
Cheminformatics Identification of Phenolics as Modulators of Penicillin-Binding Protein 2a of Staphylococcus aureus: A Structure–Activity-Relationship-Based Study


Abstract
:1. Introduction
2. Materials and Methods
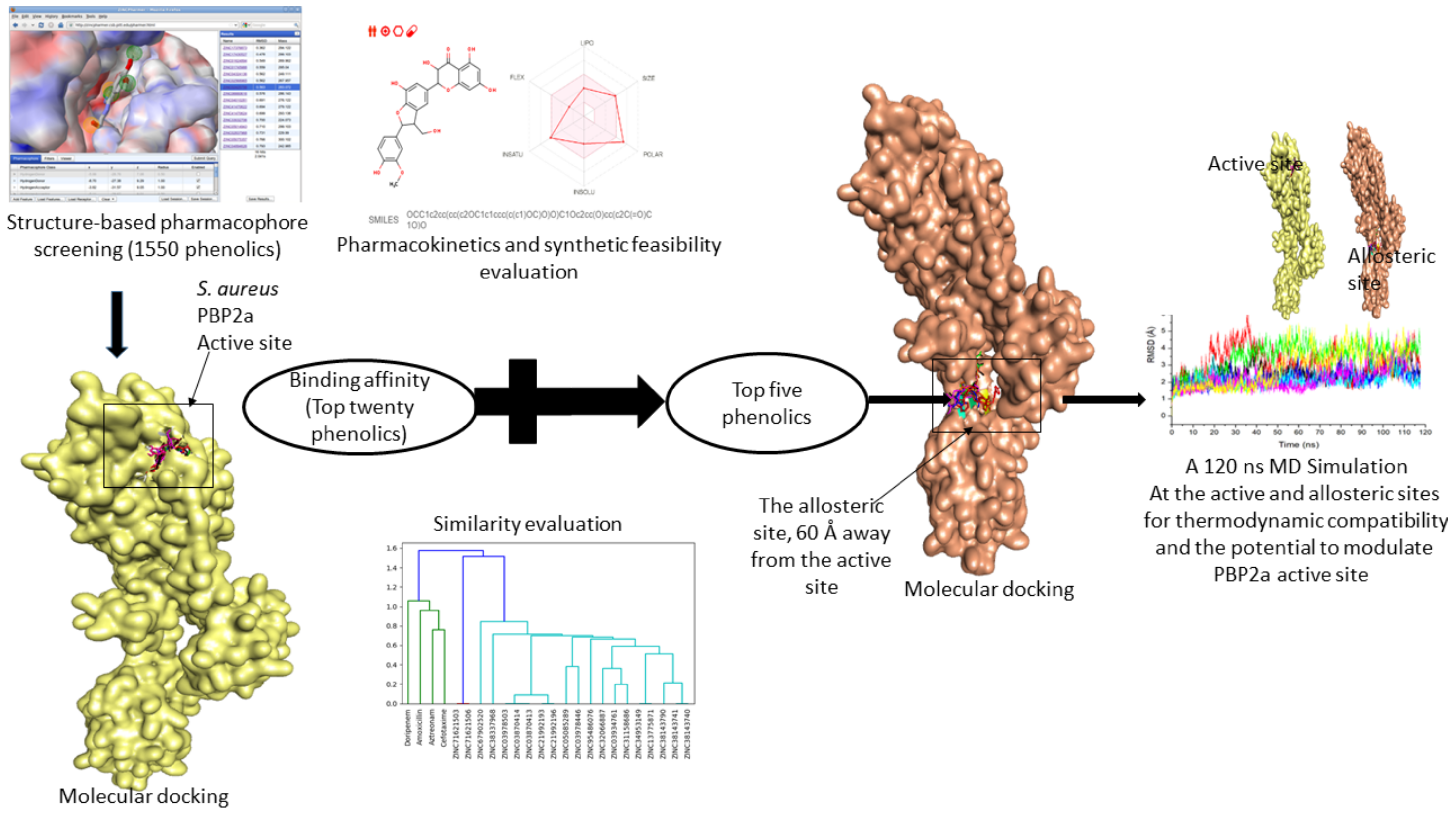
2.1. Druggable Target Acquisition, Preparation, and Identification of Binding Sites
2.2. Structure-Based Pharmacophore Screening of Phenolic Compounds
2.3. Ligand Retrieval, Optimization, and Molecular Docking at the Active and Allosteric Site of PBP2a
2.4. Top-Twenty Phenolics Pharmacokinetic Properties Prediction
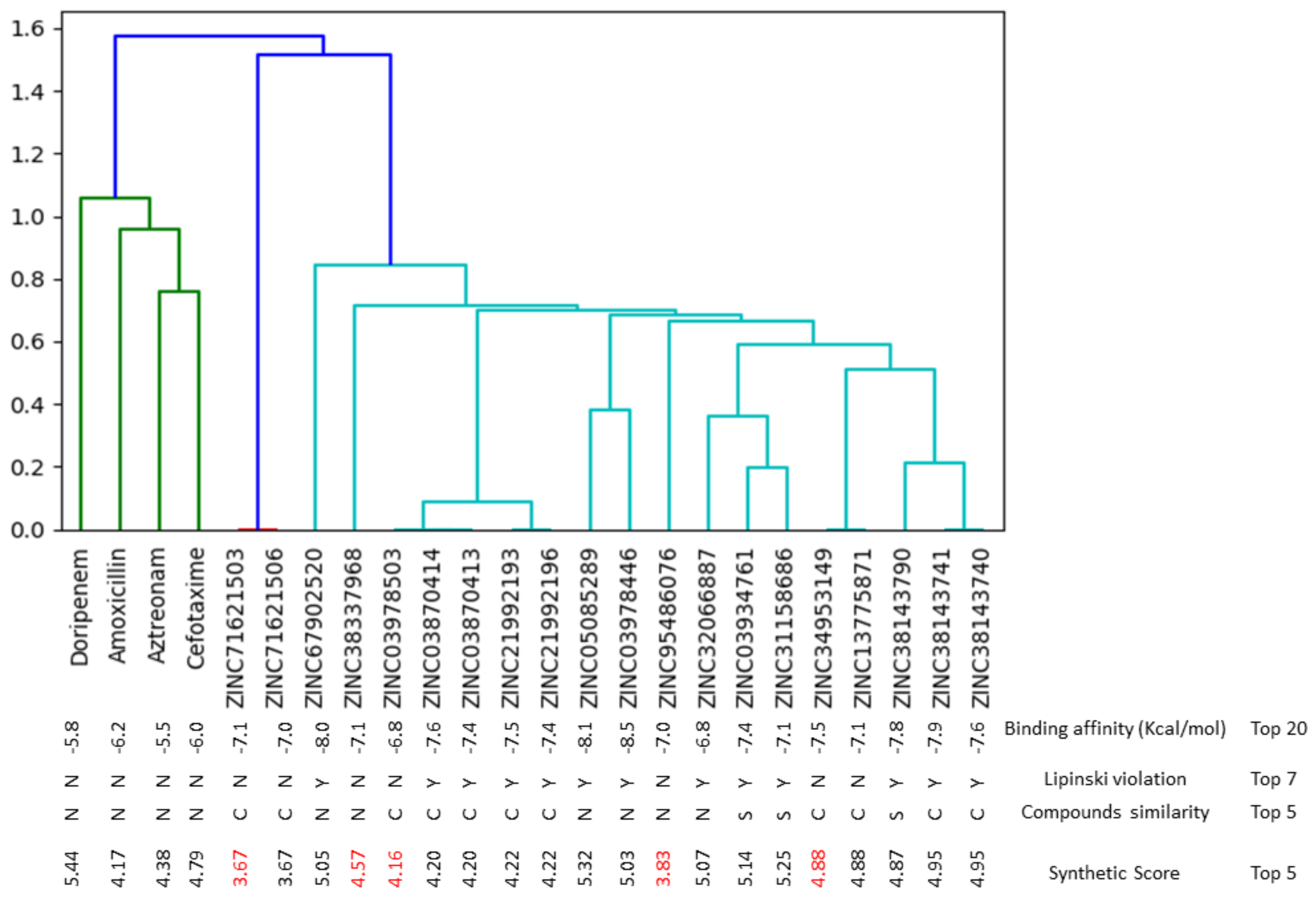
2.5. Molecular Fingerprinting of the Top-Twenty Phenolics
2.6. Molecular Dynamic (MD) Simulations of Top-Five Hit Phenolics
2.7. Post-Dynamic Analysis
3. Results and Discussion
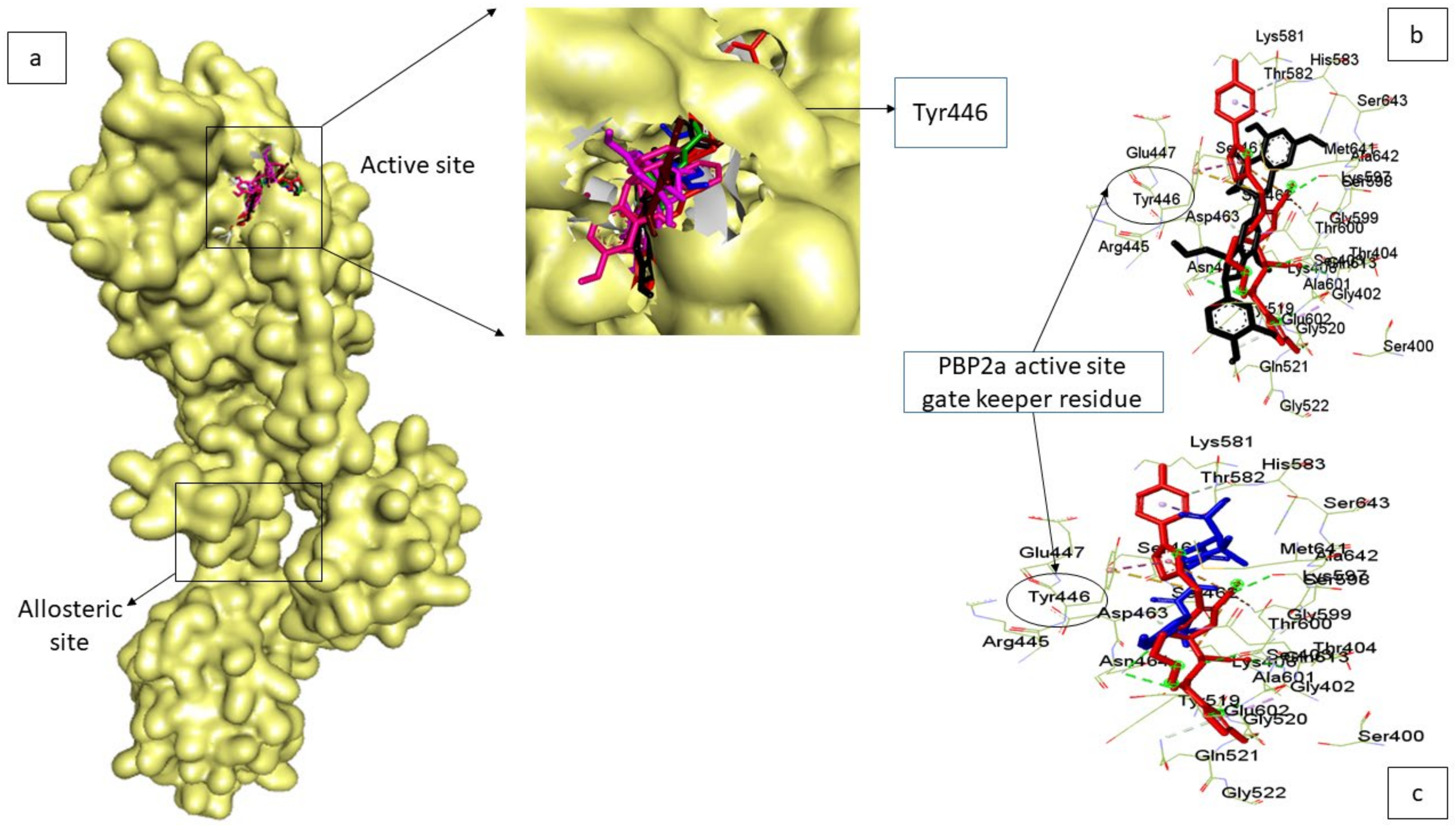
3.1. Ranking of Phenolics against the Active Site of PBP2a of S. aureus
3.2. Thermodynamic Binding Free Energy of Top-Five Phenolics at the Active Site of PBP2a of S. aureus
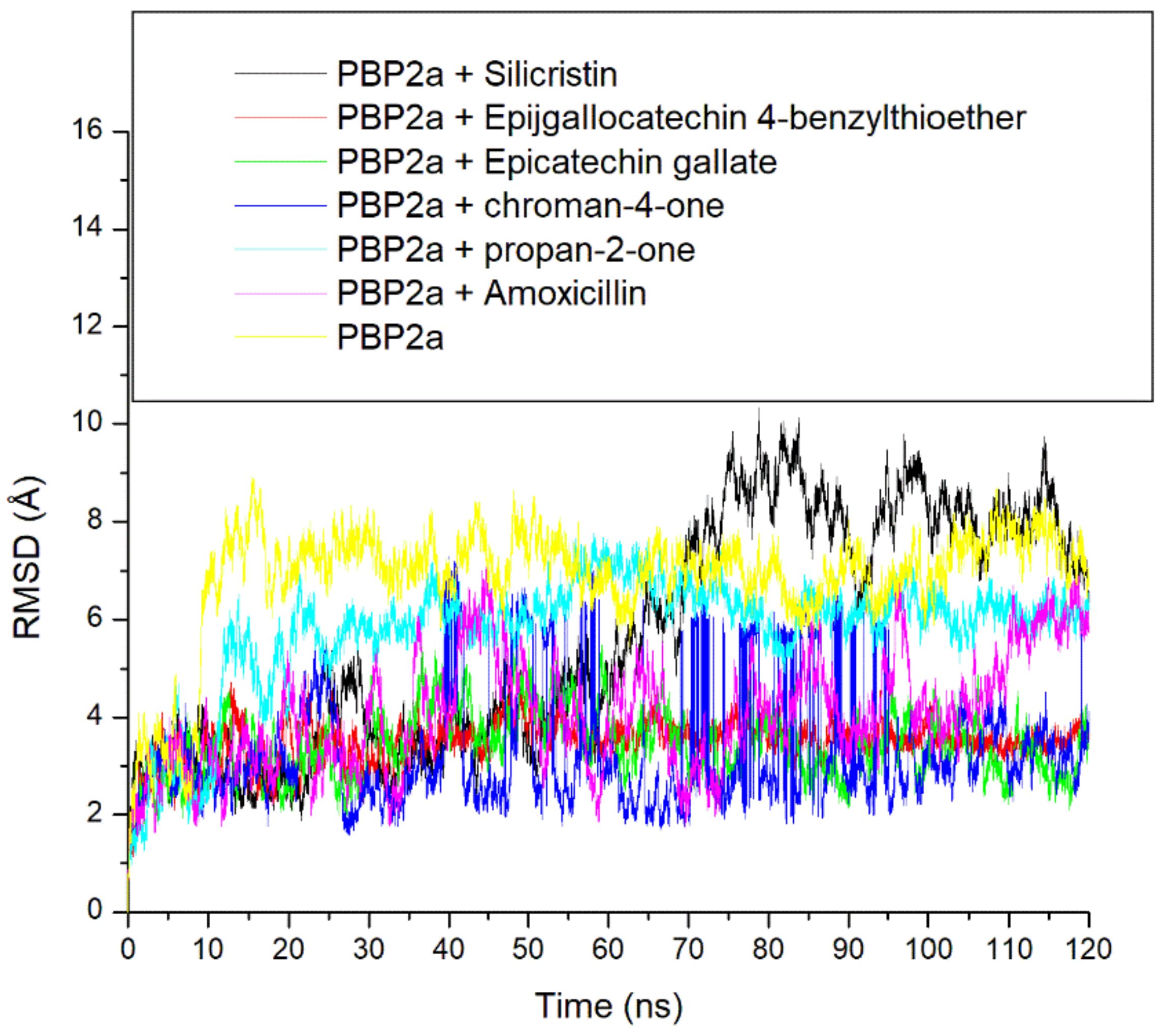
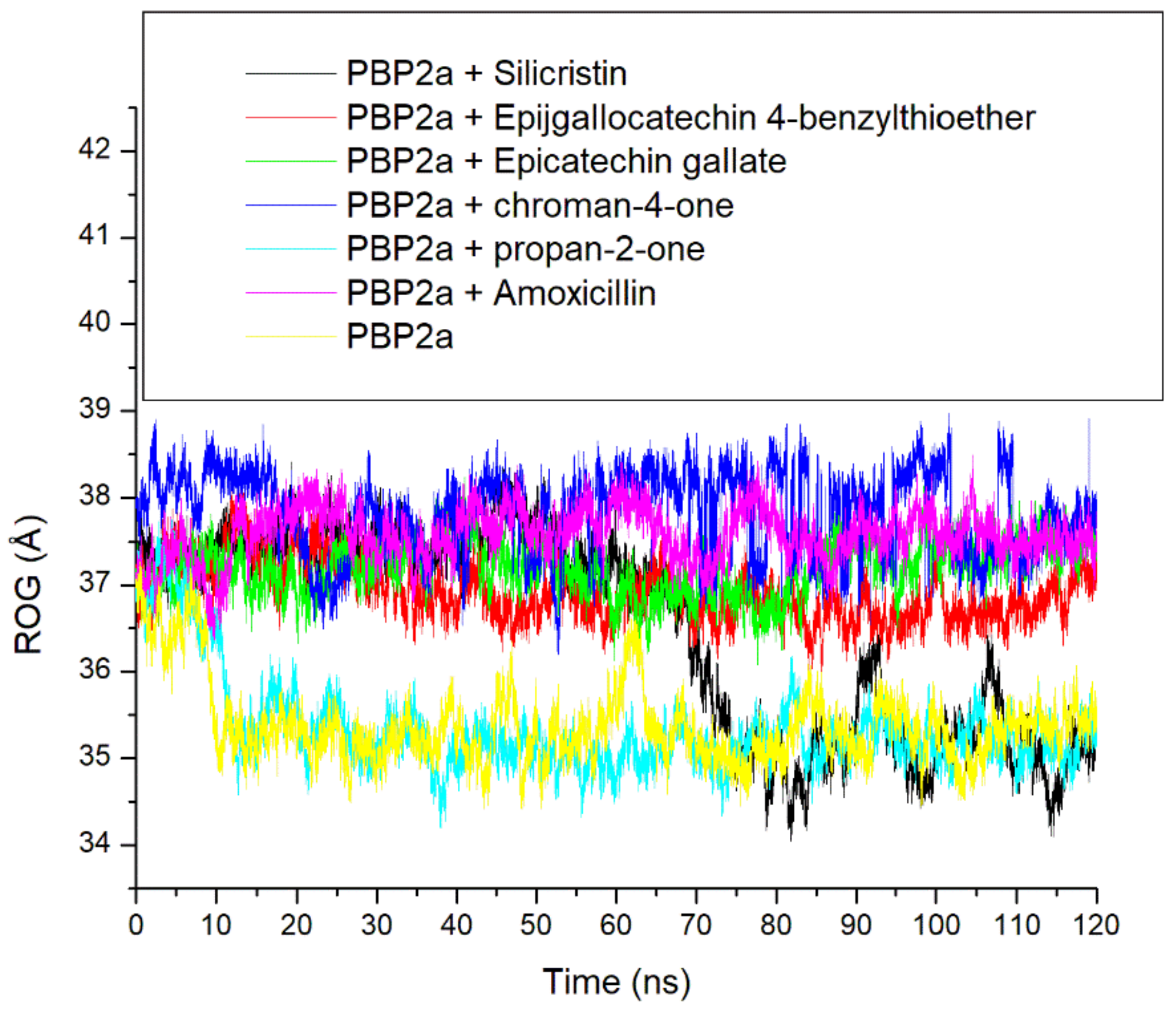
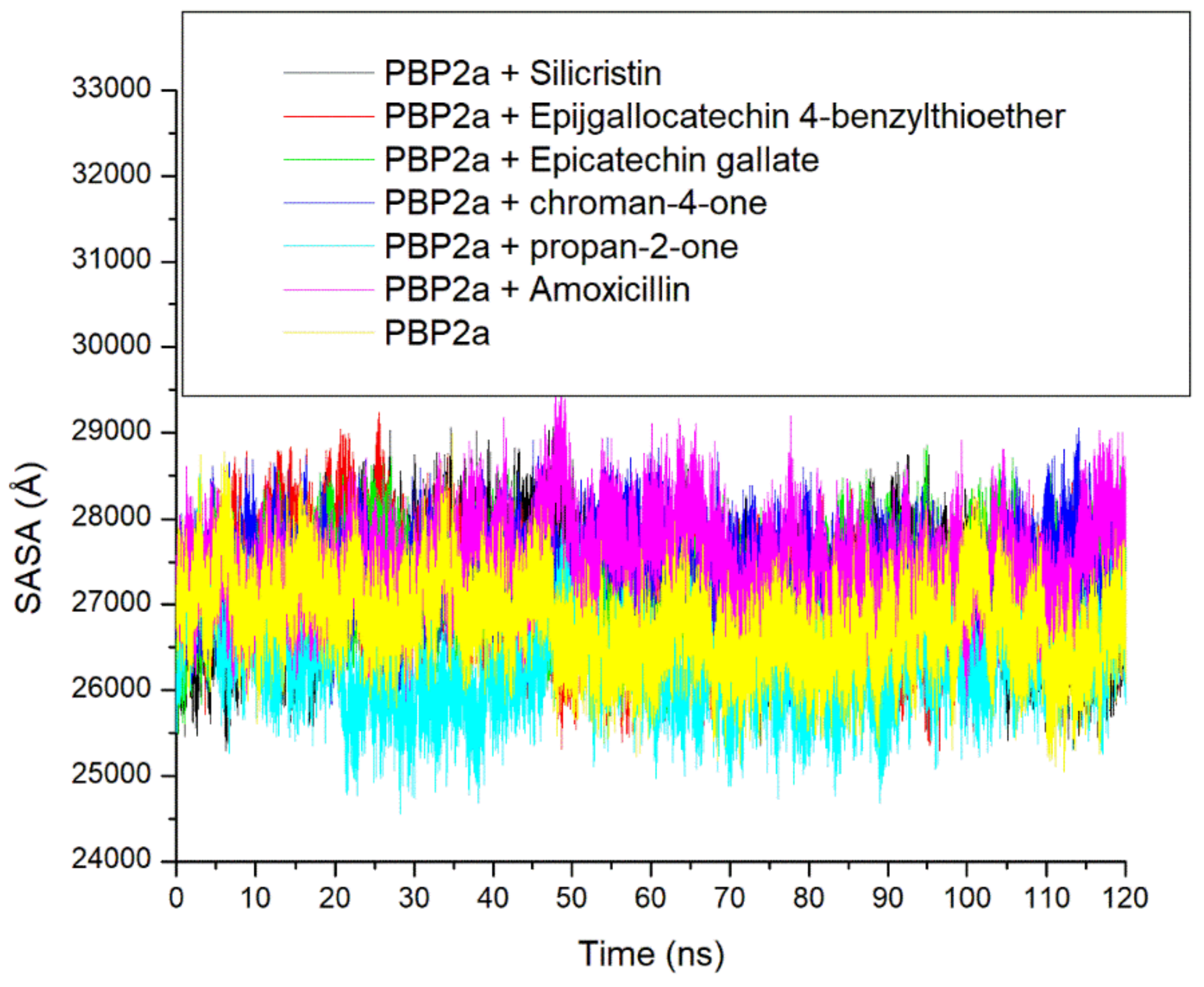
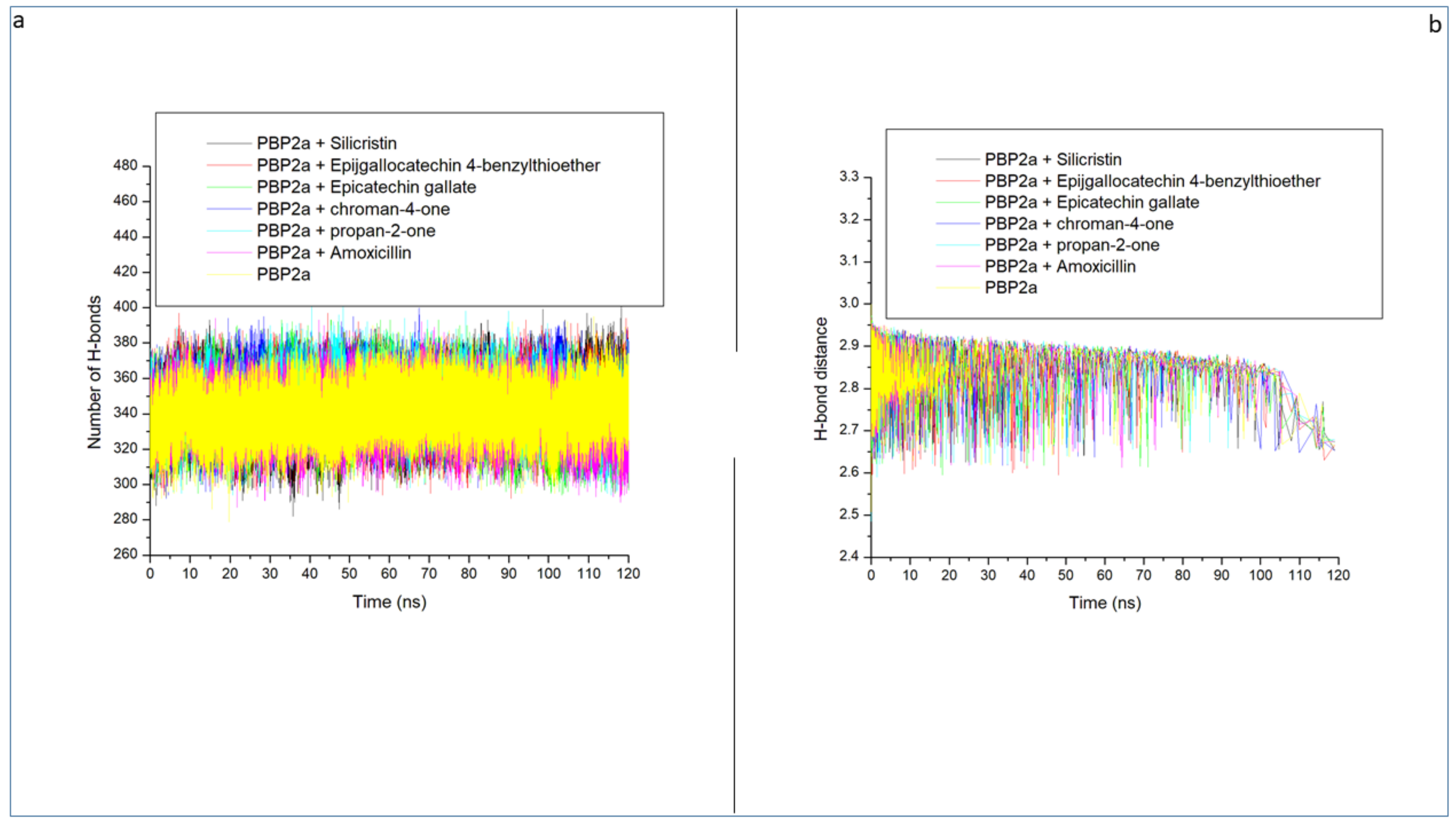
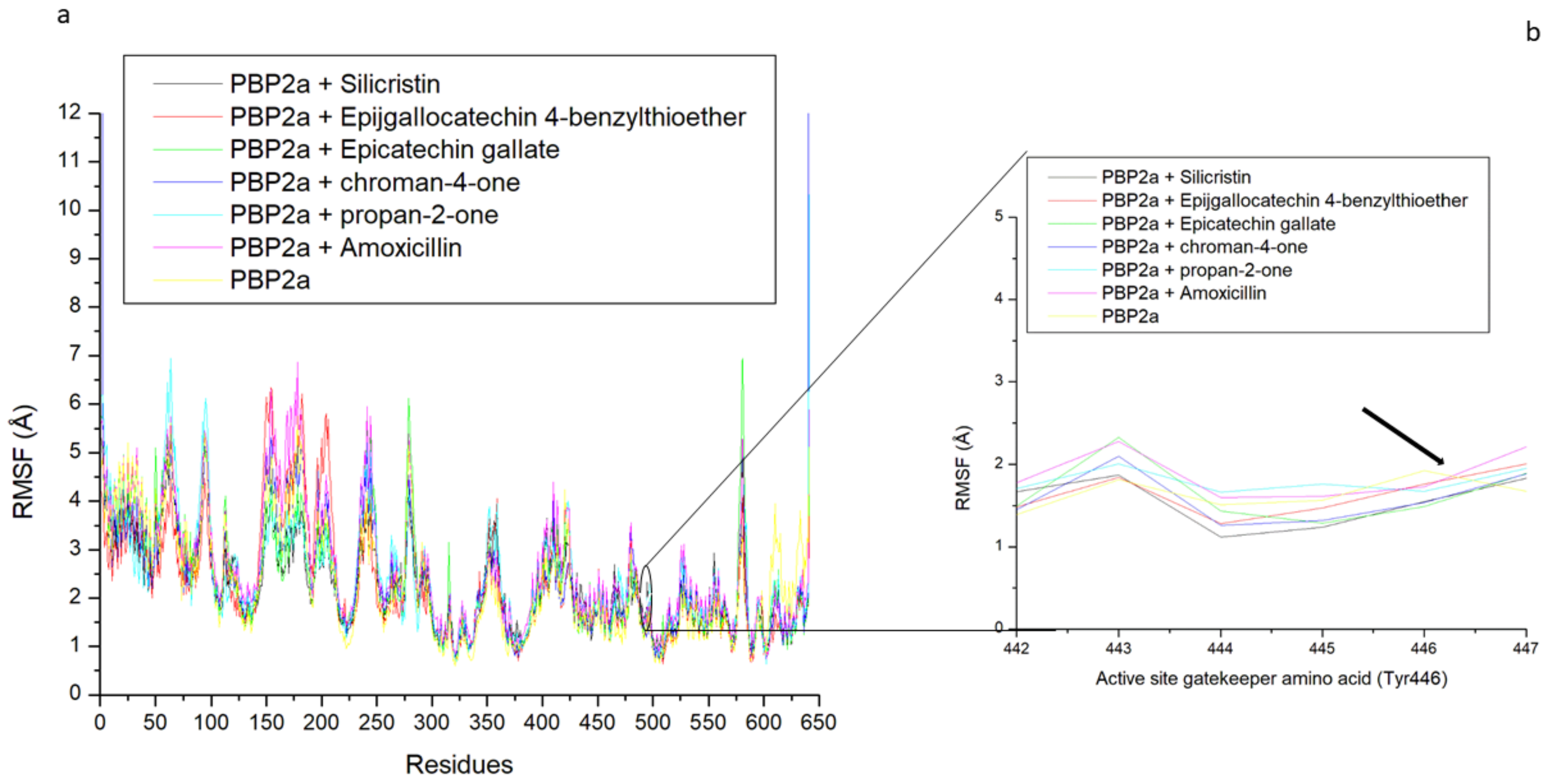
3.3. Thermodynamic Stability, Compactness and Flexibility of the Top-Five Phenolics at the Active Site of PBP2a of S. aureus
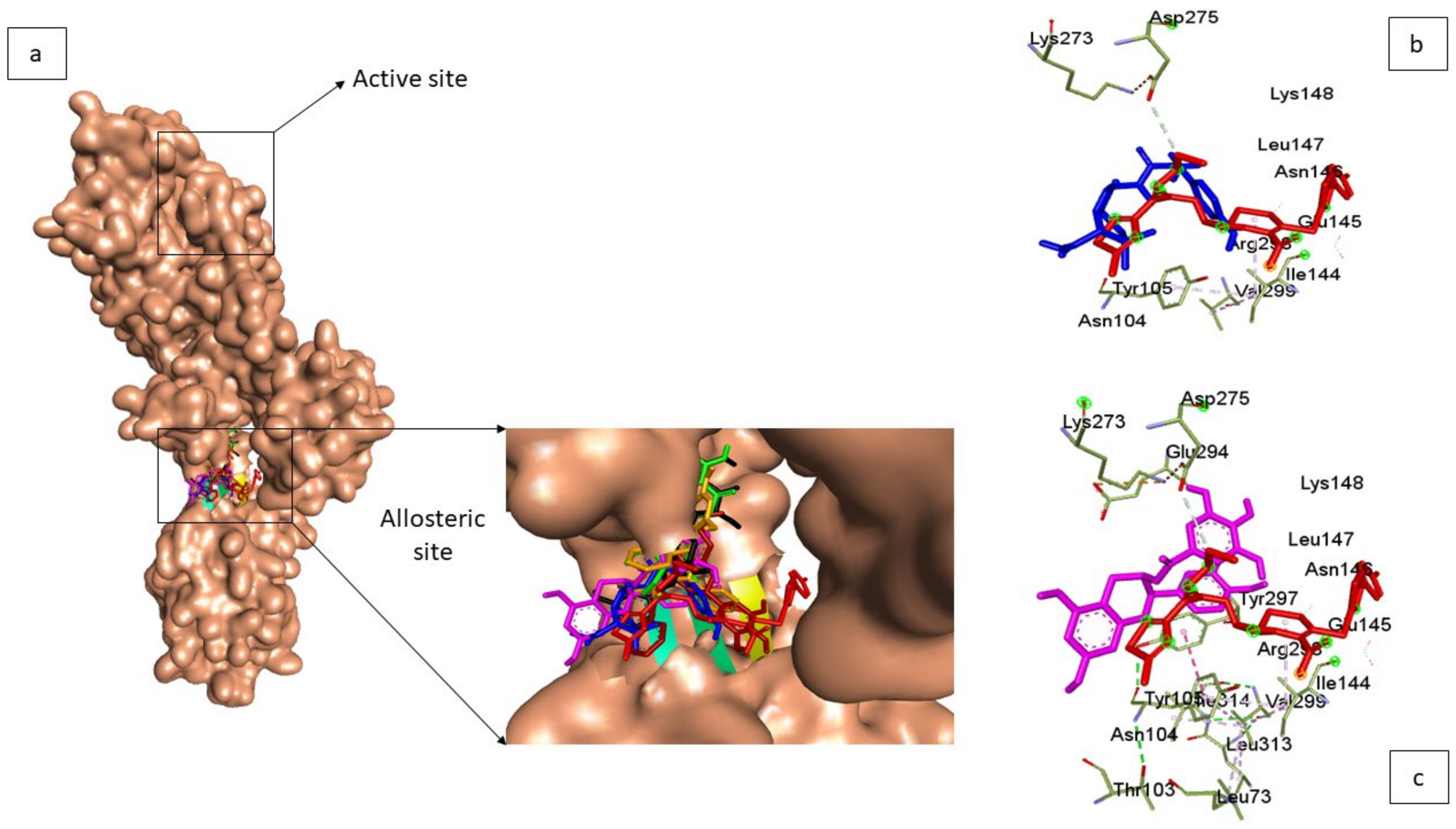
3.4. Molecular Docking of Top-Five Phenolics at the Allosteric Site of PBP2a of S. aureus
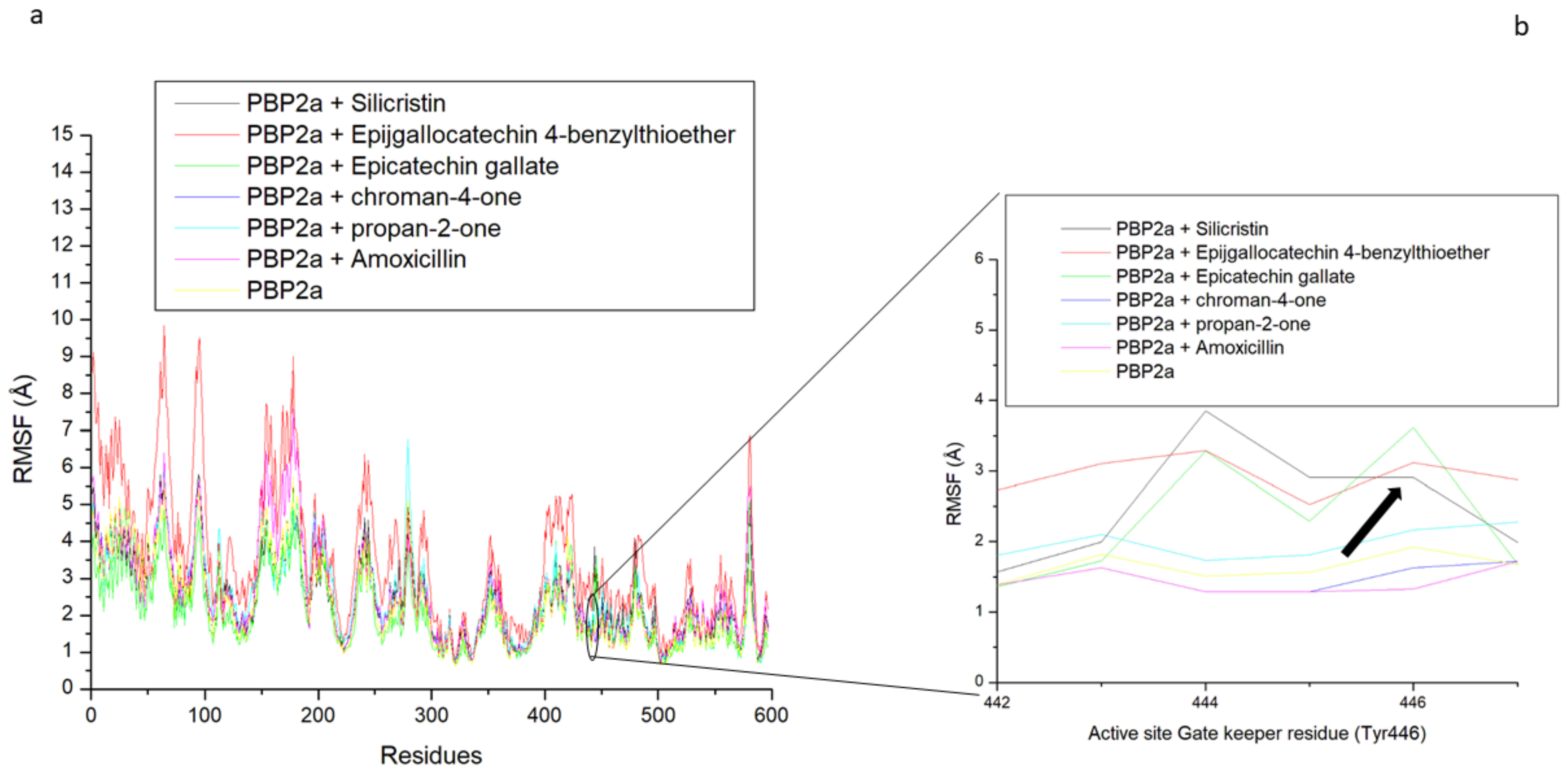
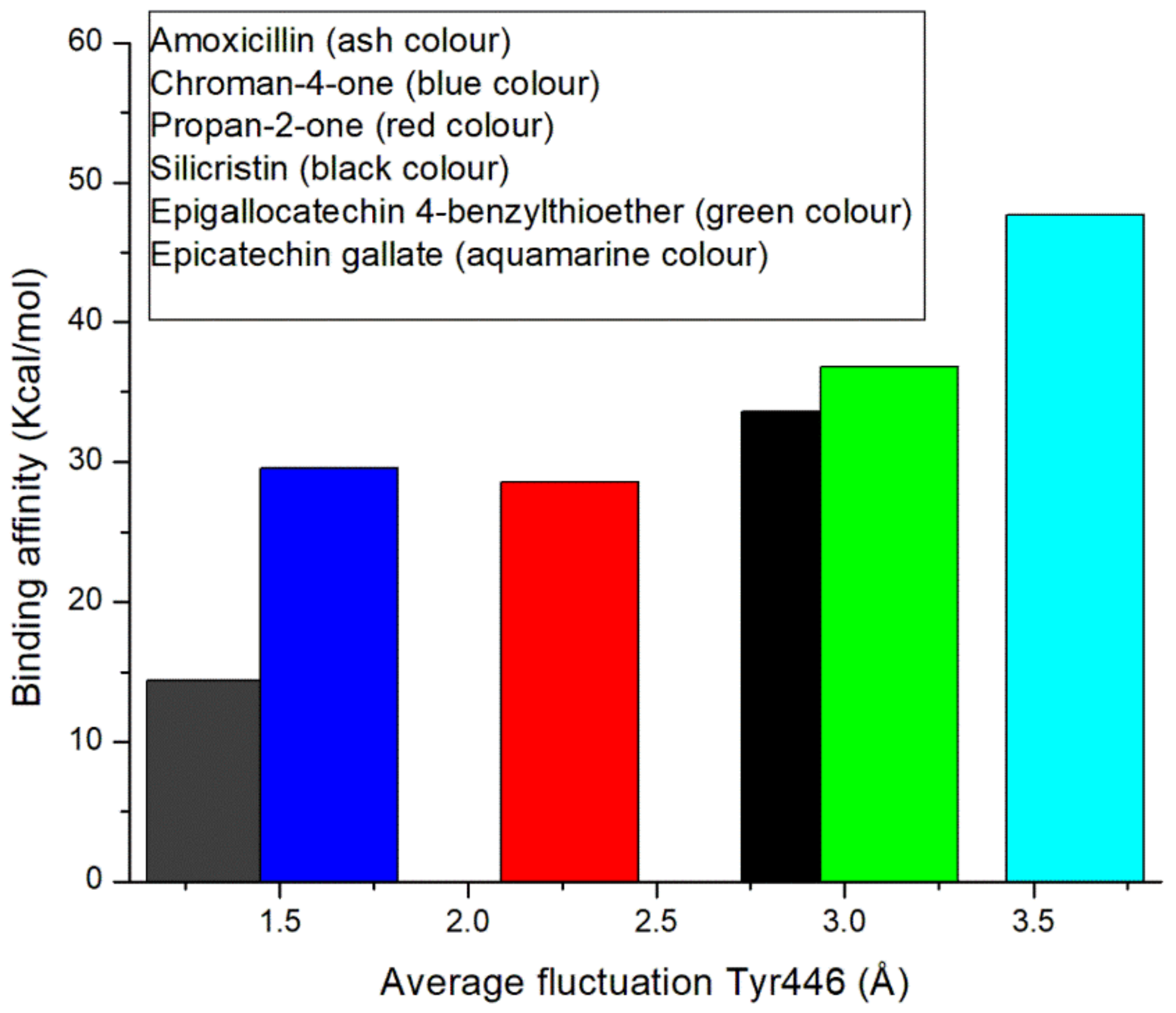
3.5. Thermodynamic Binding Free Energy Following 120 ns Simulation of the Top-Five Phenolics at the Allosteric Site of PBP2a of S. aureus
3.6. Allosteric Modulation of PBP2a Active Site Amino Acid Residues Following 120 ns Simulation at the Allosteric Site
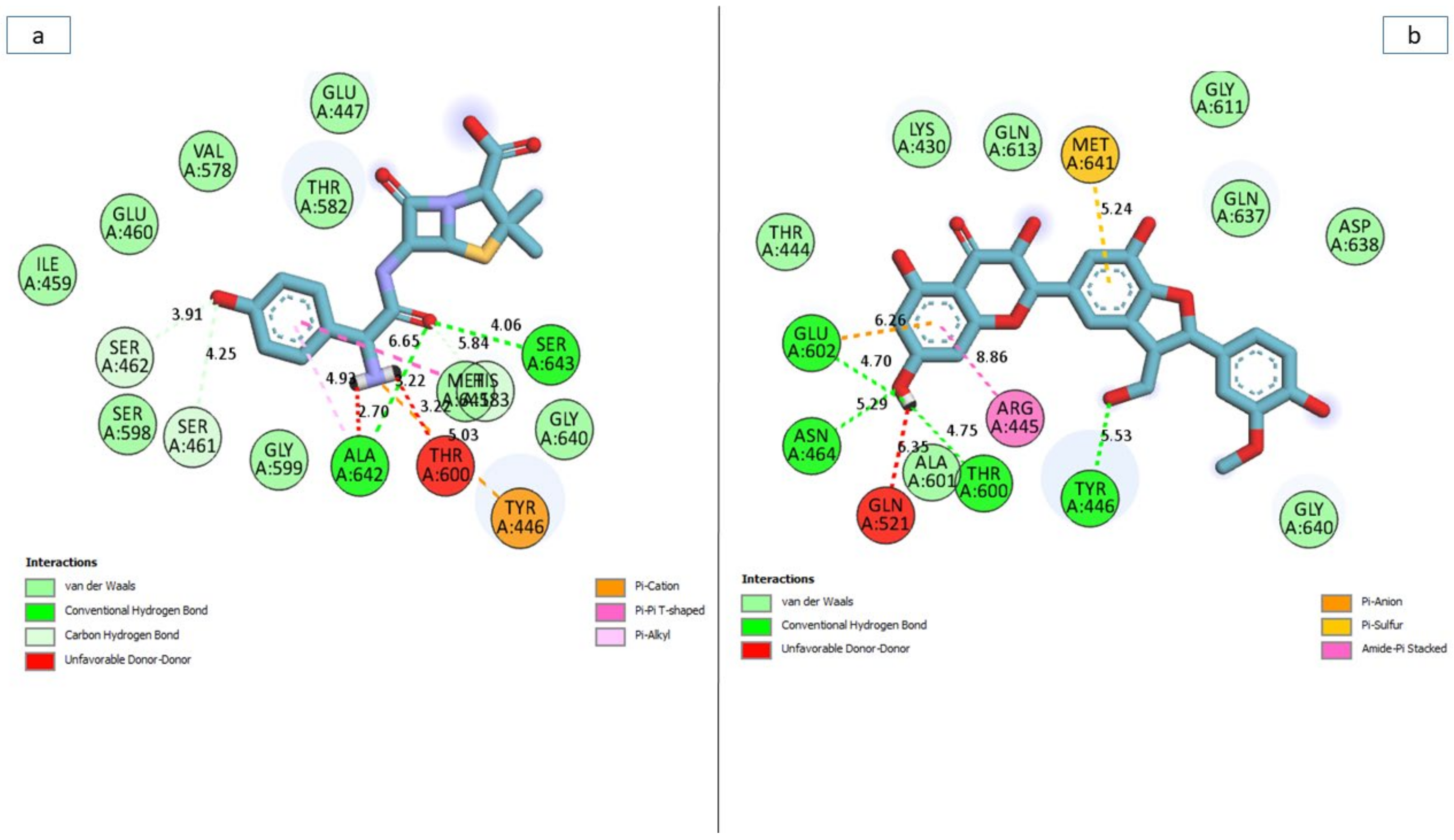
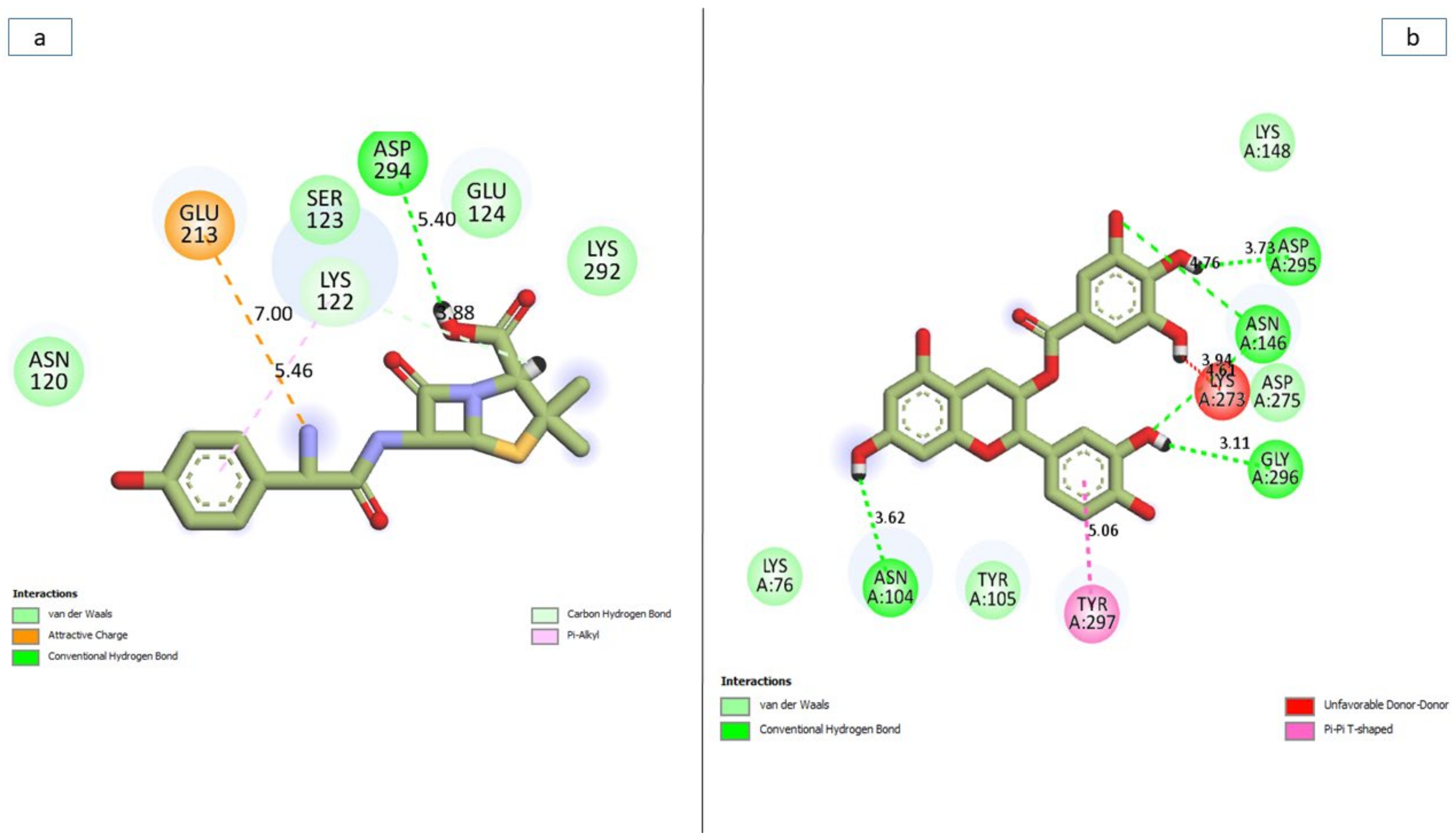
3.7. Bonds Analysis of the Interaction Plots of the Top-Five Phenolics against the Active and Allosteric Sites of PBP2a of S. aureus
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalalo, M.J.; Fatimawali, F.; Kalalo, T.; Imanuel, C.; Rambi, J. Tea bioactive compounds as inhibitor of MRSA penicillin binding protein 2a (PBP2a): A molecular docking study. Pharm. Med. J. 2020, 20, 70–75. [Google Scholar] [CrossRef]
