
Pharmacokinetics and Anti-Diabetic Studies of Gliclazide Nanosuspension
 , and
, and
Abstract
:1. Introduction
2. Materials
3. Methods
3.1. Solvent–Antisolvent Precipitation Technique for GCZ NS
3.2. Formulation by Design (FbD) Approach
3.2.1. Defining the QTPP
3.2.2. CQA Identification
3.2.3. CFAs and CPP Identification
3.2.4. Prescreening Studies
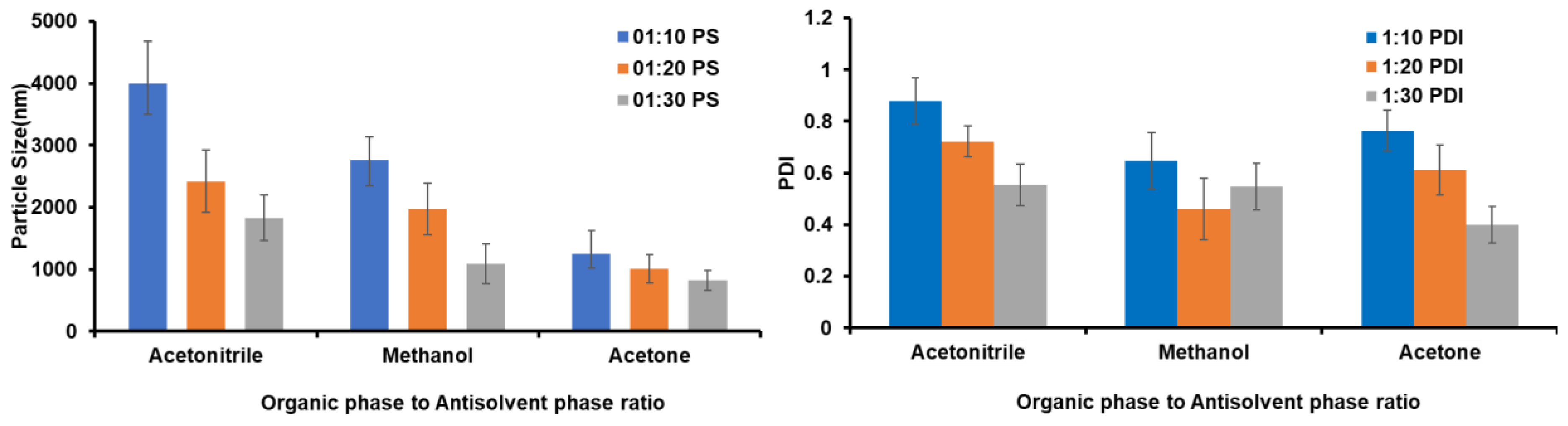
3.2.5. The Proportion of Organic Phase (OP) to Antisolvent Phase (AP) Optimization
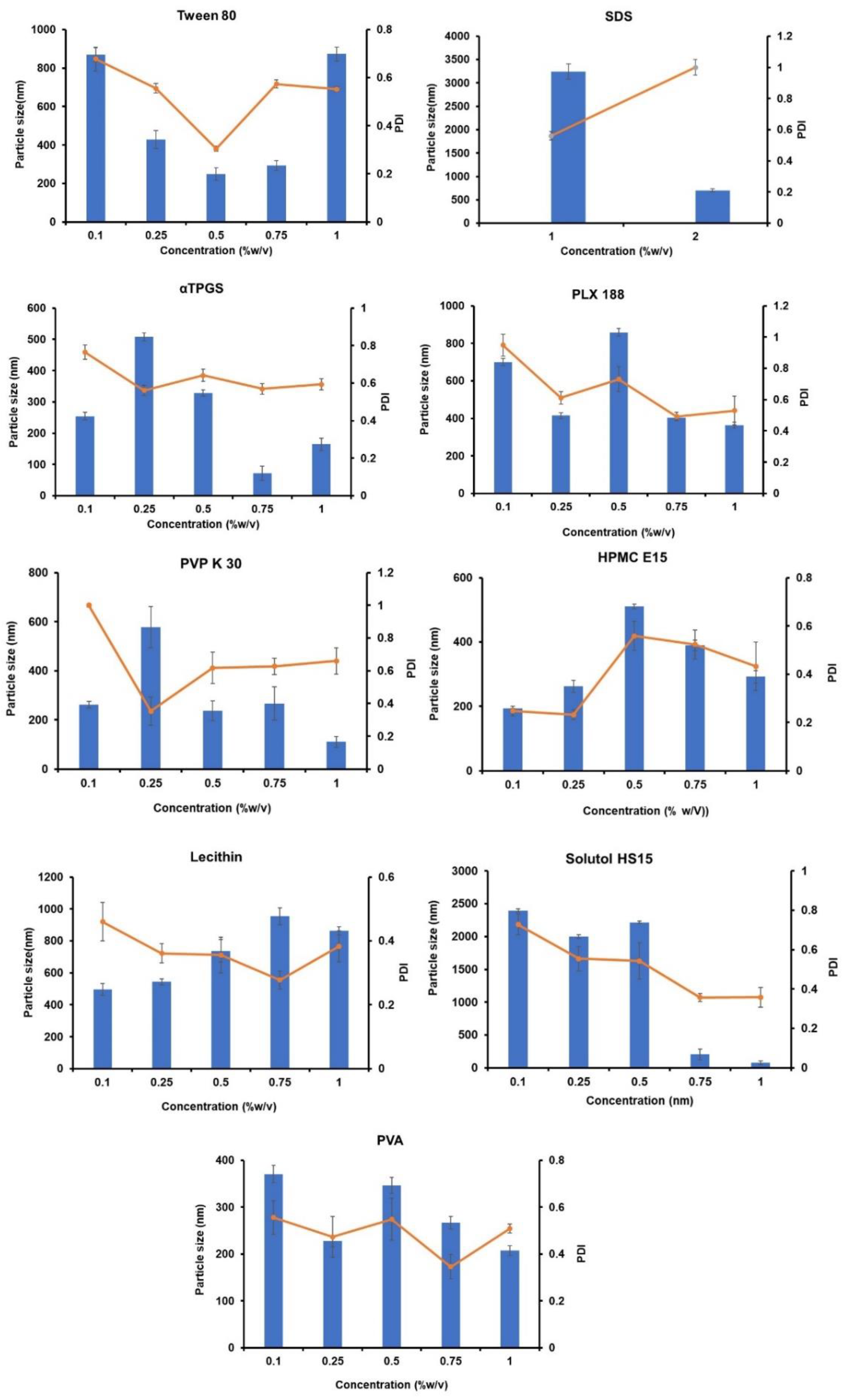
3.2.6. Screening of Stabilizers
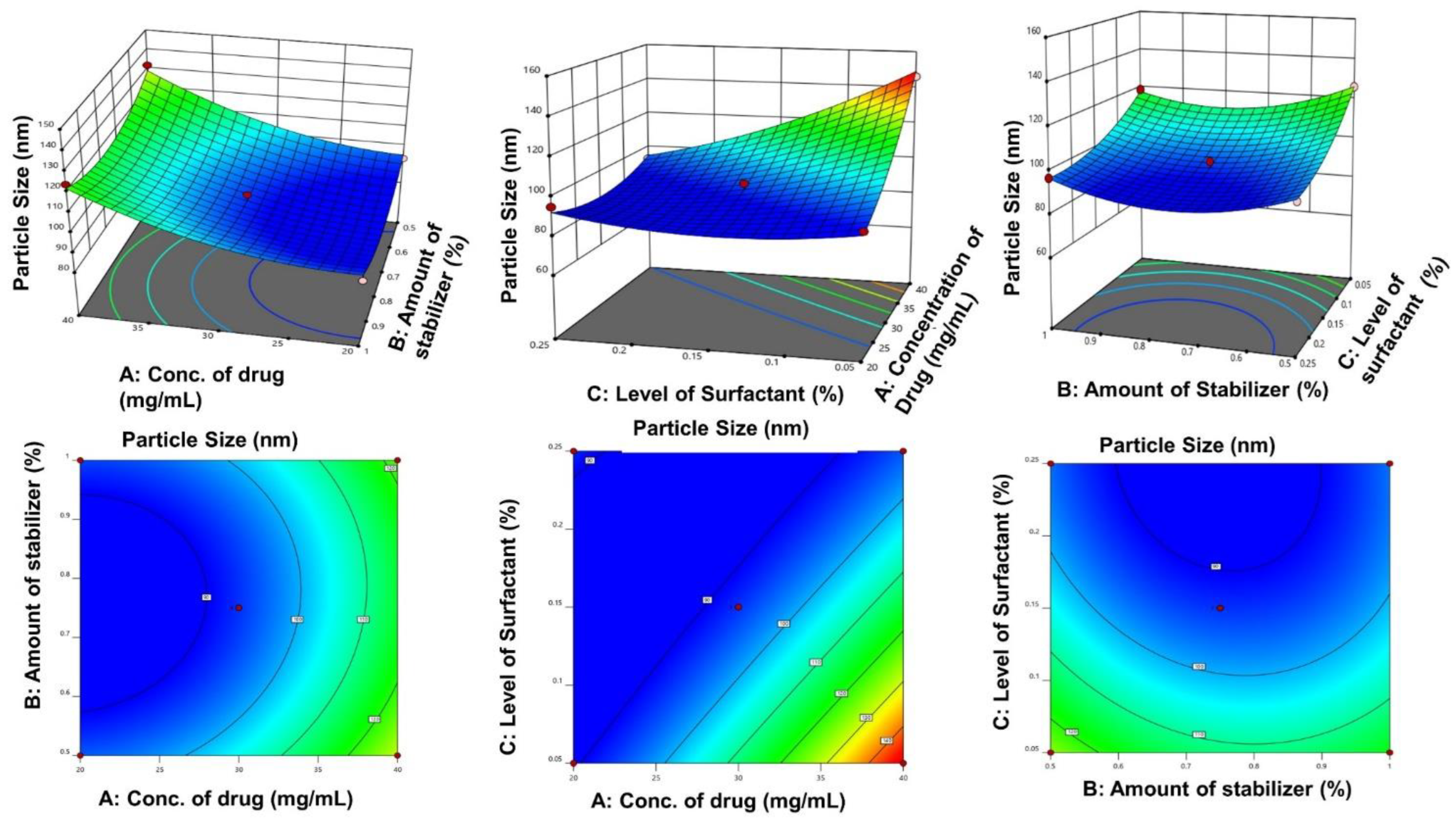
3.2.7. Experimental Design
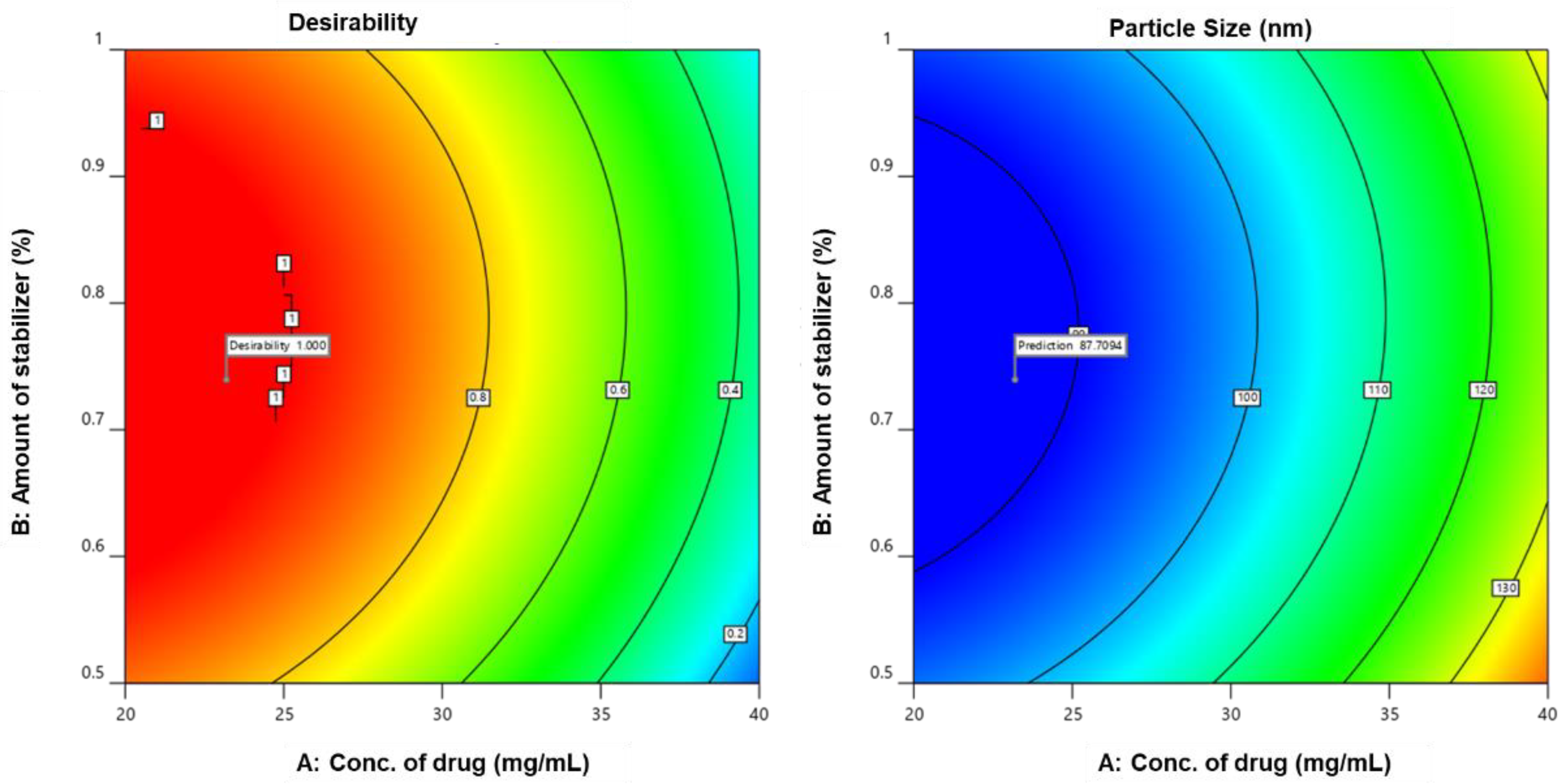
3.2.8. Search for Optimized Preparation
3.2.9. Design Validation
3.3. Physicochemical Characterization of NS
3.3.1. Particle Size, Polydispersity Index (PDI) and Zeta Potential (ZP)
3.3.2. Lyophilization and Redispersibility Index (RDI)
3.3.3. Scanning Electron Microscopy (SEM)
3.3.4. Saturation Solubility
3.3.5. In Vitro Release of GCZ NS
3.3.6. Stability Studies
3.4. Noneverted Intestinal Sac Study
3.5. In Situ Single-Pass Intestinal Perfusion (SPIP) Method
3.6. Pharmacokinetic Studies
3.7. In Vivo Antidiabetic Study
4. Results and Discussion
4.1. Formulation of GCZ NS
4.2. FbD-Based Approach
4.2.1. Defining the QTPP
4.2.2. Identification of CQAs
4.2.3. Identification of CFAs and CPPs
4.2.4. Prescreening
4.2.5. Selection of OP and Optimization of the OP-to-AP Ratio
4.2.6. Selection of Stabilizer
4.2.7. Design of Experiments
4.2.8. Search for Optimized Formulation
4.2.9. Validation of the Design
4.3. Physicochemical Characterization of NS
4.3.1. Particle Size, PDI and ZP
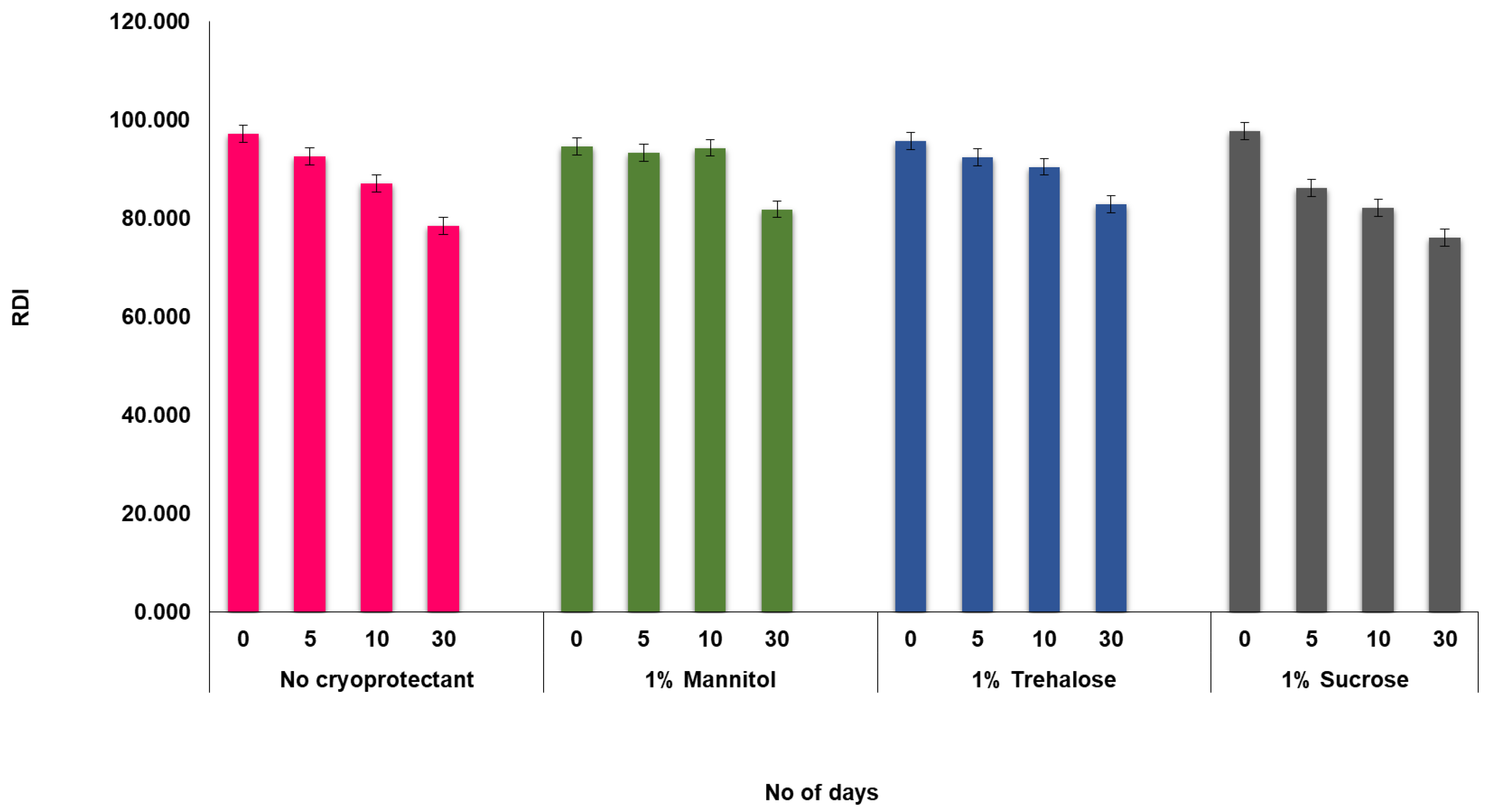
4.3.2. Lyophilization and Redispersity Index (RDI)
4.3.3. Scanning Electron Microscopy
4.3.4. Saturation Solubility
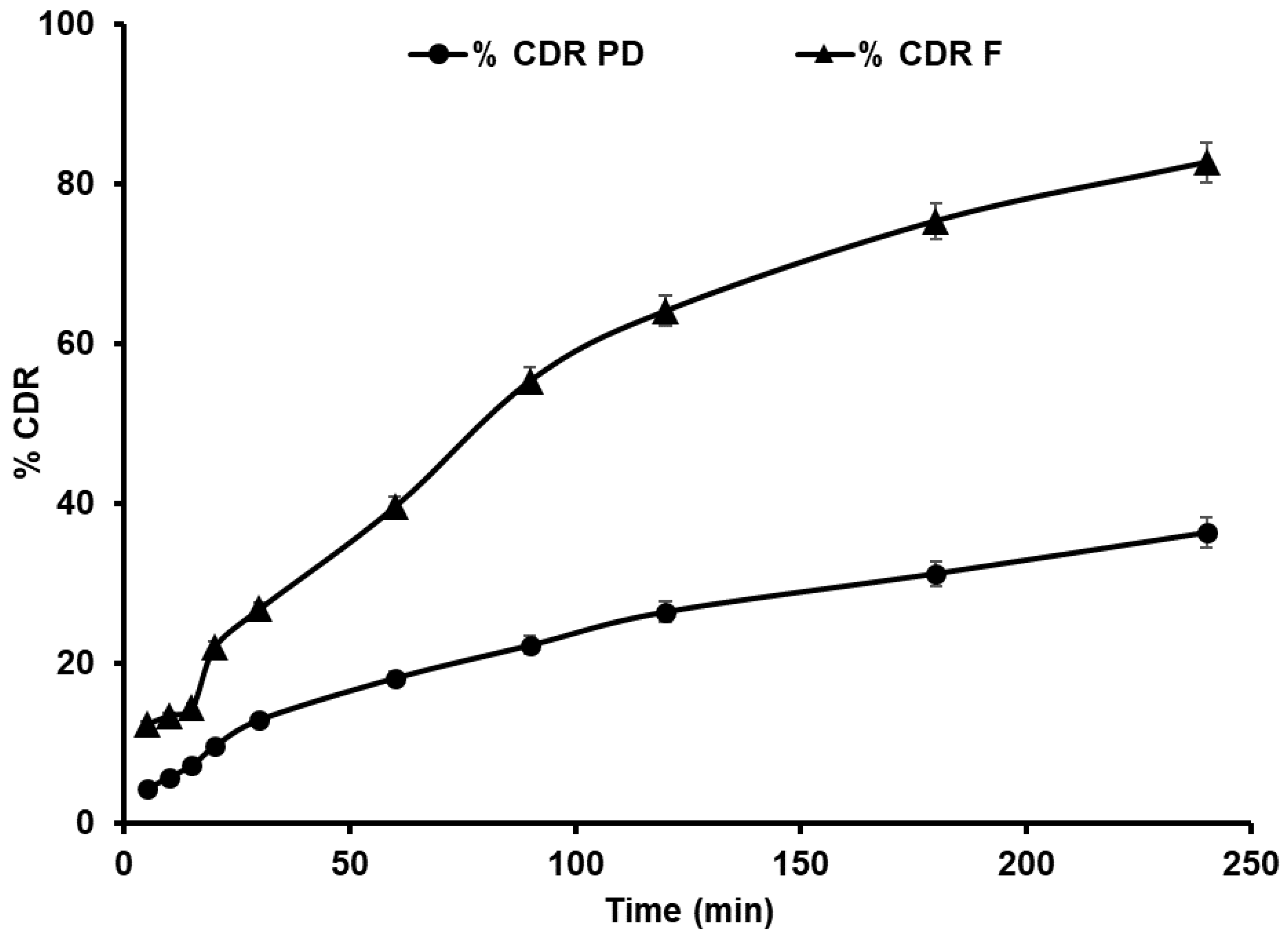
4.3.5. In Vitro Release of GCZ NS
4.3.6. Stability Studies
4.4. Noneverted Intestinal Sac Permeation Study
4.5. In Situ Single-Pass Intestinal Perfusion Method (SPIP)
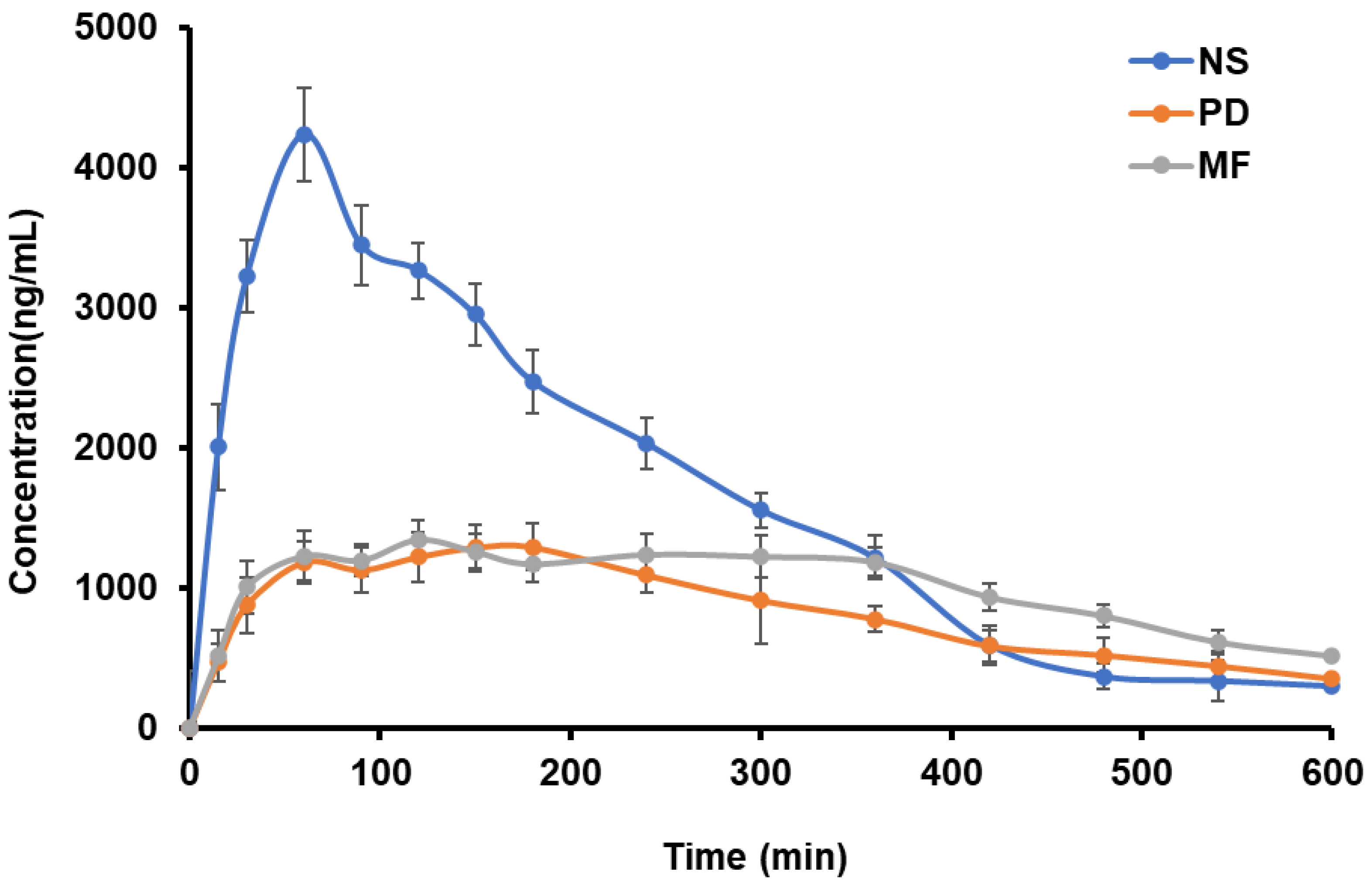
4.6. Pharmacokinetic Studies
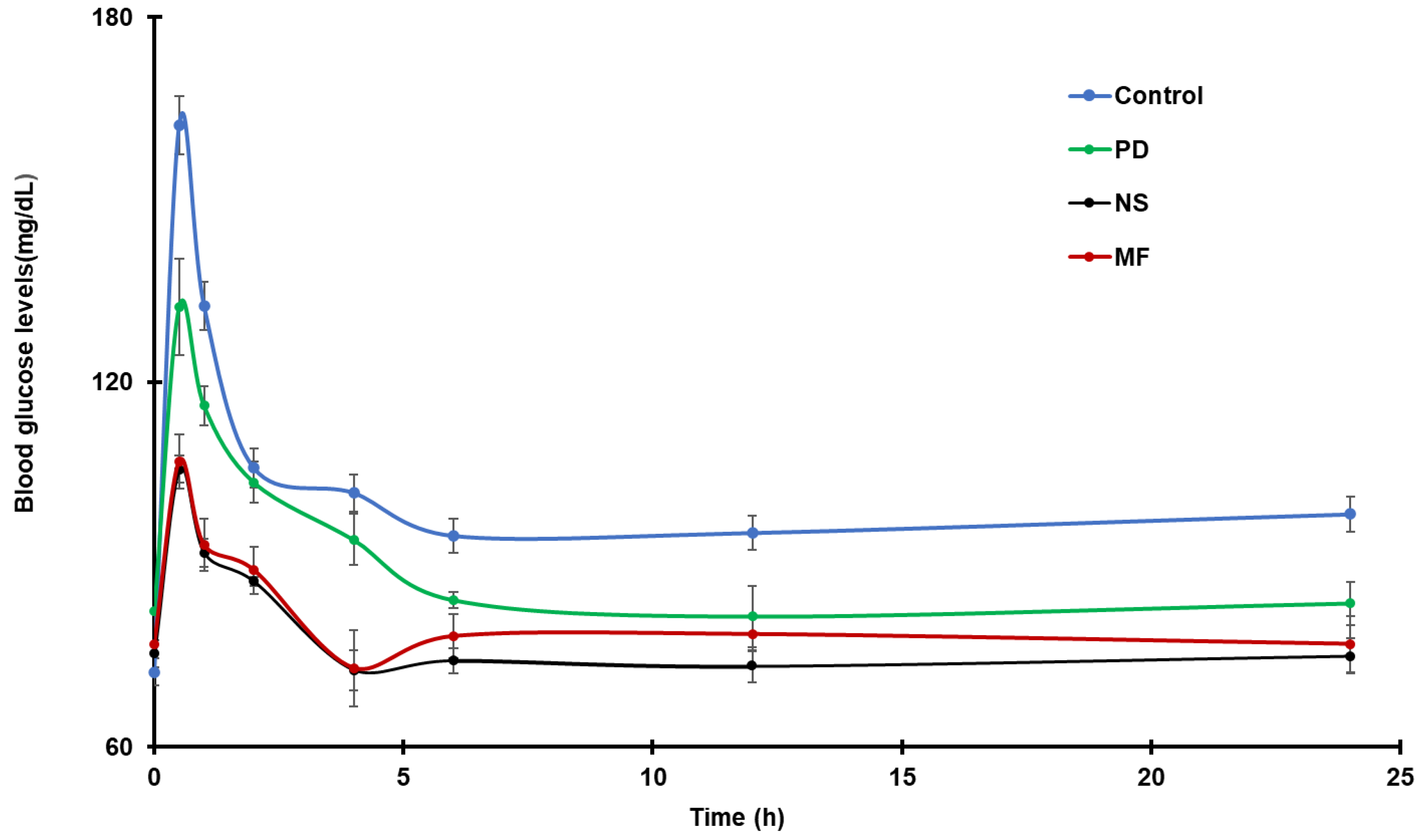
4.7. In Vivo Antidiabetic Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dressman, J.B.; Amidon, G.L.; Reppas, C.; Shah, V.P. Dissolution testing as a prognostic tool for oral drug absorption: Immediate release dosage forms. Pharm. Res. 1998, 15, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and In Vivo Bioavailability. Pharm. Res. An Off. J. Am. Assoc. Pharm. Sci. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- Löbenberg, R.; Amidon, G.L. Modern bioavailability, bioequivalence and biopharmaceutics classification system. New scientific approaches to international regulatory standards. Eur. J. Pharm. Biopharm. 2000, 50, 3–12. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef]
- Alkhamis, K.A.; Allaboun, H.; Al-Momani, W.Y. Study of the solubilization of gliclazide by aqueous micellar solutions. J. Pharm. Sci. 2003, 92, 839–846. [Google Scholar] [CrossRef]
- Varshosaz, J.; Talari, R.; Mostafavi, S.A.; Nokhodchi, A. Dissolution enhancement of gliclazide using in situ micronization by solvent change method. Powder Technol. 2008, 187, 222–230. [Google Scholar] [CrossRef]
