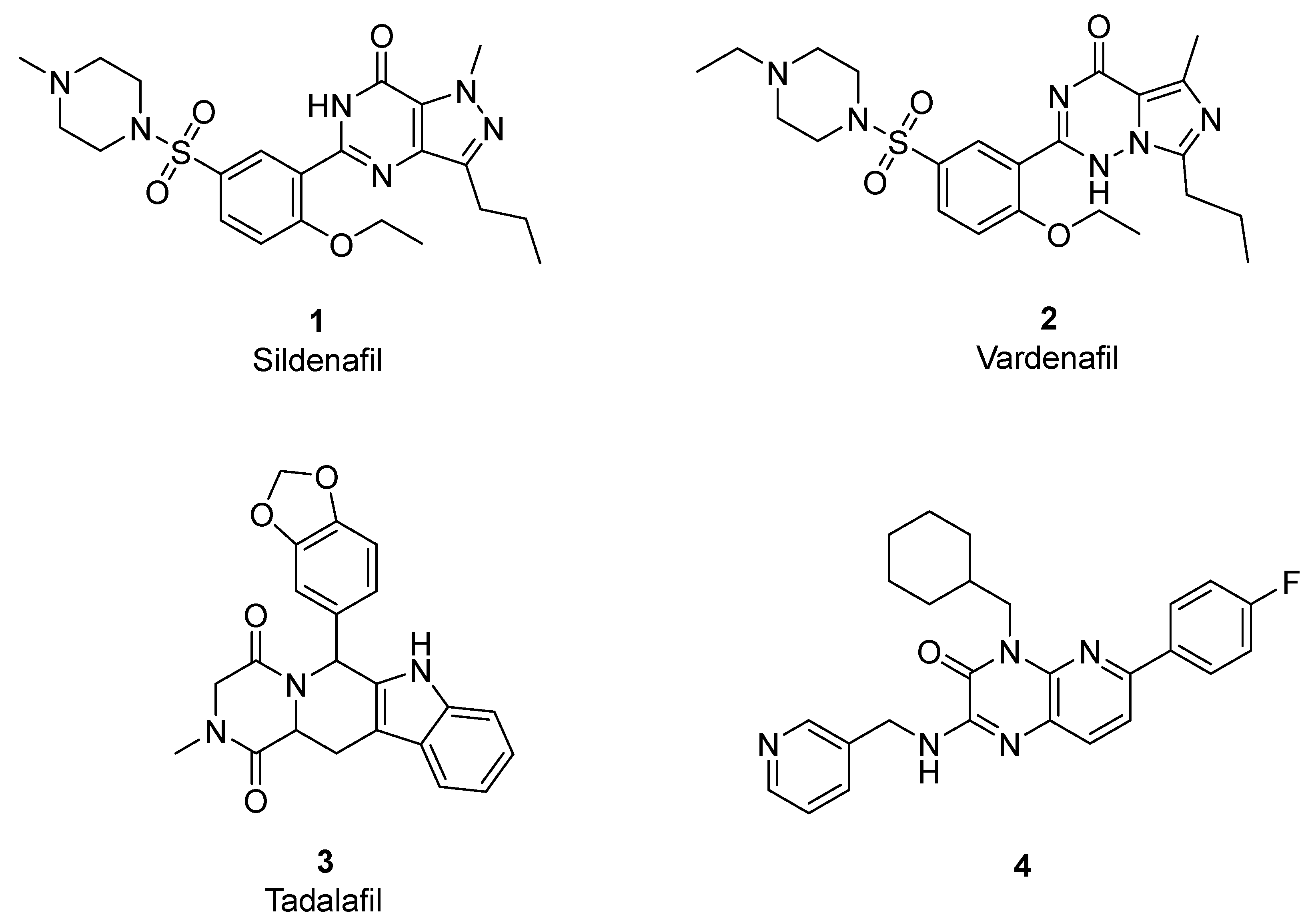{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Predicted IC50 (nM) | Compound | Predicted IC50 (nM) |
|---|---|---|---|
| 5a | 202.71 | 8i | 71.38 |
| 5b | 262.99 | 8j | 137.24 |
| 5c | 147.43 | 9a | 39.33 |
| 6a | 51.38 | 9b | 99.61 |
| 6b | 70.36 | 9c | 58.31 |
| 6c | 7.83 | 9d | −4.21 |
| 7a | 90.20 | 9e | 66.24 |
| 7b | 120.98 | 10a | 125.77 |
| 7c | 147.98 | 10b | 186.05 |
| 8a | 27.49 | 10c | 70.48 |
| 8b | 26.75 | 11a | 49.54 |
| 8c | 38.97 | 11b | 68.52 |
| 8d | 81.56 | 11c | 6.00 |
| 8e | 66.27 | 12a | 111.41 |
| 8f | 66.27 | 12b | 171.69 |
| 8g | 66.27 | 12c | 130.39 |
| 8h | 141.84 | 12d | 67.86 |
| Compound | Docking Score (S) a | Hydrophobic Interactions | Hydrogen Bond (Å) |
|---|---|---|---|
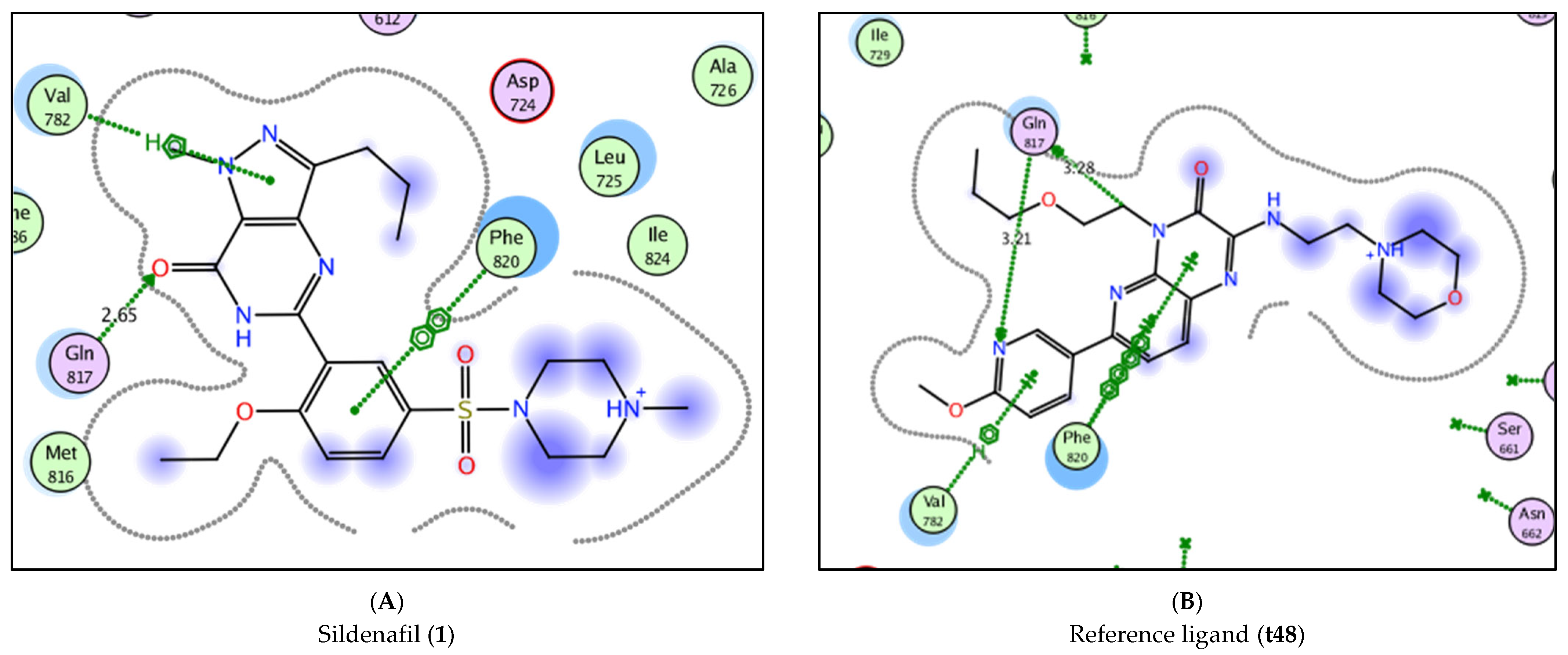
| 1 | −8.3805 | Val 782, Phe 820 | Gln 817 (2.65) |
| t48 | −9.8829 | Val 782, Phe 820 | Gln 817 (3.21), Gln 817 (3.28) |
| 5a | −6.7026 | - | - |
| 5b | −7.4526 | Leu 804 | - |
| 5c | −6.6958 | His 613 | Asp 764 (2.83) |
| 6a | −6.3718 | His 613 | - |
| 6b | −6.9287 | Phe 820 | His 685 (3.40), Asp 764 (3.19), Gln 817 (2.88) |
| 6c | −6.6302 | - | - |
| 7a | −6.4060 | Leu 804, Phe 820 | Leu 765 (3.55) |
| 7b | −6.3842 | Val 782, Phe 820 | - |
| 7c | −6.4797 | His 613 | - |
| 8a | −5.9536 | - | Gln 817 (3.65) |
| 8b | −6.4089 | Val 782, Phe 820 | Gln 817 (2.99) |