- Rahman, M.M.; Amin, K.B.; Rahman, S.M.M.; Khair, A.; Rahman, M.; Hossain, A.; Rahman, A.K.M.A.; Parvez, M.S.; Miura, N.; Alam, M.M.; et al. Investigation of methicillin-resistant Staphylococcus aureus among clinical isolates from humans and animals by culture methods and multiplex PCR. BMC Veter. Res. 2018, 14, 300. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Moein, K.A.; Zaher, H.M. Occurrence of multidrug-resistant methicillin-resistant Staphylococcus aureus among healthy farm animals: A public health concern. Int. J. Veter. Sci. Med. 2019, 7, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Stryjewski, M.; Corey, G.R. Methicillin-Resistant Staphylococcus aureus: An Evolving Pathogen. Clin. Infect. Dis. 2013, 58, S10–S19. [Google Scholar] [CrossRef]
- Malik, B.; Bhattacharyya, S. Antibiotic drug-resistance as a complex system driven by socio-economic growth and antibiotic misuse. Sci. Rep. 2019, 9, 9788. [Google Scholar] [CrossRef]
- Verma, A.K.; Ahmed, S.F.; Hossain, S.; Bhojiya, A.A.; Mathur, A.; Upadhyay, S.K.; Srivastava, A.K.; Vishvakarma, N.K.; Barik, M.; Rahaman, M.; et al. Molecular docking and simulation studies of flavonoid compounds against PBP-2a of methicillin-resistant Staphylococcus aureus. J. Biomol. Struct. Dyn. 2021, 1–17. [Google Scholar] [CrossRef]
- Larsson, D.G.J.; Flach, C.-F. Antibiotic resistance in the environment. Nat. Rev. Genet. 2021, 20, 257–269. [Google Scholar] [CrossRef]
- Josephine, H.R.; Charlier, P.; Davies, C.; Nicholas, R.A.; Pratt, R.F. Reactivity of penicillin-binding proteins with peptidoglycan-mimetic β-lactams: What’s wrong with these enzymes? ACS Chem. Biol. 2006, 45, 15873–15883. [Google Scholar] [CrossRef]
- Zapun, A.; Contreras-Martel, C.; Vernet, T. Penicillin-binding proteins and β-lactam resistance. FEMS Microbiol. Rev. 2008, 32, 361–385. [Google Scholar] [CrossRef]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J.A.; Charlier, P. The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. [Google Scholar] [CrossRef] [Green Version]
- Scheffers, D.-J.; Pinho, M.G. Bacterial Cell Wall Synthesis: New Insights from Localization Studies. Microbiol. Mol. Biol. Rev. 2005, 69, 585–607. [Google Scholar] [CrossRef] [PubMed]
