
Development and Evaluation of a Physiologically Based Pharmacokinetic Model for Predicting Haloperidol Exposure in Healthy and Disease Populations
 , ,
, ,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Software
2.2. Literature Search
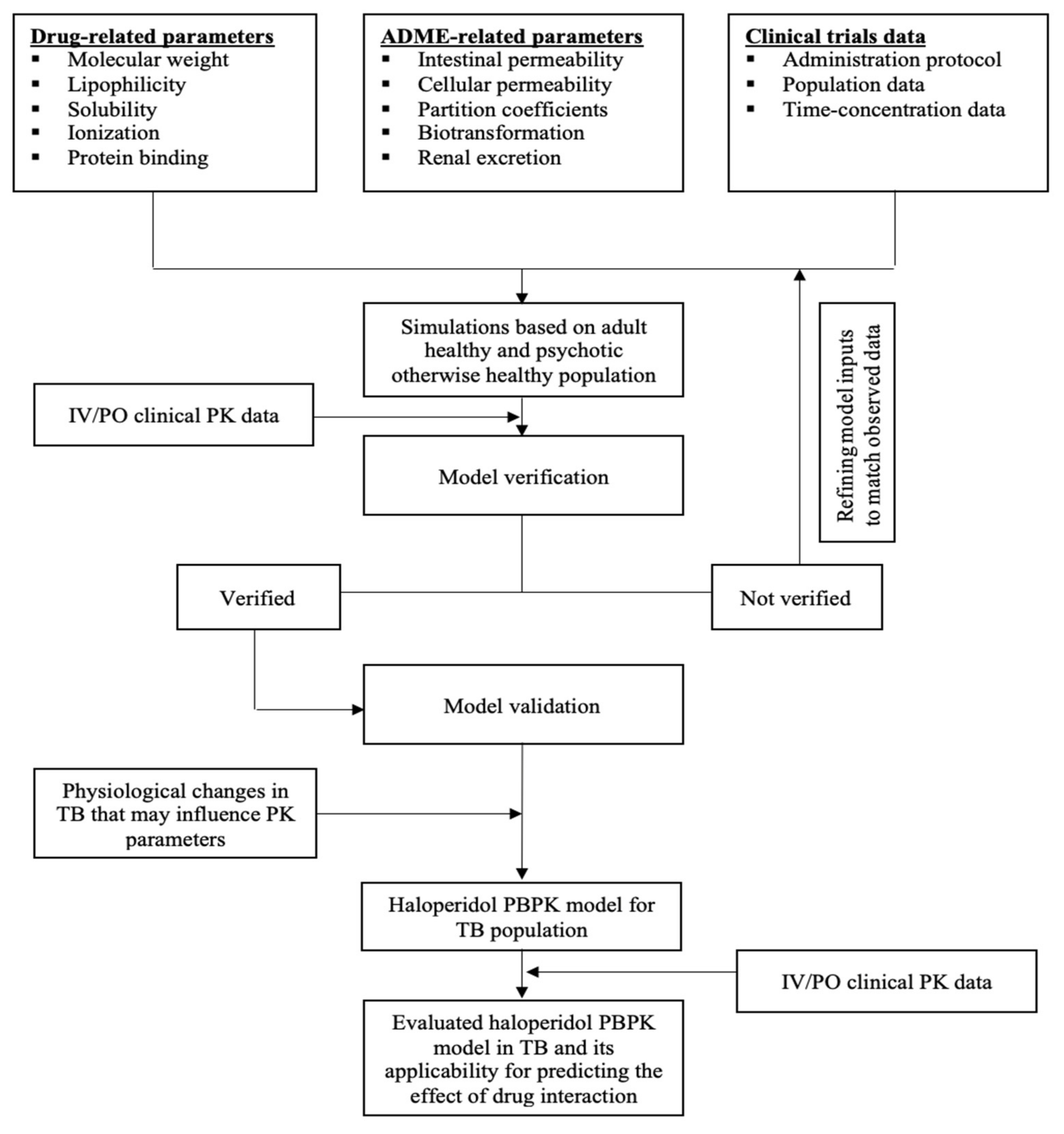
2.3. General Schematic Pathway
2.4. Absorption, Distribution, Elimination, and Absorption (ADME)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Unit | Input Value | Reported Value(s) (Reference) | Source of Input |
|---|---|---|---|---|
| Physicochemical Properties | ||||
| Molecular weight | g/mol | 376 | 376 [39] | [39] |