- Saharan, V.A.; Choudhury, P.K. Dissolution rate enhancement of gliclazide by ordered mixing. Acta Pharm. 2011, 61, 323–334. [Google Scholar] [CrossRef]
- Palmer, K.J.; Brogden, R.N. Gliclazide: An Update of its Pharmacological Properties and Therapeutic Efficacy in Non-Insulin-Dependent Diabetes Mellitus. Drugs 1993, 46, 92–125. [Google Scholar] [CrossRef]
- Desai, K.G.H.; Kulkarni, A.R.; Aminabhavi, T.M. Solubility of rofecoxib in the presence of methanol, ethanol, and sodium lauryl sulfate at (298.15, 303.15, and 308.15) K. J. Chem. Eng. Data 2003, 48, 942–945. [Google Scholar] [CrossRef]
- Hosmani, A.H.; Thorat, Y.S. Optimization and pharmacodynamic evaluation of solid dispersion of gliclazide for dissolution rate enhancement. Lat. Am. J. Pharm. 2011, 30, 1590–1595. [Google Scholar]
- Khattab, I.S.; Nada, A.; Zaghloul, A.A. Physicochemical characterization of gliclazidemacrogol solid dispersion and tablets based on optimized dispersion. Drug Dev. Ind. Pharm. 2010, 36, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Barzegar-Jalali, M.; Valizadeh, H.; Shadbad, M.R.S.; Adibkia, K.; Mohammadi, G.; Farahani, A.; Arash, Z.; Nokhodchi, A. Cogrinding as an approach to enhance dissolution rate of a poorly water-soluble drug (gliclazide). Powder Technol. 2010, 197, 150–158. [Google Scholar] [CrossRef]
- Biswal, S.; Sahoo, J.; Murthy, P.N.; Giradkar, R.P.; Avari, J.G. Enhancement of dissolution rate of gliclazide using solid dispersions with polyethylene glycol 6000. AAPS PharmSciTech 2008, 9, 563–570. [Google Scholar] [CrossRef]
- Moyano, J.R.; Arias-Blanco, M.J.; Ginésa, J.M.; Giordano, F. Solid-state characterization and dissolution characteristics of gliclazide-β-cyclodextrin inclusion complexes. Int. J. Pharm. 1997, 148, 211–217. [Google Scholar] [CrossRef]
- Aggarwal, S.; Singh, P.N.; Mishra, B. Studies on solubility and hypoglycemic activity of gliclazide β-cyclodextrin-hydroxypropylmethylcellulose complexes. Pharmazie 2002, 57, 191–193. [Google Scholar] [PubMed]
- Hong, S.S.; Lee, S.H.; Lee, Y.J.; Chung, S.J.; Lee, M.H.; Shim, C.K. Accelerated oral absorption of gliclazide in human subjects from a soft gelatin capsule containing a PEG 400 suspension of gliclazide. J. Control. Release 1998, 51, 185–192. [Google Scholar] [CrossRef]
- Devarajan, P.V.; Sonavane, G.S. Preparation and in vitro/in vivo evaluation of gliclazide loaded Eudragit nanoparticles as a sustained release carriers. Drug Dev. Ind. Pharm. 2007, 33, 101–111. [Google Scholar] [CrossRef]
- Averineni, R.K.; Shavi, G.V.; Ranjan, O.P.; Balavant Deshpande, P.; Aravind Kumar, G.; Yogendra Nayak, U.; Sreenivasa Reddy, M.; Udupa, N. Formulation of Gliclazide Encapsulated Chitosan Nanoparticles: In Vitro and In Vivo Evaluation. In NanoFormulation; Royal Society of Chemistry: London, UK, 2012; pp. 77–85. [Google Scholar]
- Awasthi, R.; Kulkarni, G.T. Development of Novel Gastroretentive Floating Particulate Drug Delivery System of Gliclazide. Curr. Drug Deliv. 2012, 9, 437–451. [Google Scholar] [CrossRef]
- Patel, P.; Pailla, S.R.; Rangaraj, N.; Cheruvu, H.S.; Dodoala, S.; Sampathi, S. Quality by Design Approach for Developing Lipid-Based Nanoformulations of Gliclazide to Improve Oral Bioavailability and Anti-Diabetic Activity. AAPS PharmSciTech 2019, 20, 45. [Google Scholar] [CrossRef] [PubMed]