- Meisel, J.E.; Fisher, J.F.; Chang, M.; Mobashery, S. Allosteric Inhibition of Bacterial Targets: An Opportunity for Discovery of Novel Antibacterial Classes. In Antibacterials; Fisher, J.F., Mobashery, S., Miller, M.J., Eds.; Springer: Cham, Switzerland, 2017; pp. 119–147. [Google Scholar] [CrossRef]
- Otero, L.H.; Rojas-Altuve, A.; Llarrull, L.I.; Carrasco-López, C.; Kumarasiri, M.; Lastochkin, E.; Fishovitz, J.; Dawley, M.; Hesek, D.; Lee, M.; et al. How allosteric control of Staphylococcus aureus penicillin binding protein 2a enables methicillin resistance and physiological function. Proc. Natl. Acad. Sci. USA 2013, 110, 16808–16813. [Google Scholar] [CrossRef] [PubMed]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34, W116–W118. [Google Scholar] [CrossRef]
- Fisher, J.F.; Mobashery, S. β-lactam resistance mechanisms: Gram-positive bacteria and Mycobacterium tuberculosis. Cold Spring Harb. Perspect Med. 2020, 6, a025221. [Google Scholar] [CrossRef]
- Alhadrami, H.A.; Hamed, A.A.; Hassan, H.M.; Belbahri, L.; Rateb, M.E.; Sayed, A.M. Flavonoids as Potential anti-MRSA Agents through Modulation of PBP2a: A Computational and Experimental Study. Antibiotics 2020, 9, 562. [Google Scholar] [CrossRef] [PubMed]
- Aldulaimi, O. General overview of phenolics from plant to laboratory, good antibacterials or not. Pharmacogn. Rev. 2017, 11, 123–127. [Google Scholar] [CrossRef]
- Araya-Cloutier, C.; Vincken, J.-P.; van Ederen, R.; Besten, H.M.D.; Gruppen, H. Rapid membrane permeabilization of Listeria monocytogenes and Escherichia coli induced by antibacterial prenylated phenolic compounds from legumes. Food Chem. 2018, 240, 147–155. [Google Scholar] [CrossRef]
- Weinreb, O.; Mandel, S.; Amit, T.; Youdim, M.B. Neurological mechanisms of green tea polyphenols in Alzheimer’s and Parkinson’s diseases. J. Nutr. Biochem. 2004, 15, 506–516. [Google Scholar] [CrossRef]
- Panat, N.A.; Singh, B.G.; Maurya, D.K.; Sandur, S.K.; Ghaskadbi, S.S. Troxerutin, a natural flavonoid binds to DNA minor groove and enhances cancer cell killing in response to radiation. Chem. Interact. 2016, 251, 34–44. [Google Scholar] [CrossRef]