| Effective molecular weight | g/mol | 336.86 | PK-Sim | |
| Lipophilicity | Log | 3.66 | 2.9 [64], 3.01 [28], 3.20 [37], 3.23 [65], 3.66 [38], 3.7 [38], 4 [39], 4.3 [37] | [38] |
| Water solubility | mg/mL | 0.0045 | 0.0045 [38,39], 0.014 [37], 0.023 [37] | [39] |
| pKa-monoprotic base | 8.65 | 7.8 [65], 8.65 [66], 8.05 [38,39] | [66] | |
| Absorption | ||||
| Intestinal permeability | cm/min | 3.09 × 10−4 | 14.5 × 10−6 (cm/s) [28] | Calculated |
| Distribution | ||||
| Fraction unbound | % | 8.5 | 7.5–11.6 [19], 12.5 [65], 16.6 [28] | Parameter identification |
| Partition coefficient model | Rodger and Roland | PK-Sim | ||
| Cellular permeability model | PK-Sim | PK-Sim | ||
| Renal Clearance | ||||
| GFR fraction | % | 1 | 1 | [19] |
| Enzymatic Biotransformation | ||||
| Enzyme: CYP3A4 Metabolite: dealkylation to 4-FBPA In vitro metabolic system: Human liver microsome | ||||
| Km * | µmol/L | 21.7 | 62 [19] | [19] |
| Vmax | pmol/min/mg | 289 | 289 [19] | [19] |
| Enzyme: CYP3A4 Metabolite: oxidation to HPP+ In vitro metabolic system: Human liver microsome | ||||
| Km * | µmol/L | 28 | 80 [19] | [19] |
| Vmax | pmol/min/mg | 243.8 | 0.53 pmol/min/pmol [19] | Calculated based on 460 pmol/mg [19] |