- Dyawanapelly, S.; Junnuthula, V.; Singh, A. The Holy Grail of Polymer Therapeutics for Cancer Therapy: An Overview on the Pharmacokinetics and Bio Distribution. Curr. Drug Metab. 2015, 16, 522–537. [Google Scholar] [CrossRef]
- Sarkar, A.; Junnuthula, V.; Dyawanapelly, S. Ocular therapeutics and molecular delivery strategies for neovascular age-related macular degeneration (Namd). Int. J. Mol. Sci. 2021, 22, 10594. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Jayesh Sodha, S.; Junnuthula, V.; Kolimi, P.; Dyawanapelly, S. Novel and investigational therapies for wet and dry age-related macular degeneration. Drug Discov. Today 2022, 27, 2322–2332. [Google Scholar] [CrossRef] [PubMed]
- Xiong, R.; Xu, R.X.; Huang, C.; De Smedt, S.; Braeckmans, K. Stimuli-responsive nanobubbles for biomedical applications. Chem. Soc. Rev. 2021, 50, 5746–5776. [Google Scholar] [CrossRef] [PubMed]
- Xiong, R.; Hua, D.; Van Hoeck, J.; Berdecka, D.; Léger, L.; De Munter, S.; Fraire, J.C.; Raes, L.; Harizaj, A.; Sauvage, F.; et al. Photothermal nanofibres enable safe engineering of therapeutic cells. Nat. Nanotechnol. 2021, 16, 1281–1291. [Google Scholar] [CrossRef]
- Delrat, P.; Paraire, M.; Jochemsen, R. Complete bioavailability and lack of food-effect on pharmacokinetics of gliclazide 30 mg modified release in healthy volunteers. Biopharm. Drug Dispos. 2002, 23, 151–157. [Google Scholar] [CrossRef]
- Talari, R.; Varshosaz, J.; Mostafavi, S.A.; Nokhodchi, A. Dissolution enhancement of gliclazide using pH change approach in presence of twelve stabilizers with various physico-chemical properties. J. Pharm. Pharm. Sci. 2009, 12, 250–265. [Google Scholar] [CrossRef]
- Ravouru, N.; Venna, R.S.A.; Penjuri, S.C.B.; Damineni, S.; Kotakadi, V.S.; Poreddy, S.R. Fabrication and characterization of gliclazide nanocrystals. Adv. Pharm. Bull. 2018, 8, 419–427. [Google Scholar] [CrossRef]
- Rubina Reichal, C.; Pius, C.R.; Manju, S.; Shobana, M. Formulation and characterization of gliclazide nanosuspension. Res. J. Pharm. Technol. 2021, 14, 779–786. [Google Scholar] [CrossRef]
- Gera, S.; Talluri, S.; Rangaraj, N.; Sampathi, S. Formulation and Evaluation of Naringenin Nanosuspensions for Bioavailability Enhancement. AAPS PharmSciTech 2017, 18, 3151–3162. [Google Scholar] [CrossRef]
- Rangaraj, N.; Pailla, S.R.; Chowta, P.; Sampathi, S. Fabrication of Ibrutinib Nanosuspension by Quality by Design Approach: Intended for Enhanced Oral Bioavailability and Diminished Fast Fed Variability. AAPS PharmSciTech 2019, 20, 326. [Google Scholar] [CrossRef]
- Rangaraj, N.; Sampathi, S.; Junnuthula, V.; Kolimi, P.; Mandati, P.; Narala, S.; Nyavanandi, D.; Dyawanapelly, S. Fast-Fed Variability: Insights into Drug Delivery, Molecular Manifestations, and Regulatory Aspects. Pharmaceutics 2022, 14, 1807. [Google Scholar] [CrossRef]
- Müller, R.H.; Jacobs, C.; Kayser, O. Nanosuspensions as particulate drug formulations in therapy: Rationale for development and what we can expect for the future. Adv. Drug Deliv. Rev. 2001, 47, 3–19. [Google Scholar] [CrossRef]
- He, J.; Han, Y.; Xu, G.; Yin, L.; Ngandeu Neubi, M.; Zhou, J.; Ding, Y. Preparation and evaluation of celecoxib nanosuspensions for bioavailability enhancement. RSC Adv. 2017, 7, 13053–13064. [Google Scholar] [CrossRef]