- Dai, Q.; Borenstein, A.R.; Wu, Y.; Jackson, J.C.; Larson, E.B. Fruit and Vegetable Juices and Alzheimer’s Disease: The Kame Project. Am. J. Med. 2006, 119, 751–759. [Google Scholar] [CrossRef]
- Chiorcea-Paquim, A.; Enache, T.A.; De Souza Gil, E.; Oliveira-Brett, A.M. Natural phenolic antioxidants electrochemistry: Towards a new food science methodology. Compr. Rev. Food Sci. Food Saf. 2020, 19, 1680–1726. [Google Scholar] [CrossRef] [PubMed]
- Aribisala, J.O.; Abdulsalam, R.A.; Dweba, Y.; Madonsela, K.; Sabiu, S. Identification of secondary metabolites from Crescentia cujete as promising antibacterial therapeutics targeting type 2A topoisomerases through molecular dynamics simulation. Comput. Biol. Med. 2022, 145, 105432. [Google Scholar] [CrossRef] [PubMed]
- MubarakAli, D.; MohamedSaalis, J.; Sathya, R.; Irfan, N.; Kim, J.-W. An evidence of microalgal peptides to target spike protein of COVID-19: In silico approach. Microb. Pathog. 2021, 160, 105189. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Camacho, C.J. ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 2012, 40, W409–W414. [Google Scholar] [CrossRef]
- Fahad, M.; Al-Khodairy, M.; Kalim, A.; Khan, M.K.; Manogaran, S.P.; Salman, A.; Jamal, M.A. In Silico Prediction of Mechanism of Erysolin-induced Apoptosis in Human Breast Cancer Cell Lines, American. J. Bioinform. 2013, 3, 62–71. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Methods of Protein Structure Comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [CrossRef]
- Sabiu, S.; Balogun, F.O.; Amoo, S.O. Phenolics Profiling of Carpobrotus edulis (L.) N.E.Br. and Insights into Molecular Dynamics of Their Significance in Type 2 Diabetes Therapy and Its Retinopathy Complication. Molecules 2021, 26, 4867. [Google Scholar] [CrossRef]
- Aribisala, J.O.; Nkosi, S.; Idowu, K.; Nurain, I.O.; Makolomakwa, G.M.; Shode, F.O.; Sabiu, S. Astaxanthin-Mediated Bacterial Lethality: Evidence from Oxidative Stress Contribution and Molecular Dynamics Simulation. Oxidative Med. Cell. Longev. 2021, 2021, e7159652. [Google Scholar] [CrossRef]