| 8c | −6.0430 | Val 782 | Gln 817 (2.97) |
| 8d | −6.0887 | - | - |
| 8e | −6.3671 | Phe 820 | Gln 817 (2.88) |
| 8f | −6.5098 | Val 782 | Gln 817 (2.89) |
| 8g | −6.4050 | Leu 804, Phe 820 | - |
| 8h | −6.2434 | His 613, Phe 820 | Gln 817 (3.10) |
| 8i | −6.5529 | Leu 804, Phe 820 | - |
| 8j | −7.1926 | - | - |
| 9a | −6.4902 | Val 782, Phe 820 | Gln 817 (3.48) |
| 9b | −6.6590 | - | - |
| 9c | −6.4239 | - | Tyr 612 (3.20) |
| 9d | −6.9129 | Leu 804 | - |
| 9e | −6.8623 | Val 782, Phe 820 | Gln 817 (3.33) |
| 10a | −6.7878 | - | - |
| 10b | −6.6090 | - | - |
| 10c | −6.7319 | - | Tyr 612 (3.33), Asp 764 (2.96) |
| 11a | −6.8508 | - | - |
| 11b | −7.0486 | Phe 786, Phe 820 | Met 816 (3.35), Gln 817 (2.69) |
| 11c | −7.4782 | Ile 768, Leu 804 | - |
| 12a | −6.1199 | Val 782 | Gln 817 (3.35) |
| 12b | −6.1236 | Phe 820 | - |
| 12c | −6.4532 | Phe 820 | - |
| 12d | −6.6629 | Phe 786, Phe 820 | - |
| 9 | IC50 (nM) | Compound | IC50 (nM) | ||
|---|---|---|---|---|---|
| Exp | Pred | Exp | Pred | ||
| 6a | 59.13 | 51.38 | 8i | 137.31 | 71.38 |
| 6b | 67.91 | 70.36 | 8j | ND | 137.24 |
| 6c | 47.00 | 7.83 | 9a | 32.20 | 39.33 |
| 8a | 93.83 | 27.49 | 9b | 30.42 | 99.61 |
| 8b | 44.63 | 26.75 | 9c | 41.41 | 58.31 |
| 8c | 89.01 | 38.97 | 9d | 32.34 | −4.21 |
| 8d | 101.32 | 81.56 | 9e | 29.40 | 66.24 |
| 8e | ND | 66.27 | 11a | 26.33 | 49.54 |
| 8f | 21.01 | 66.27 | 11b | 18.13 | 68.52 |