- He, S.; Yang, H.; Zhang, R.; Li, Y.; Duan, L. Preparation and in vitro-in vivo evaluation of teniposide nanosuspensions. Int. J. Pharm. 2015, 478, 131–137. [Google Scholar] [CrossRef]
- Matteucci, M.E.; Hotze, M.A.; Johnston, K.P.; Williams, R.O. Drug nanoparticles by antisolvent precipitation: Mixing energy versus surfactant stabilization. Langmuir 2006, 22, 8951–8959. [Google Scholar] [CrossRef]
- Rahim, H.; Sadiq, A.; Khan, S.; Khan, M.A.; Shah, S.M.H.; Hussain, Z.; Ullah, R.; Shahat, A.A.; Ibrahim, K. Aceclofenac nanocrystals with enhanced in vitro, in vivo performance: Formulation optimization, characterization, analgesic and acute toxicity studies. Drug Des. Dev. Ther. 2017, 11, 2443–2452. [Google Scholar] [CrossRef]
- Verma, S.; Gokhale, R.; Burgess, D.J. A comparative study of top-down and bottom-up approaches for the preparation of micro/nanosuspensions. Int. J. Pharm. 2009, 380, 216–222. [Google Scholar] [CrossRef]
- Kassem, M.A.A.; ElMeshad, A.N.; Fares, A.R. Enhanced Solubility and Dissolution Rate of Lacidipine Nanosuspension: Formulation Via Antisolvent Sonoprecipitation Technique and Optimization Using Box–Behnken Design. AAPS PharmSciTech 2017, 18, 983–996. [Google Scholar] [CrossRef]
- Wang, L.; Du, J.; Zhou, Y.; Wang, Y. Safety of nanosuspensions in drug delivery. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 455–469. [Google Scholar] [CrossRef]
- Shah, P.P.; Mashru, R.C.; Rane, Y.M.; Badhan, A.C. Design and optimization of artemether microparticles for bitter taste masking. Acta Pharm. 2008, 58, 379–392. [Google Scholar] [CrossRef]
- Gera, S.; Pooladanda, V.; Godugu, C.; Swamy Challa, V.; Wankar, J.; Dodoala, S.; Sampathi, S. Rutin nanosuspension for potential management of osteoporosis: Effect of particle size reduction on oral bioavailability, in vitro and in vivo activity. Pharm. Dev. Technol. 2020, 25, 971–988. [Google Scholar] [CrossRef]
- Colombo, M.; Staufenbiel, S.; Rühl, E.; Bodmeier, R. In situ determination of the saturation solubility of nanocrystals of poorly soluble drugs for dermal application. Int. J. Pharm. 2017, 521, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Kho, K.; Cheow, W.S.; Lie, R.H.; Hadinoto, K. Aqueous re-dispersibility of spray-dried antibiotic-loaded polycaprolactone nanoparticle aggregates for inhaled anti-biofilm therapy. Powder Technol. 2010, 203, 432–439. [Google Scholar] [CrossRef]
- Singh, M.K.; Pooja, D.; Ravuri, H.G.; Gunukula, A.; Kulhari, H.; Sistla, R. Fabrication of surfactant-stabilized nanosuspension of naringenin to surpass its poor physiochemical properties and low oral bioavailability. Phytomedicine 2018, 40, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guan, J.; Ni, R.; Li, L.C.; Mao, S. Preparation and solidification of redispersible nanosuspensions. J. Pharm. Sci. 2014, 103, 2166–2176. [Google Scholar] [CrossRef]
- Liu, D.; Pan, H.; He, F.; Wang, X.; Li, J.; Yang, X.; Pan, W. Effect of particle size on oral absorption of carvedilol nanosuspensions: In vitro and in vivo evaluation. Int. J. Nanomed. 2015, 10, 6425–6434. [Google Scholar] [CrossRef]
- Tang, X.J.; Fu, Y.H.; Meng, Q.H.; Li, L.M.; Ying, X.Y.; Han, M.; He, Q.J.; Yang, B.; Zeng, S.; Hu, Y.Z.; et al. Evaluation of pluronic nanosuspensions loading a novel insoluble anticancer drug both in vitro and in vivo. Int. J. Pharm. 2013, 456, 243–250. [Google Scholar] [CrossRef]