- Uhomoibhi, J.O.-O.; Shode, F.O.; Idowu, K.A.; Sabiu, S. Molecular modelling identification of phytocompounds from selected African botanicals as promising therapeutics against druggable human host cell targets of SARS-CoV-2. J. Mol. Graph. Model. 2022, 114, 108185. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Khumbulani, M.; Alayande, K.A.; Sabiu, S. Orientin Enhances Colistin-Mediated Bacterial Lethality through Oxidative Stress Involvement. Evid. -Based Complement. Altern. Med. 2022, 2022, 3809232. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Ramirex, D.; Caballero, J. Is it reliable to use common molecular docking methods for comparing the binding affinities of Enantiomer pairs for their protein target. Int. J. Mol. Sci. 2016, 17, 525. [Google Scholar] [CrossRef] [PubMed]
- Stella, L.; Melchionna, S. Equilibration and sampling in molecular dynamics simulations of biomolecules. J. Chem. Phys. 1998, 109, 10115–10117. [Google Scholar] [CrossRef]
- Baig, M.H.; Sudhakar, D.R.; Kalaiarasan, P.; Subbarao, N.; Wadhawa, G.; Lohani, M.; Khan, M.K.A.; Khan, A.U. Insight into the Effect of Inhibitor Resistant S130G Mutant on Physico-Chemical Properties of SHV Type BetaLactamase: A Molecular Dynamics Study. PLoS ONE 2014, 9, e112456. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Mousavi, S.S.; Karami, A.; Haghighi, T.M.; Tumilaar, S.G.; Idroes, R.; Mahmud, S.; Celik, I.; Ağagündüz, D.; Tallei, T.E.; Fatimawali; et al. In Silico Evaluation of Iranian Medicinal Plant Phytoconstituents as Inhibitors against Main Protease and the Receptor-Binding Domain of SARS-CoV-2. Molecules 2021, 26, 5724. [Google Scholar] [CrossRef]
- Izadi, H.; Stewart, K.M.E.; Penlidis, A. Role of contact electrification and electrostatic interactions in gecko adhesion. J. R. Soc. Interface 2014, 11, 20140371. [Google Scholar] [CrossRef] [Green Version]
- Mahasenan, K.V.; Molina, R.; Bouley, R.; Batuecas, M.T.; Fisher, J.F.; Hermoso, J.A.; Chang, M.; Mobashery, S. Conformational Dynamics in Penicillin-Binding Protein 2a of Methicillin-Resistant Staphylococcus aureus, Allosteric Communication Network and Enablement of Catalysis. J. Am. Chem. Soc. 2017, 139, 2102–2110. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Nasution, M.A.F.; Toepak, E.P.; Alkaff, A.H.; Tambunan, U.S.F. Flexible docking-based molecular dynamics simulation of natural product compounds and Ebola virus Nucleocapsid (EBOV NP): A computational approach to discover new drug for combating Ebola. BMC Bioinform. 