| Enzyme: Carbonyl Reductase Metabolite: cytosolic reduction to reduced haloperidol In vitro metabolic system: Human liver cytosol | ||||
| Km * | µmol/L | 88 | 250 [19] | [19] |
| Vmax | nmol/min/mg | 1.30 | 1.30 [19] | [19] |
| Reductase content in liver cytosol | pmol/mg | 75 | 75 [58] | [58] |
| Enzyme: UGT1A4 Metabolite: O-glucuronidation In vitro metabolic system: Recombinant system | ||||
| Km * | µmol/L | 22.4 | 64 [29] | [29] |
| Vmax | pmol/min/mg | 600 × ISEFUGT1A4 | 600 [29] | [29] |
| Content in liver microsome | pmol/mg | 33 | 33 [67] | [67] |
| ISEFUGT1A4 | 0.16 | Calculated | ||
| Enzyme: UGT1A9 Metabolite: O-glucuronidation In vitro metabolic system: Recombinant system | ||||
| Km * | µmol/L | 61 | 174 | [29] |
| Vmax | pmol/min/mg | 2300 × ISEFUGT1A9 | 2300 [29] | [29] |
| Content in liver microsome | pmol/mg | 22.7 | 22.7 [67] | [67] |
| ISEFUGT1A9 | 0.06 | Calculated | ||
| Enzyme: UGT2B7 Metabolite: O-glucuronidation In vitro metabolic system: Recombinant system | ||||
| Km * | µmol/L | 16 | 45.0 | [29] |
| Vmax | pmol/min/mg | 1000 × ISEFUGT2B7 | 1000 [29] | [29] |
| Content in liver microsome | pmol/mg | 69.4 | 69.4 [67] | [67] |
| ISEFUGT2B7 | 0.05 | Calculated | ||