- Guo, L.; Kang, L.; Liu, X.; Lin, X.; Di, D.; Wu, Y.; Kong, D.; Deng, Y.; Song, Y. A novel nanosuspension of andrographolide: Preparation, characterization and passive liver target evaluation in rats. Eur. J. Pharm. Sci. 2017, 104, 13–22. [Google Scholar] [CrossRef]
- Neerati, P.; Kumar Bedada, S. Effect of diosmin on the intestinal absorption and pharmacokinetics of fexofenadine in rats. Pharmacol. Rep. 2015, 67, 339–344. [Google Scholar] [CrossRef]
- Srikanth, C.H.; Chaira, T.; Sampathi, S.; Sreekumar, V.B.; Bambal, R.B. Correlation of in vitro and in vivo plasma protein binding using ultracentrifugation and UPLC-tandem mass spectrometry. Analyst 2013, 138, 6106–6116. [Google Scholar] [CrossRef]
- Pailla, S.; Sampathi, S.; Junnuthula, V.; Maddukuri, S.; Dodoala, S.; Dyawanapelly, S. Brain-Targeted Intranasal Delivery of Zotepine Microemulsion: Pharmacokinetics and Pharmacodynamics. Pharmaceutics 2022, 14, 978. [Google Scholar] [CrossRef] [PubMed]
- Dalvi, S.V.; Dave, R.N. Controlling particle size of a poorly water-soluble drug using ultrasound and stabilizers in antisolvent precipitation. Ind. Eng. Chem. Res. 2009, 48, 7581–7593. [Google Scholar] [CrossRef]
- Singh, A.; Neupane, Y.R.; Panda, B.P.; Kohli, K. Lipid Based nanoformulation of lycopene improves oral delivery: Formulation optimization, ex vivo assessment and its efficacy against breast cancer. J. Microencapsul. 2017, 34, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Kakran, M.; Sahoo, N.G.; Tan, I.L.; Li, L. Preparation of nanoparticles of poorly water-soluble antioxidant curcumin by antisolvent precipitation methods. J. Nanopart. Res. 2012, 14, 757. [Google Scholar] [CrossRef]
- Junnuthula, V.; Kolimi, P.; Nyavanandi, D.; Sampathi, S.; Vora, L.K.; Dyawanapelly, S. Polymeric Micelles for Breast Cancer Therapy: Recent Updates, Clinical Translation and Regulatory Considerations. Pharmaceutics 2022, 14, 1860. [Google Scholar] [CrossRef]
- Gera, S.; Sampathi, S.; Maddukuri, S.; Dodoala, S.; Junnuthula, V.; Dyawanapelly, S. Therapeutic Potential of Naringenin Nanosuspension: In Vitro and In Vivo Anti-Osteoporotic Studies. Pharmaceutics 2022, 14, 1449. [Google Scholar] [CrossRef]
- Afifi, S.A.; Hassan, M.A.; Abdelhameed, A.S.; Elkhodairy, K.A. Nanosuspension: An emerging trend for bioavailability enhancement of etodolac. Int. J. Polym. Sci. 2015, 2015, 938594. [Google Scholar] [CrossRef]
- Ghosh, I.; Bose, S.; Vippagunta, R.; Harmon, F. Nanosuspension for improving the bioavailability of a poorly soluble drug and screening of stabilizing agents to inhibit crystal growth. Int. J. Pharm. 2011, 409, 260–268. [Google Scholar] [CrossRef]
- Lu, S.; Yu, P.P.; He, J.H.; Zhang, S.S.; Xia, Y.L.; Zhang, W.L.; Liu, J.P. Enhanced dissolution and oral bioavailability of lurasidone hydrochloride nanosuspensions prepared by antisolvent precipitation-ultrasonication method. RSC Adv. 2016, 6, 49052–49059. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, Y.; Zheng, Y.; Song, J.; Yang, X.; Bi, C.; Zhang, D.; Zhang, Q. In vitro and in vivo anticancer activity of a novel puerarin nanosuspension against colon cancer, with high efficacy and low toxicity. Int. J. Pharm. 2013, 441, 728–735. [Google Scholar] [CrossRef]