2018, 19, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M.A. Medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 14, 5061–5084. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands Zinc Code | Popular Name | B A (kcal/mol) | MW < 500 (g/mol) | HB-A ≤ 10 | HB-D ≤ 5 | Log Po/w ≤ 5 | RT-B ≤ 9 | LV < 2 | WS | GI -A | BS | pgp | Inhibitor of CYP 450s | H | C | IM | M | CY | TC | SA | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CYP 1A2 | CYP 2C19 | CYP 2C9 | CYP 2D6 | CYP 3A4 | ||||||||||||||||||||
| Amoxicillin | Amoxicillin | −6.2 | 365.40 | 6 | 4 | −0.39 | 5 | N | VS | L | 0.55 | N | N | N | N | N | N | I | I | I | I | I | 6 | 4.17 |
| ZINC34953149 | Silicristin | −7.5 | 482.44 | 10 | 6 | 1.49 | 4 | N | S | L | 0.55 | N | N | N | N | N | Y | I | I | A | I | I | 4 | 4.88 |
| ZINC71621503 | Propan-2-one | −7.1 | 458.46 | 7 | 6 | 3.3 | 6 | N | MS | L | 0.55 | N | N | N | Y | N | Y | I | I | I | I | I | 4 | 3.67 |
| ZINC38337968 | Epigallocatechin 4-benzylthioether | −7.1 | 428.46 | 7 | 6 | 2.23 | 4 | N | MS | L | 0.55 | N | N | N | N | N | Y | I | I | I | I | I | 5 | 4.57 |
| ZINC95486076 | Chroman-4-one | −7.0 | 356.37 | 6 | 3 | 3.09 | 3 | N | MS | H | 0.55 | N | Y | N | Y | N | Y | I | I | A | I | I | 4 | 3.83 |
| ZINC03978503 | Epicatechin gallate | −6.8 | 442.37 | 10 | 7 | 1.3 | 4 | N | S | L | 0.55 | N | N | N | N | N | N | I | I | I | I | I | 4 | 4.16 |
| Systems | ΔEvdW | ΔEelec | ΔGgas | ΔGsolv | ΔGbind |
|---|---|---|---|---|---|
| PBP2a + amoxicillin | −22.30 ± 7.99 | −74.82 ± 26.08 | −97.12 ± 28.39 | 75.58 ± 23.67 | −21.54 ± 6.59 |
| PBP2a + silicristin | −35.15 ± 8.08 | −32.24 ± 12.41 | −67.42 ± 15.90 | 41.80 ± 10.24 | −25.61 ± 7.08 |
| PBP2a + propan-2-one | −23.27 ± 5.46 | −35.71 ± 16.25 | −58.99 ± 18.62 | 39.92 ± 12.55 | −19.06 ± 7.26 |
| PBP2a + epigallocatechin 4-benzylthioether | −19.36 ± 4.64 | −49.56 ± 13.09 | −68.93 ± 11.38 | 44.18 ± 7.98 | −24.75 ± 4.72 |
| PBP2a + chroman-4-one | −22.23 ± 4.56 | −47.43 ± 10.23 | −65.56 ± 12.34 | −42.78 ± 23.34 | −22.34 ± 5.23 |
| PBP2a + epicatechin gallate | −20.61 ± 4.45 | −43.07 ± 16.16 | −63.69 ± 16.86 | 40.58 ± 10.45 | −23.11 ± 7.45 |
| Systems | RMSD (Å) | RMSF(Å) | ROG (Å) | SASA (Å) | Number of Intramolecular H-Bond | Intramolecular H-Bond Distance (Å) |