| Enzyme: UGT1A4 Metabolite: N-glucuronidation In vitro metabolic system: Recombinant system | ||||
| Km * | µmol/L | 64 | 64 | |
| Vmax | pmol/min/mg | 440 × ISEFUGT1A4 | 210 [29] | Calculated based on reported V [29] |
| Incubational fraction unbound | 0.35 | 0.34 [31], 0.35 [64], 0.75 [28] | [64] | |
| Parameter | Value | Source |
|---|---|---|
| Physicochemical Properties | ||
| Molecular weight | 378 | Drug-Bank |
| Effective molecular weight | 339 | |
| LogP | 3.52 | |
| pKa | 8.66 | |
| Solubility | 0.0131 mg/mL | |
| Absorption | ||
| Intestinal permeability | 2.08 × 10−4 (cm/min) | Calculated |
| Distribution | ||
| Partition coefficient model | Rodger and Roland | PK-Sim |
| Cellular permeability model | PK-Sim | PK-Sim |
| Fraction unbound | 24.4% | [68] |
| Elimination | ||
| Enzyme: CYP3A4 Metabolic pathway: oxidation to haloperidol In vitro metabolic system: Human liver microsome | ||
| Km | 46 μmol/L | [19] |
| Vmax | 98 pmol/min/mg | [19] |
| Additional hepatic clearance | 5 mL/min/kg | Estimated |
2.5. The Predictability Assessment
2.6. PK of Haloperidol in Disease Population
3. Results
3.1. Clinical PK Studies That Were Used to Build and Evaluate the Haloperidol PBPK Model