- Soroushnia, A.; Ganji, F.; Vasheghani-Farahani, E.; Mobedi, H. Effect of combined stabilizers on midazolam nanosuspension properties. Iran. Polym. J. 2022, 31, 215–222. [Google Scholar] [CrossRef]
- Voorhees, P.W. The theory of Ostwald ripening. J. Stat. Phys. 1985, 38, 231–252. [Google Scholar] [CrossRef]
- Jacob, S.; Nair, A.B.; Shah, J. Emerging role of nanosuspensions in drug delivery systems. Biomater. Res. 2020, 24, 3. [Google Scholar] [CrossRef] [PubMed]
- Kocbek, P.; Baumgartner, S.; Kristl, J. Preparation and evaluation of nanosuspensions for enhancing the dissolution of poorly soluble drugs. Int. J. Pharm. 2006, 312, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Van Eerdenbrugh, B.; Van den Mooter, G.; Augustijns, P. Top-down production of drug nanocrystals: Nanosuspension stabilization, miniaturization and transformation into solid products. Int. J. Pharm. 2008, 364, 64–75. [Google Scholar] [CrossRef]
- Chouhan, P.; Saini, T.R. D-optimal design and development of microemulsion based transungual drug delivery formulation of ciclopirox olamine for treatment of onychomycosis. Indian J. Pharm. Sci. 2016, 78, 498–511. [Google Scholar] [CrossRef]
- Lin, Y.; Alexandridis, P. Temperature-dependent adsorption of Pluronic F127 block copolymers onto carbon black particles dispersed in aqueous media. J. Phys. Chem. B 2002, 106, 10834–10844. [Google Scholar] [CrossRef]
- Shah, B.; Khunt, D.; Bhatt, H.; Misra, M.; Padh, H. Application of quality by design approach for intranasal delivery of rivastigmine loaded solid lipid nanoparticles: Effect on formulation and characterization parameters. Eur. J. Pharm. Sci. 2015, 78, 54–66. [Google Scholar] [CrossRef]
- Patravale, V.B.; Date, A.A.; Kulkarni, R.M. Nanosuspensions: A promising drug delivery strategy. J. Pharm. Pharmacol. 2010, 56, 827–840. [Google Scholar] [CrossRef]
- Kesisoglou, F.; Panmai, S.; Wu, Y. Nanosizing—Oral formulation development and biopharmaceutical evaluation. Adv. Drug Deliv. Rev. 2007, 59, 631–644. [Google Scholar] [CrossRef]
- Saez, A.; Guzmán, M.; Molpeceres, J.; Aberturas, M.R. Freeze-drying of polycaprolactone and poly(d,l-lactic-glycolic) nanoparticles induce minor particle size changes affecting the oral pharmacokinetics of loaded drugs. Eur. J. Pharm. Biopharm. 2000, 50, 379–387. [Google Scholar] [CrossRef]
- Ma, Y.Q.; Zhang, Z.Z.; Li, G.; Zhang, J.; Xiao, H.Y.; Li, X.F. Solidification drug nanosuspensions into nanocrystals by freeze-drying: A case study with ursodeoxycholic acid. Pharm. Dev. Technol. 2016, 21, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.; Junghanns, J.-U.A.H. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.; Müller, R.H.; Möschwitzer, J.P. Bottom-up approaches for preparing drug nanocrystals: Formulations and factors affecting particle size. Int. J. Pharm. 2013, 453, 126–141. [Google Scholar] [CrossRef]
- Belkacem, N.; Sheikh Salem, M.A.; AlKhatib, H.S. Effect of ultrasound on the physico-chemical properties of poorly soluble drugs: Antisolvent sonocrystallization of ketoprofen. Powder Technol. 2015, 285, 16–24. [Google Scholar] [CrossRef]
- Verma, S.; Kumar, S.; Gokhale, R.; Burgess, D.J. Physical stability of nanosuspensions: Investigation of the role of stabilizers on Ostwald ripening. Int. J. Pharm. 2011, 406, 145–152. [Google Scholar] [CrossRef]