|---|---|---|---|---|---|---|
| Unbound PBP2a | 6.86 ± 1.18 | 2.71 ± 0.91 | 35.37 ± 0.45 | 26786.05 ± 473.91 | 339.24 ± 12.45 | 2.85 ± 0.06 |
| PBP2a + Amoxicillin | 4.07 ± 1.15 | 2.85 ± 1.24 | 37.57 ± 0.29 | 27484.50 ± 458.31 | 341.44 ± 13.23 | 2.85 ± 0.06 |
| PBP2a + Silicristin | 5.65 ± 2.34 | 2.26 ± 2.09 | 36.47 ± 1.15 | 27160.02 ± 545.40 | 346.00 ± 12.15 | 2.85 ± 0.05 |
| PBP2a + Propan-2-one | 5.78 ± 1.21 | 2.45 ± 1.13 | 35.34 ± 0.55 | 26303.34 ± 464.78 | 342.94 ± 12.13 | 2.85 ± 0.06 |
| PBP2a + Epigallocatechin 4-benzylthioether | 3.49 ± 0.42 | 2.27 ± 1.14 | 36.92 ± 0.32 | 27060.31 ± 531.98 | 344.00 ± 12.22 | 2.85 ± 0.06 |
| PBP2a + Chroman-4-one | 3.26 ± 1.16 | 2.49 ± 3.22 | 37.84 ± 0.46 | 27311.74 ± 408.97 | 345.47 ± 12.13 | 2.85 ± 0.05 |
| PBP2a + Epicatechin gallate | 3.42 ± 0.67 | 2.38 ± 1.14 | 37.19 ± 0.29 | 27187.53 ± 411.42 | 343.16 ± 12.12 | 2.85 ± 0.06 |
| PBP2a Active Site Gatekeeper Residue | Top-Five Phenolics | ||||||
|---|---|---|---|---|---|---|---|
| Silicristin | Epigallocatechin 4-benzylthioether | Epicatechin gallate | Chroman-4-one | Propan-2-one | Amoxicillin | Unbound PBP2a | |
| Tyr446 | 1.55 | 1.76 | 1.49 | 1.53 | 1.67 | 1.72 | 1.92 |
| Systems | Binding Affinity (Kcal/mol) | ΔGbind (Kcal/mol) | RMSF (Å) |
|---|---|---|---|
| Unbound PBP2a | 2.71 ± 1.17 | ||
| PBP2a + Amoxicillin | −7.7 | −14.36 ± 6.00 | 2.56 ± 1.32 |
| PBP2a + Silicristin | −8.4 | −33.57 ± 5.38 | 2.45 ± 1.11 |
| PBP2a + Propan-2-one | −8.3 | −28.55 ± 8.07 | 2.49 ± 1.14 |
| PBP2a + Epigallocatechin 4-benzylthioether | −8.1 | −36.83 ± 8.04 | 3.48 ± 1.84 |
| PBP2a + Chroman-4-one | −8.0 | −29.52 ± 4.20 | 2.55 ± 1.32 |
| PBP2a + Epicatechin gallate | −8.5 | −47.65 ± 8.42 | 2.06 ± 0.95 |
| PBP2a Active Site Gatekeeper Residue | Top-Five Phenolics | ||||||
|---|---|---|---|---|---|---|---|
| Silicristin | Epigallocatechin 4-benzylthioether | Epicatechin gallate | Chroman-4-one | Propan-2-one | Amoxicillin | Unbound PBP2a | |
| Tyr446 | 2.91 | 3.12 | 3.61 | 1.63 | 2.27 | 1.33 | 1.93 |
| Top-Five Phenolics and Standard | Total Number of Interactions (Average Distances in Å) | Number of Hydrogen Bonds and Interaction Residues | Other Important Interactions and Residues | Unfavourable Bonds (Bond Length in Å) |