| 8g | ND | 66.27 | 11c | 31.03 | 6.00 |
| 8h | ND | 141.84 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amin, K.M.; El-Badry, O.M.; Abdel Rahman, D.E.; Abdellattif, M.H.; Abourehab, M.A.S.; El-Maghrabey, M.H.; Elsaid, F.G.; El Hamd, M.A.; Elkamhawy, A.; Ammar, U.M. Scaffold Repurposing Reveals New Nanomolar Phosphodiesterase Type 5 (PDE5) Inhibitors Based on Pyridopyrazinone Scaffold: Investigation of In Vitro and In Silico Properties. Pharmaceutics 2022, 14, 1954. https://doi.org/10.3390/pharmaceutics14091954
Amin KM, El-Badry OM, Abdel Rahman DE, Abdellattif MH, Abourehab MAS, El-Maghrabey MH, Elsaid FG, El Hamd MA, Elkamhawy A, Ammar UM. Scaffold Repurposing Reveals New Nanomolar Phosphodiesterase Type 5 (PDE5) Inhibitors Based on Pyridopyrazinone Scaffold: Investigation of In Vitro and In Silico Properties. Pharmaceutics. 2022; 14(9):1954. https://doi.org/10.3390/pharmaceutics14091954
Chicago/Turabian StyleAmin, Kamelia M., Ossama M. El-Badry, Doaa E. Abdel Rahman, Magda H. Abdellattif, Mohammed A. S. Abourehab, Mahmoud H. El-Maghrabey, Fahmy G. Elsaid, Mohamed A. El Hamd, Ahmed Elkamhawy, and Usama M. Ammar. 2022. "Scaffold Repurposing Reveals New Nanomolar Phosphodiesterase Type 5 (PDE5) Inhibitors Based on Pyridopyrazinone Scaffold: Investigation of In Vitro and In Silico Properties" Pharmaceutics 14, no. 9: 1954. https://doi.org/10.3390/pharmaceutics14091954