- Ruan, L.P.; Chen, S.; Yu, B.Y.; Zhu, D.N.; Cordell, G.A.; Qiu, S.X. Prediction of human absorption of natural compounds by the non-everted rat intestinal sac model. Eur. J. Med. Chem. 2006, 41, 605–610. [Google Scholar] [CrossRef]
- Attari, Z.; Bhandari, A.; Jagadish, P.C.; Lewis, S. Enhanced ex vivo intestinal absorption of olmesartan medoxomil nanosuspension: Preparation by combinative technology. Saudi Pharm. J. 2016, 24, 57–63. [Google Scholar] [CrossRef]
- Nazief, A.M.; Hassaan, P.S.; Khalifa, H.M.; Sokar, M.S.; El-Kamel, A.H. Lipid-Based Gliclazide Nanoparticles for Treatment of Diabetes: Formulation, Pharmacokinetics, Pharmacodynamics and Subacute Toxicity Study. Int. J. Nanomed. 2020, 15, 1129. [Google Scholar] [CrossRef]
- Nasr, M.; Almawash, S.; Al Saqr, A.; Bazeed, A.Y.; Saber, S.; Elagamy, H.I. Bioavailability and Antidiabetic Activity of Gliclazide-Loaded Cubosomal Nanoparticles. Pharmaceuticals 2021, 14, 786. [Google Scholar] [CrossRef]
- Kumar, V.; Mazumder, B.; Sharma, P.P.; Ahmed, Y. Pharmacokinetics and hypoglycemic effect of gliclazide loaded in Isabgol husk mucilage microparticles. J. Pharm. Investig. 2021, 51, 159–171. [Google Scholar] [CrossRef]
- Karakucuk, A.; Celebi, N.; Teksin, Z.S. Preparation of ritonavir nanosuspensions by microfluidization using polymeric stabilizers: I. A Design of Experiment approach. Eur. J. Pharm. Sci. 2016, 95, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Rangaraj, N.; Pailla, S.R.; Shah, S.; Prajapati, S.; Sampathi, S. QbD aided development of ibrutinib-loaded nanostructured lipid carriers aimed for lymphatic targeting: Evaluation using chylomicron flow blocking approach. Drug Deliv. Transl. Res. 2020, 10, 1476–1494. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables | Levels | |||
|---|---|---|---|---|
| Low (−1) | Medium (0) | High (+1) | ||
| A | Concentration of drug (mg/mL) | 20 | 30 | 40 |
| B | Amount of stabilizer (%) | 0.5 | 0.75 | 1 |
| C | Level of surfactant (%) | 0.05 | 0.15 | 0.25 |
| Responses | Constraints | |||
| X | Particle size | Minimize | ||
| QTPP | Target | Justification |
|---|---|---|
| Formulation | Nanosuspension (NS) | The solubility and bioavailability can be improved by NS formulation |
| Route of administration | Oral | The commercial formulation is oral, and we are working to increase oral bioavailability |
| Dissolution | Higher compared to plain drug | Increased solubility could result in accelerated dissolution |
| Pharmacokinetics | Should be better than the already available form | For increased bioavailability |
| Stability | No visible signs of aggregation/cake formation up to 120 days after formulation | The efficiency of the formulation depends on particle size. It is vital to maintain the same stability |
| CQAs | ||
| CQA | Target | Justification |
| Particle size | nm | The solubility and dissolution are both increased when size is reduced to the nanoscale because it increases surface area. Bioavailability is improved via higher solubility and dissolution |
| Formulation | Concentration of Stabilizers (% w/v) | |||||||
|---|---|---|---|---|---|---|---|---|
| HPMC | SDS | PLX 188 | Lecithin | Tween 80 | PVA | Avg. PS (nm) | Avg. PDI | |
| F1 | 0.1 | 0.1 | - | - | - | - | 825.7 ± 89.38 | 0.831 ± 0.06 |
| F2 | 0.25 | 0.25 | - | - | - | - | 2038.66 ± 17.00 | 0.769 ± 0.39 |