|---|---|---|---|---|
| Active Site | ||||
| Amoxicillin | 17 (5.14) | 4 (Ala642, Ser643, Ser461, Ser462) | 3 (Tyr446, Ala642, His583) | 2 (Thyr600, Ala642) (2.96) |
| Silicristin | 16 (5.75) | 4 (Tyr446, Thyr600, Asn464, Glu602) | 3 (Met641, Glu602, Arg445) | 1 (Gln521) (6.35) |
| Propan-2-one | 17 (5.53) | 1 (Tyr447) | 3 (His583, Tyr446, Ala642) | None |
| Epigallocatechin 4-benzylthioether | 15 (4.91) | 4 (Lys584, Glu460, Thr582, Asp586) | 3 (Ala642, His583, Tyr446) | None |
| Chroman-4-one | 15 (4.99) | 1(Ser642) | 2 (His583, Ala642) | None |
| Epicatechin gallate | 15 (4.94) | 5 (Lys581, Ser461, Glu447 (2), Gly599) | 2 (Ala642, His583) | None |
| Allosteric Site | ||||
| Amoxicillin | 8 (5.44) | 2 (Lys122, Asp294) | 2 (Lys122, Glu212) | None |
| Silicristin | 16 (5.37) | 5 (Lys273, Lys148, Asn146, Asp295(2)) | 3 (Asp275, Lys316, Tyr297) | (Tyr297) (6.06) |
| Propan-2-one | 16 (5.23) | 4 (Asp275, Lys273, Asn146 (2)) | 2 (Tyr297, Lys273) | none |
| Epigallocatechin 4-benzylthioether | 23 (4.71) | 7 (Asp269, Ser214, Glu213, Glu144, Val251, Ala250, Hie267) | 2 (Val251, Pro332) | none |
| Chroman-4-one | 12 (5.0) | 4 (Lys148, Lys273, Val277, Ala276) | 1 (Tyr297) | none |
| Epicatechin gallate | 11 (3.87) | 5 (Asn104, Gly296, Asn146 (2), Asp295) | 1 (Tyr297) | Lys273 (4.61) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aribisala, J.O.; Sabiu, S. Cheminformatics Identification of Phenolics as Modulators of Penicillin-Binding Protein 2a of Staphylococcus aureus: A Structure–Activity-Relationship-Based Study. Pharmaceutics 2022, 14, 1818. https://doi.org/10.3390/pharmaceutics14091818
Aribisala JO, Sabiu S. Cheminformatics Identification of Phenolics as Modulators of Penicillin-Binding Protein 2a of Staphylococcus aureus: A Structure–Activity-Relationship-Based Study. Pharmaceutics. 2022; 14(9):1818. https://doi.org/10.3390/pharmaceutics14091818
Chicago/Turabian StyleAribisala, Jamiu Olaseni, and Saheed Sabiu. 2022. "Cheminformatics Identification of Phenolics as Modulators of Penicillin-Binding Protein 2a of Staphylococcus aureus: A Structure–Activity-Relationship-Based Study" Pharmaceutics 14, no. 9: 1818. https://doi.org/10.3390/pharmaceutics14091818