| F3 | 0.5 | - | 0.1 | - | - | - | 4777.66 ± 29.48 | 0.873 ± 0.21 |
| F4 | 0.1 | 0.25 | - | - | - | - | 4795.66 ± 26.84 | 1.000 ± 0.00 |
| F5 | 0.1 | - | 0.1 | - | - | - | 475.30 ± 64.00 | 0.619 ± 0.12 |
| F6 | 0.1 | - | 0.5 | - | - | - | 1018.83 ± 58.38 | 0.896 ± 0.08 |
| F7 | 0.1 | - | - | 0.1 | - | - | 1399.66 ± 14.84 | 0.920 ± 0.03 |
| F8 | - | 0.25 | - | 0.1 | - | - | 939.33 ± 72.34 | 0.654 ± 0.32 |
| F9 | - | - | - | - | 1 | 1 | 247.3 ± 130.44 | 0.347 ± 0.09 |
| F10 | - | - | - | - | 0.1 | 0.1 | 999.96 ± 281.36 | 0.702 ± 0.16 |
| F11 | 0.1 | - | - | - | 0.5 | - | 1134.1 ± 158.66 | 0.697 ± 0.08 |
| F12 | - | 0.25 | - | 1.0 | - | - | 96.49 ± 15.00 | 0.326 ± 0.05 |
| F13 | - | 0.1 | - | 1.0 | - | - | 146.70 ± 2.55 | 0.386 ± 0.01 |
| Different Medias | Plain Drug (GCZ) (µg/mL) | NS Formulation (µg/mL) |
|---|---|---|
| Water | 49.6 ± 6.37 | 681.87 ± 63.24 |
| 0.1 N Hydrochloric acid (pH 1.2) | 29.61 ± 9.58 | 182.05 ± 10.36 |
| Acetate buffer (pH 4.5) | 24.88 ± 6.58 | 364.18 ± 89.31 |
| Phosphate buffer (pH 7.4) | 629.04 ± 163.05 | 2604.57 ± 321.68 |
| Temperature | Months | PS (nm) | PDI | ZP (mV) |
|---|---|---|---|---|
| 5 ± 3 °C | Initial | 87.12 ± 3.76 | 0.172 ± 0.026 | −22.19 ± 2.16 |
| 0.5 | 89.86 ± 5.56 | 0.265 ± 0.022 | −19.17 ± 1.78 | |
| 1 | 90.65 ± 5.48 | 0.271 ± 0.042 | −21.42 ± 2.95 | |
| 3 | 92.88 ± 8.13 | 0.289 ± 0.021 | −19.86 ± 2.42 | |
| 6 | 102.14 ± 16.28 | 0.308 ± 0.038 | −21.84 ± 3.01 | |
| 25 ± 2 °C | Initial | 87.12 ± 3.76 | 0.172 ± 0.026 | −21.56 ± 2.86 |
| 0.5 | 96.92 ± 8.31 | 0.297 ± 0.028 | −22.19 ± 2.16 | |
| 1 | 124.71 ± 6.18 | 0.263 ± 0.029 | −22.99 ± 2.52 | |
| 3 | 149.52 ± 6.98 | 0.304 ± 0.032 | −22.66 ± 2.65 | |
| 6 | 153.92 ± 5.73 | 0.322 ± 0.025 | −23.12 ± 2.38 | |
| 40 ± 2 °C | Initial | 87.12 ± 3.76 | 0.172 ± 0.022 | −22.19 ± 2.16 |
| 0.5 | 119.42 ± 5.21 | 0.297 ± 0.020 | −21.02 ± 3.90 | |
| 1 | 133.26 ± 7.39 | 0.303 ± 0.025 | −20.04 ± 3.12 | |
| 3 | 180.66 ± 6.98 | 0.327 ± 0.030 | −20.38 ± 2.90 | |
| 6 | 212.38 ± 8.04 | 0.322 ± 0.032 | −20.65 ± 3.44 |
| PK Parameter (Units) | Plain Drug Suspension | NS | MF |
|---|---|---|---|
| AUC(0–t) (ng/mL·h) | 8334.106 ± 102 | 16,766.277 ± 125 | 10,163.584 ± 132 |
| AUC(0–α) (ng·h/mL) | 10,238.84 ± 105 | 19,649.178 ± 128 | 12,627.599 ± 135 |
| Cmax (ng/mL) | 1290.813 ± 118 | 4234.691 ± 120 | 1346.013 ± 115 |
| Tmax (h) | 3.00 | 1.00 | 2.00 |
| Kel (1/h) | 5 ± 0.04 | 0.104 ± 0.06 | 0.209 ± 0.01 |
| t1/2 (h) | 3.744 ± 1.2 | 6.662 ± 0.15 | 3.316 ± 2.5 |
| % Relative Bioavailability (Frel) * | 100.00 | 201.175 | 121.951 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sampathi, S.; Prajapati, S.; Junnuthula, V.; Dyawanapelly, S. Pharmacokinetics and Anti-Diabetic Studies of Gliclazide Nanosuspension. Pharmaceutics 2022, 14, 1947. https://doi.org/10.3390/pharmaceutics14091947
Sampathi S, Prajapati S, Junnuthula V, Dyawanapelly S. Pharmacokinetics and Anti-Diabetic Studies of Gliclazide Nanosuspension. Pharmaceutics. 2022; 14(9):1947. https://doi.org/10.3390/pharmaceutics14091947
Chicago/Turabian StyleSampathi, Sunitha, Shubham Prajapati, Vijayabhaskarreddy Junnuthula, and Sathish Dyawanapelly. 2022. "Pharmacokinetics and Anti-Diabetic Studies of Gliclazide Nanosuspension" Pharmaceutics 14, no. 9: 1947. https://doi.org/10.3390/pharmaceutics14091947