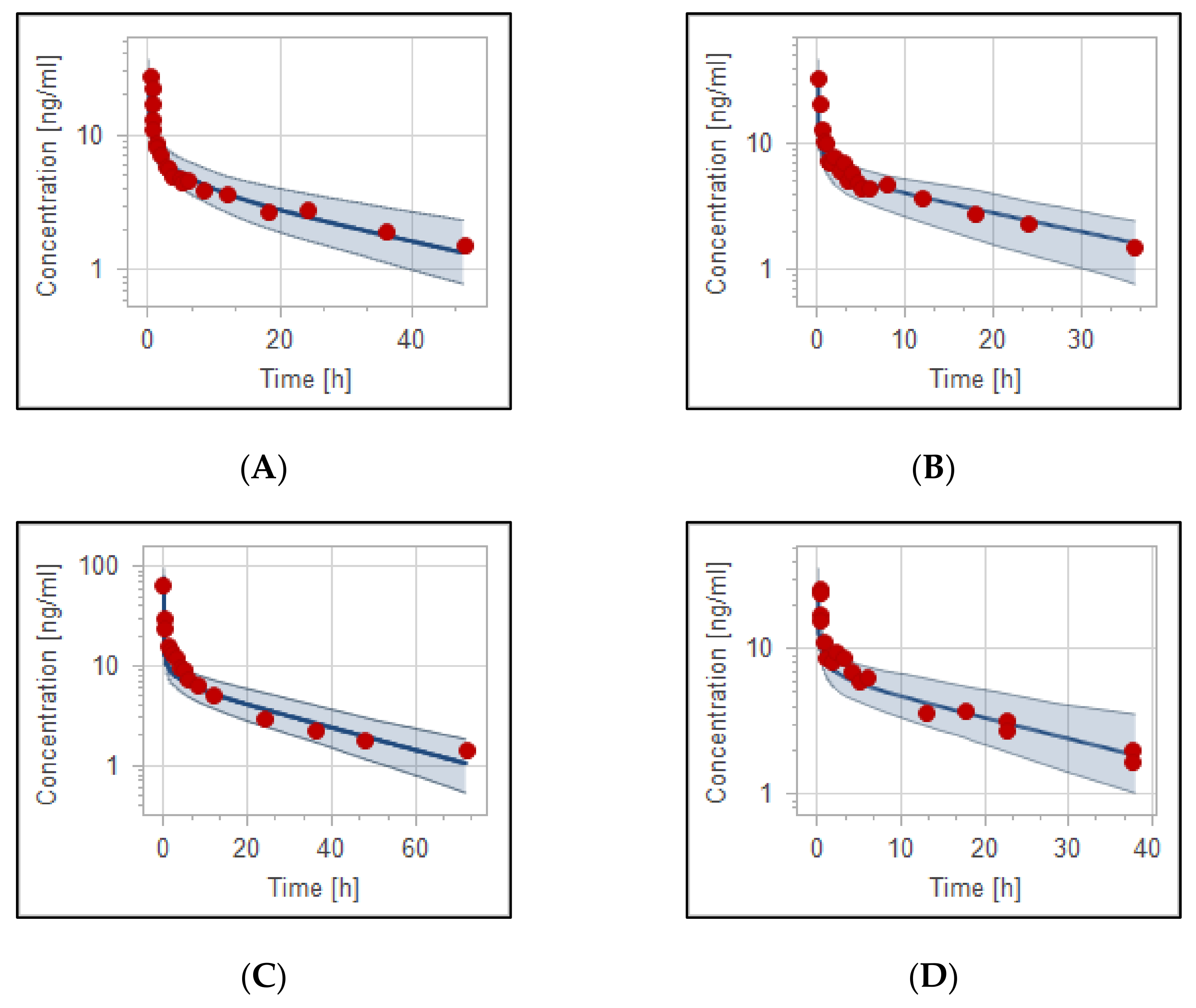
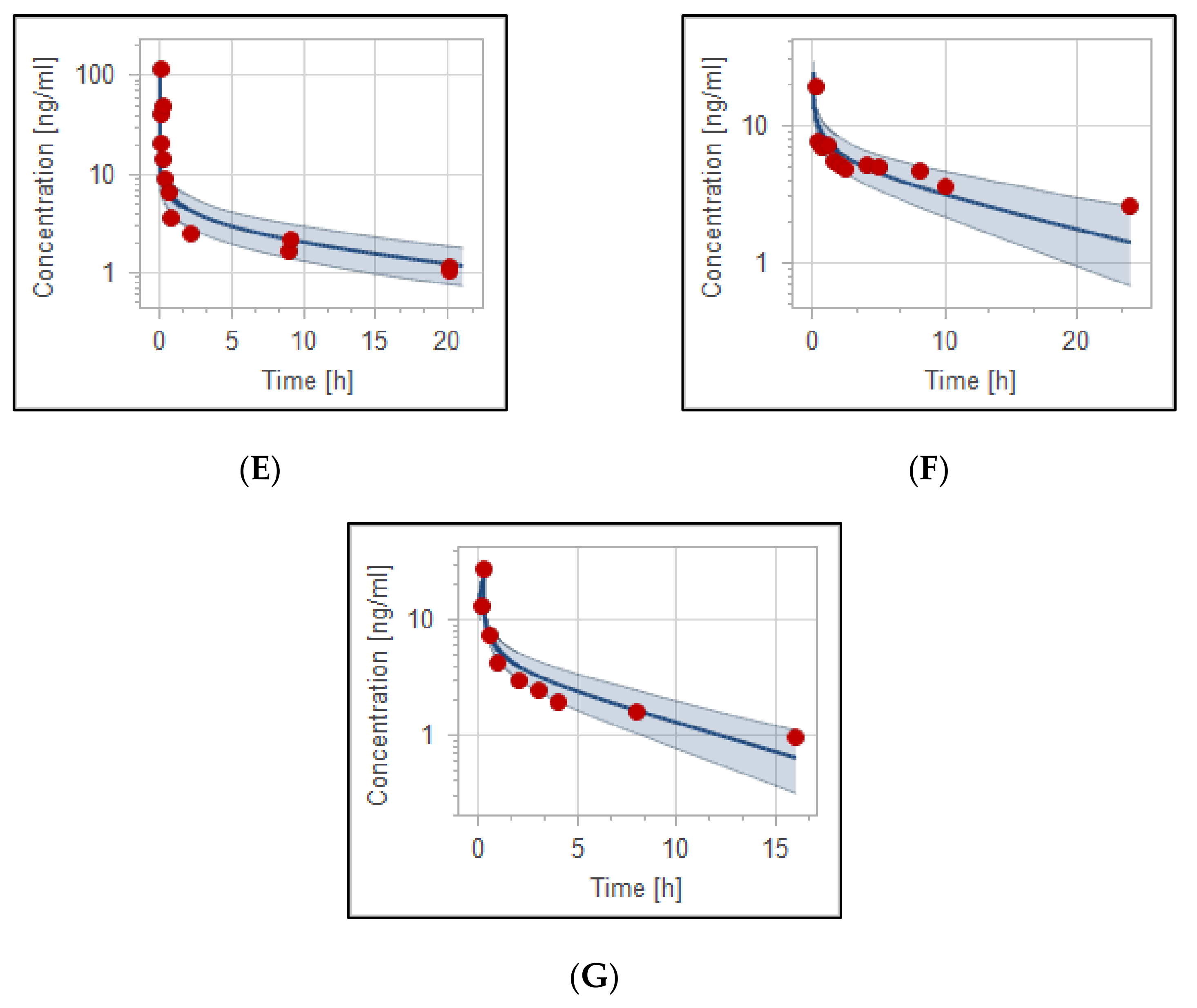
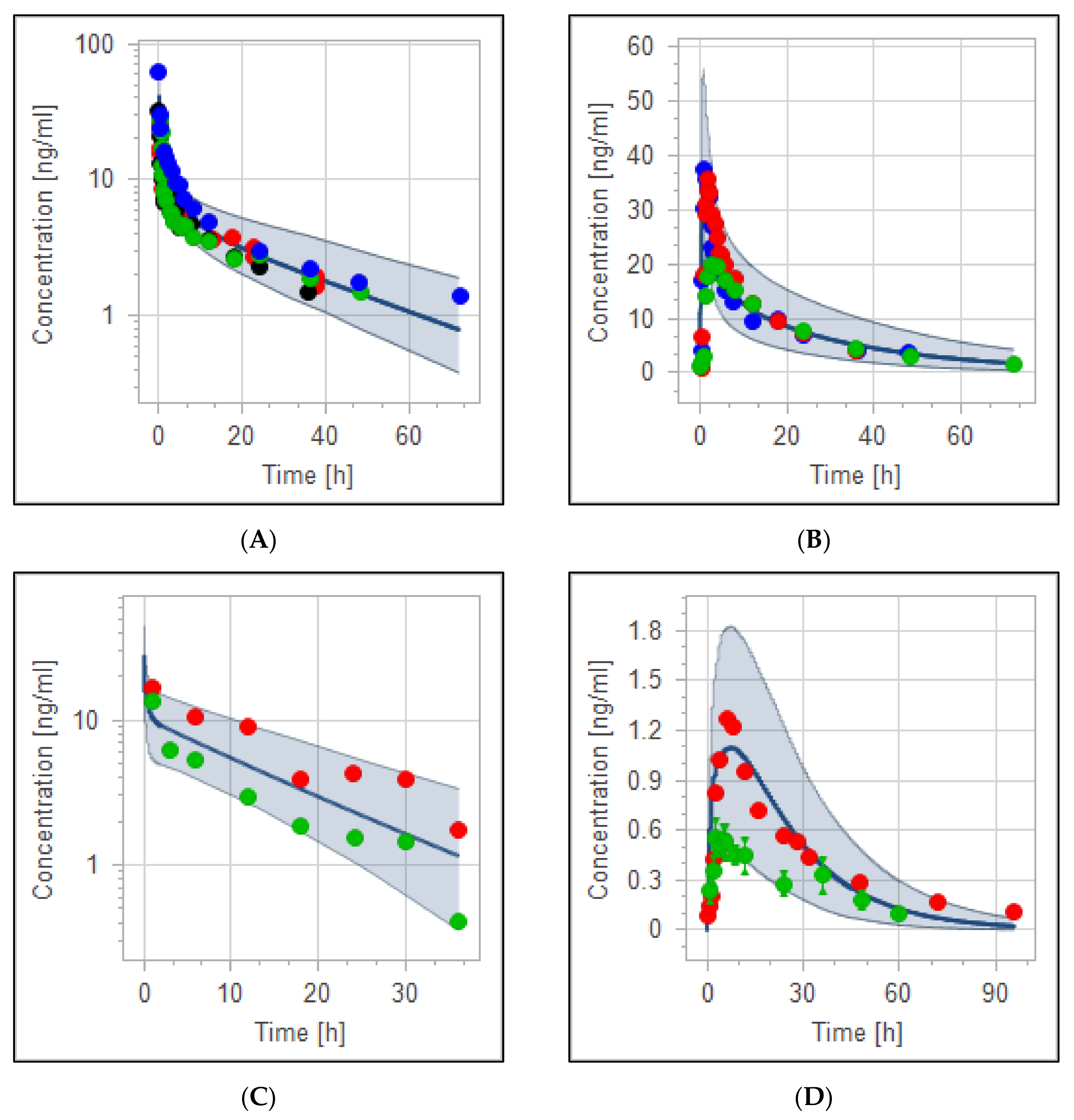
3.2. The PBPK Model for Adult Healthy and Psychotic, Otherwise Healthy, Populations after Intravenous and Oral Administration
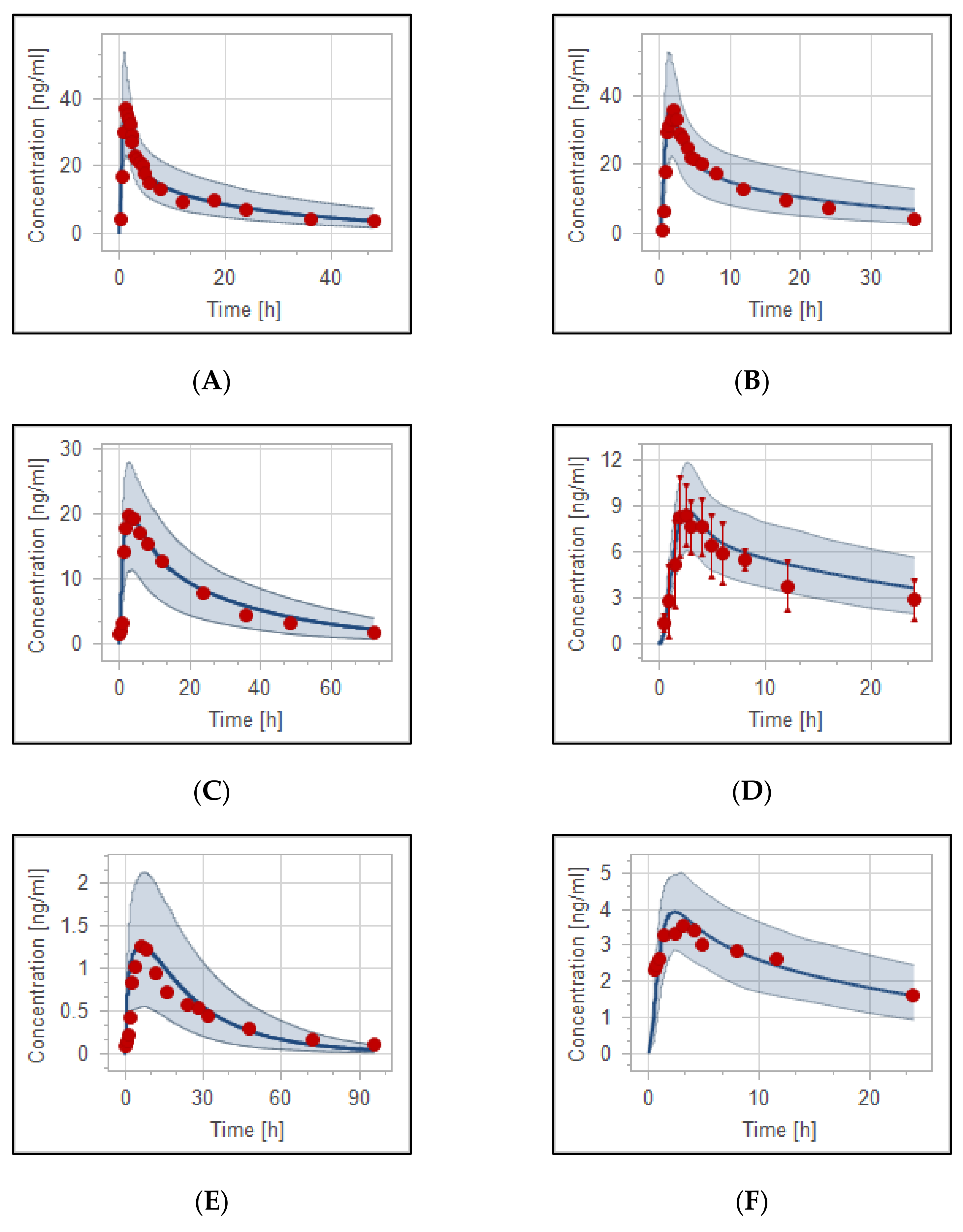
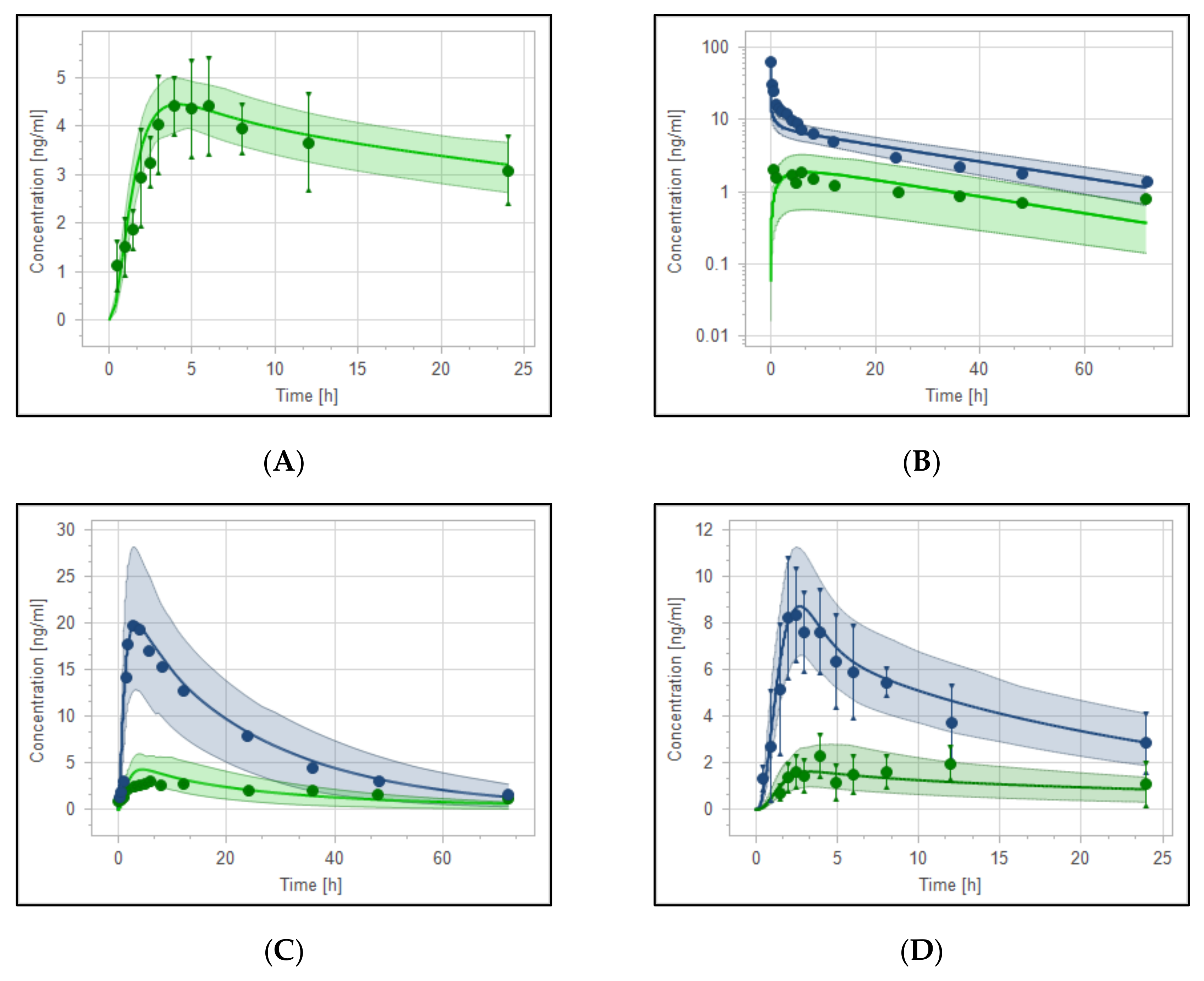
3.3. Reduced Haloperidol as a Parent and as a Metabolite
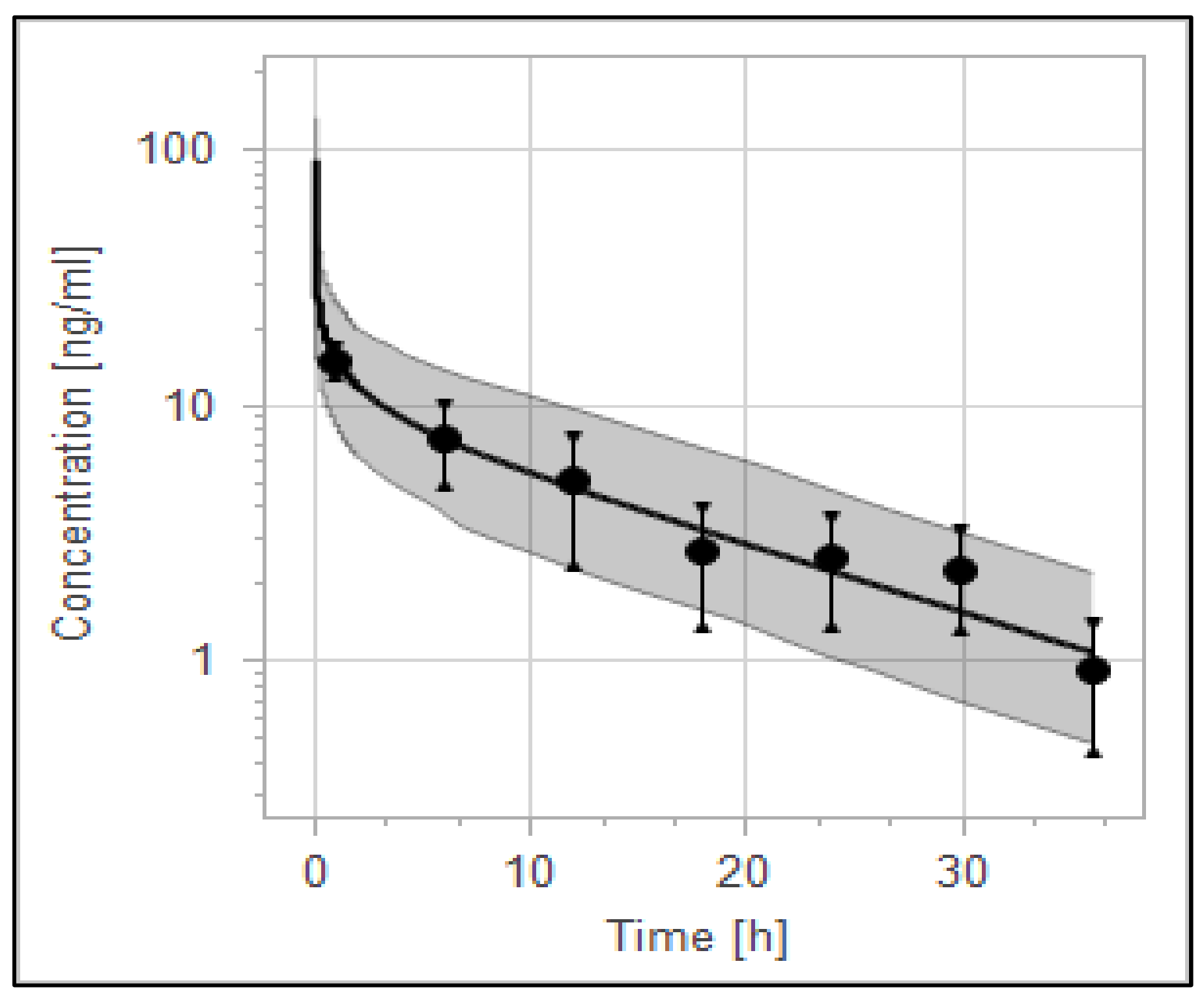
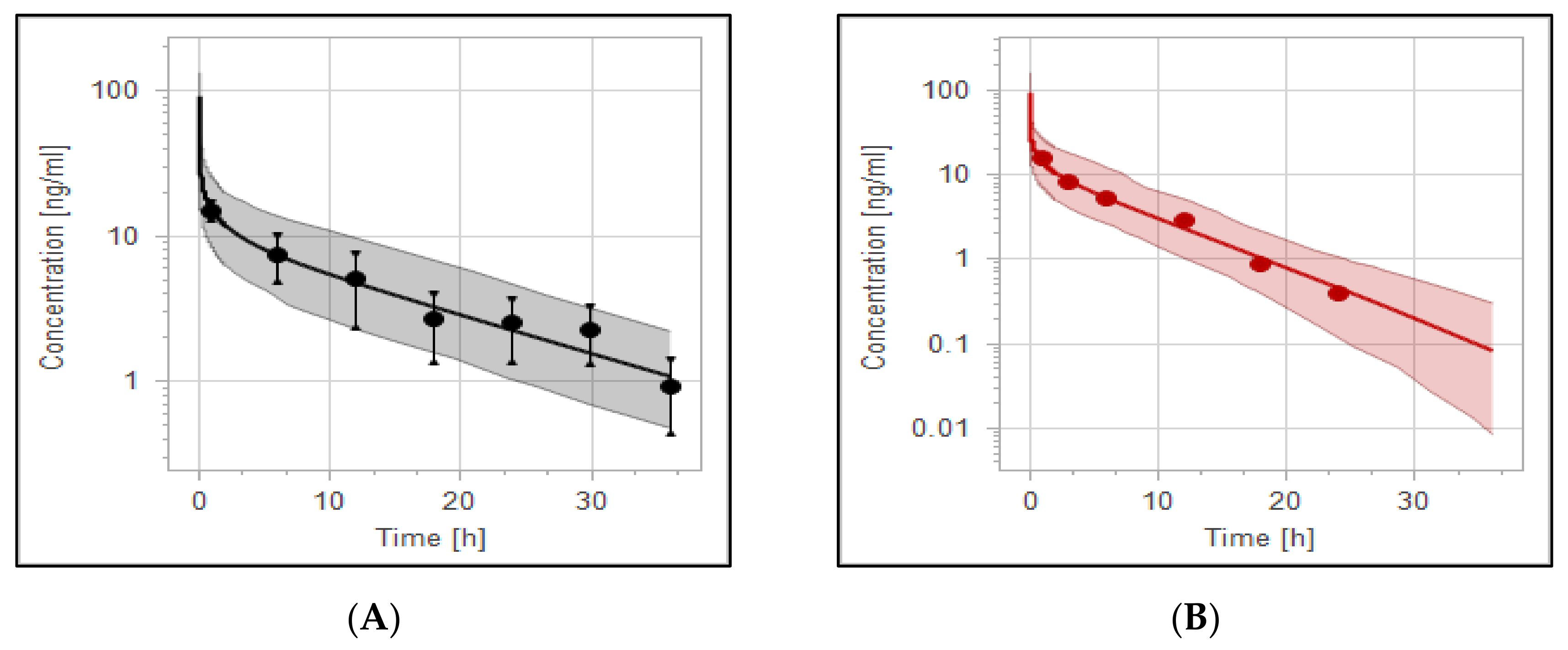
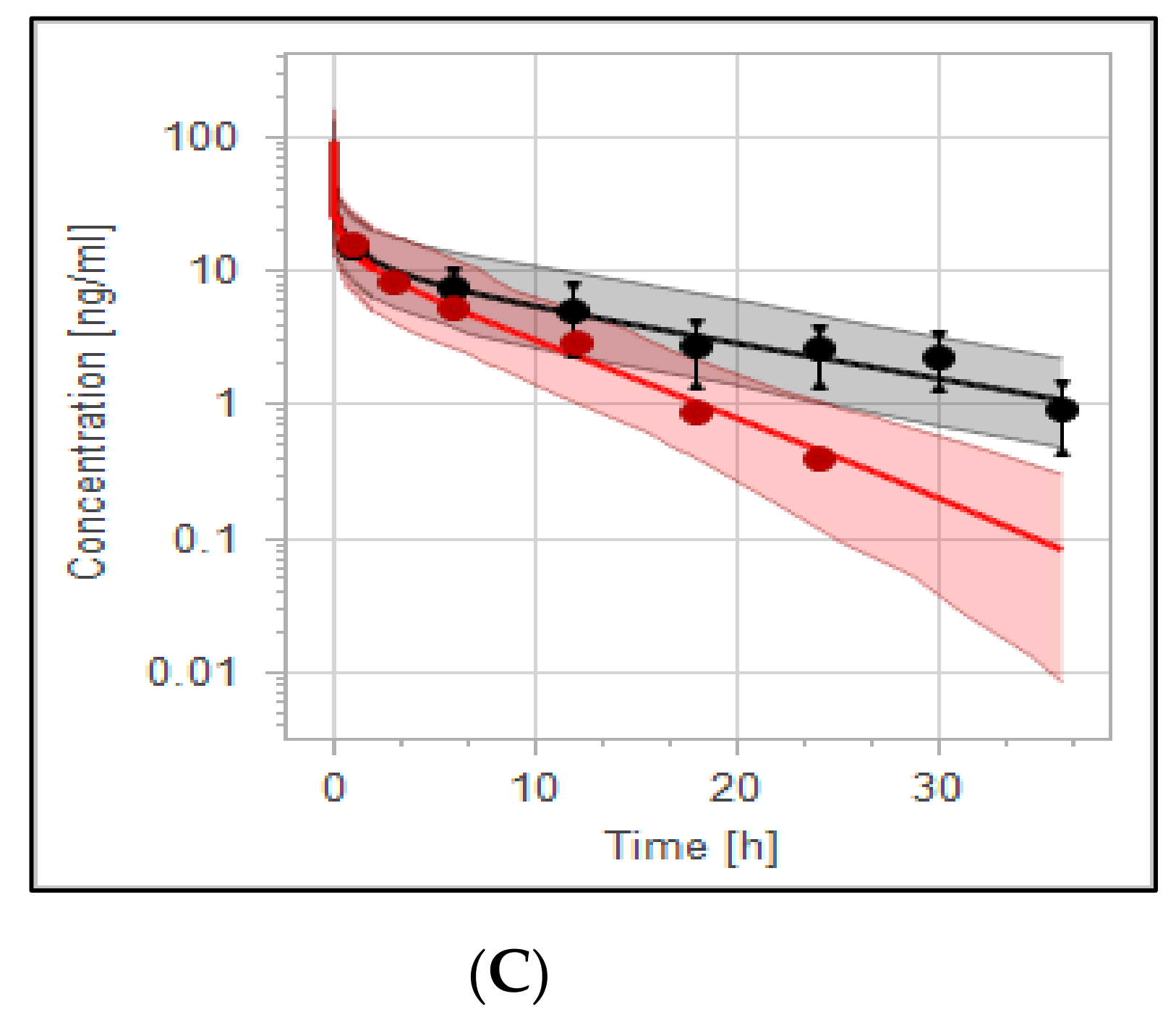
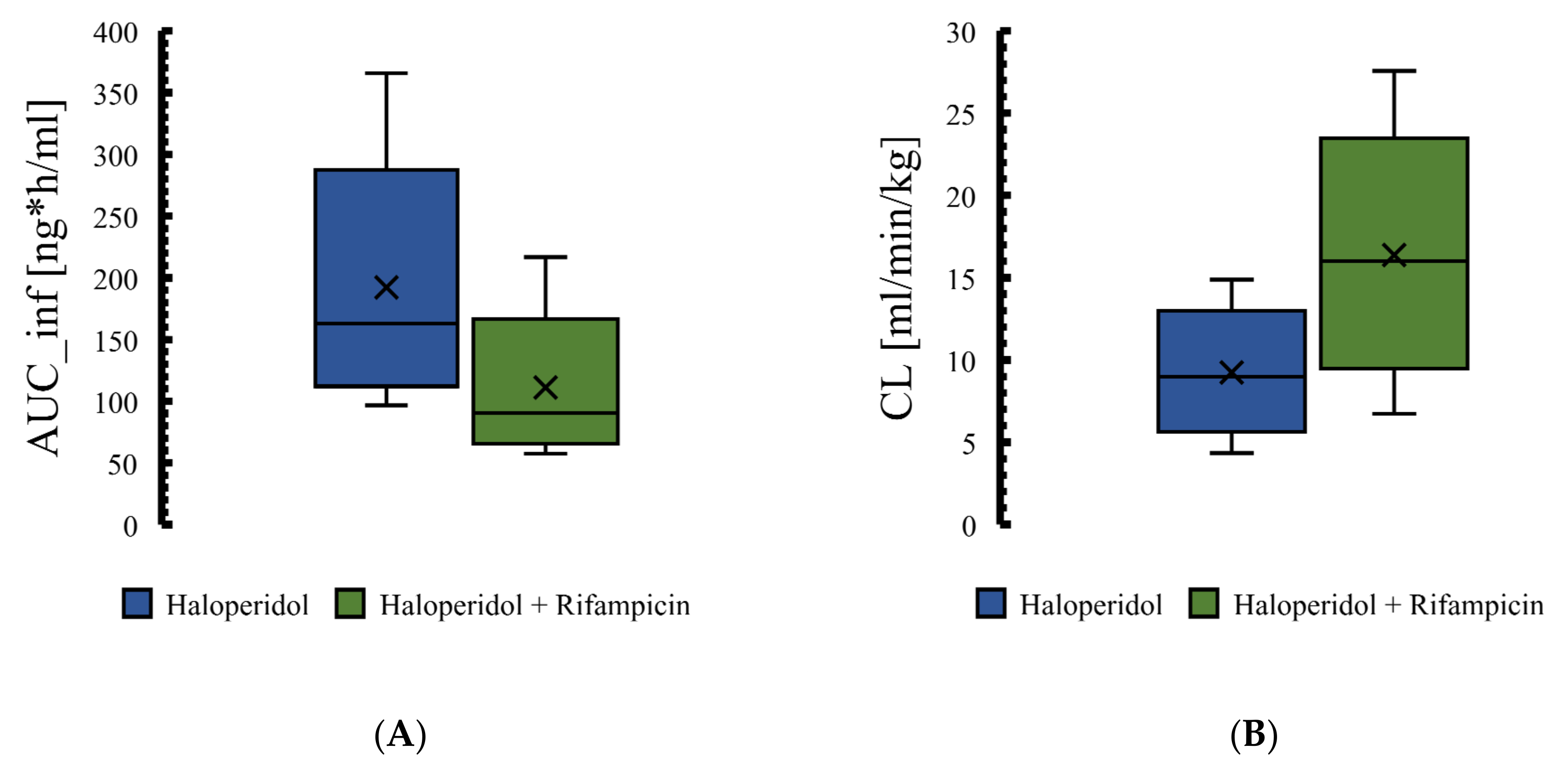
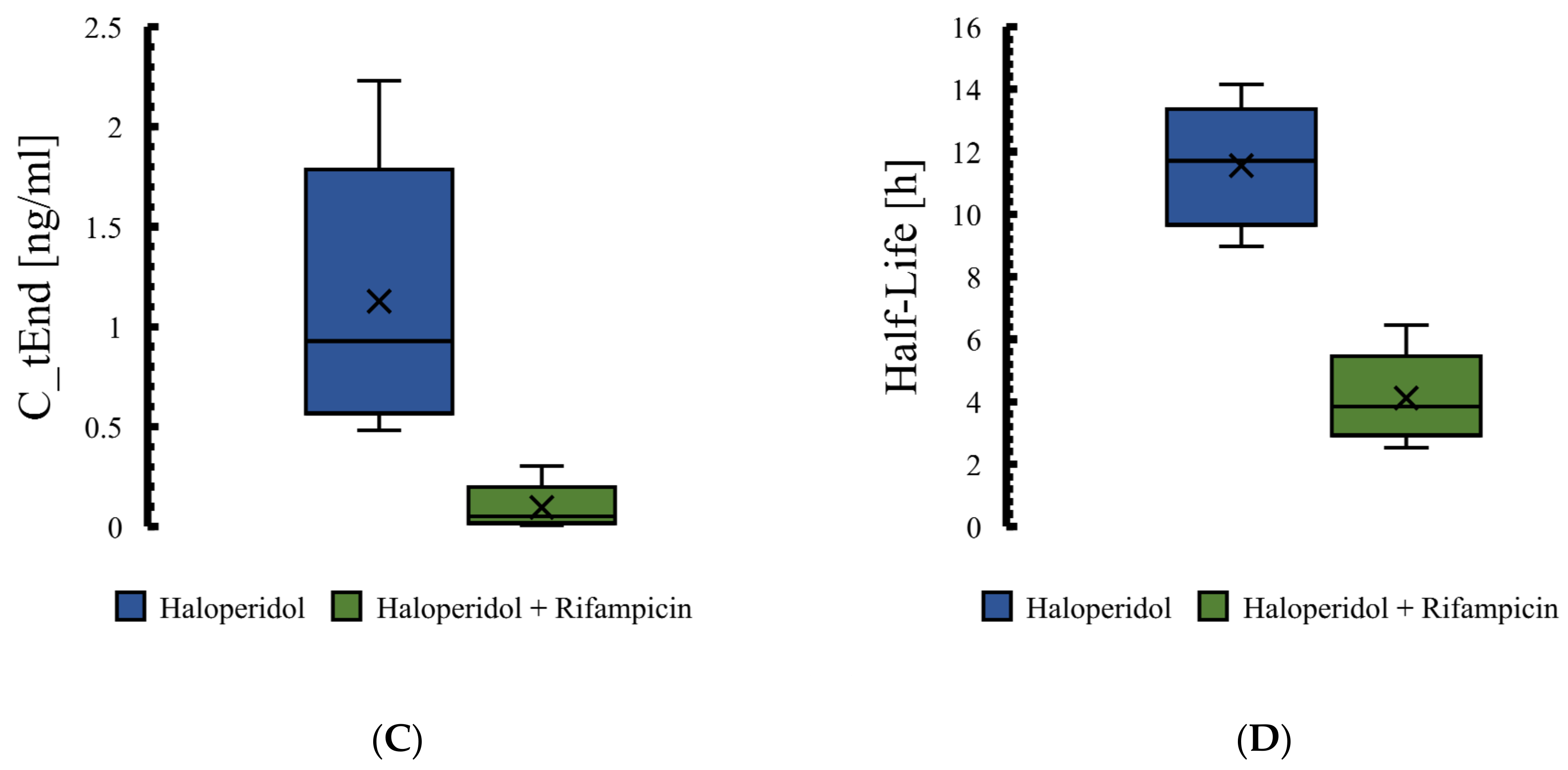
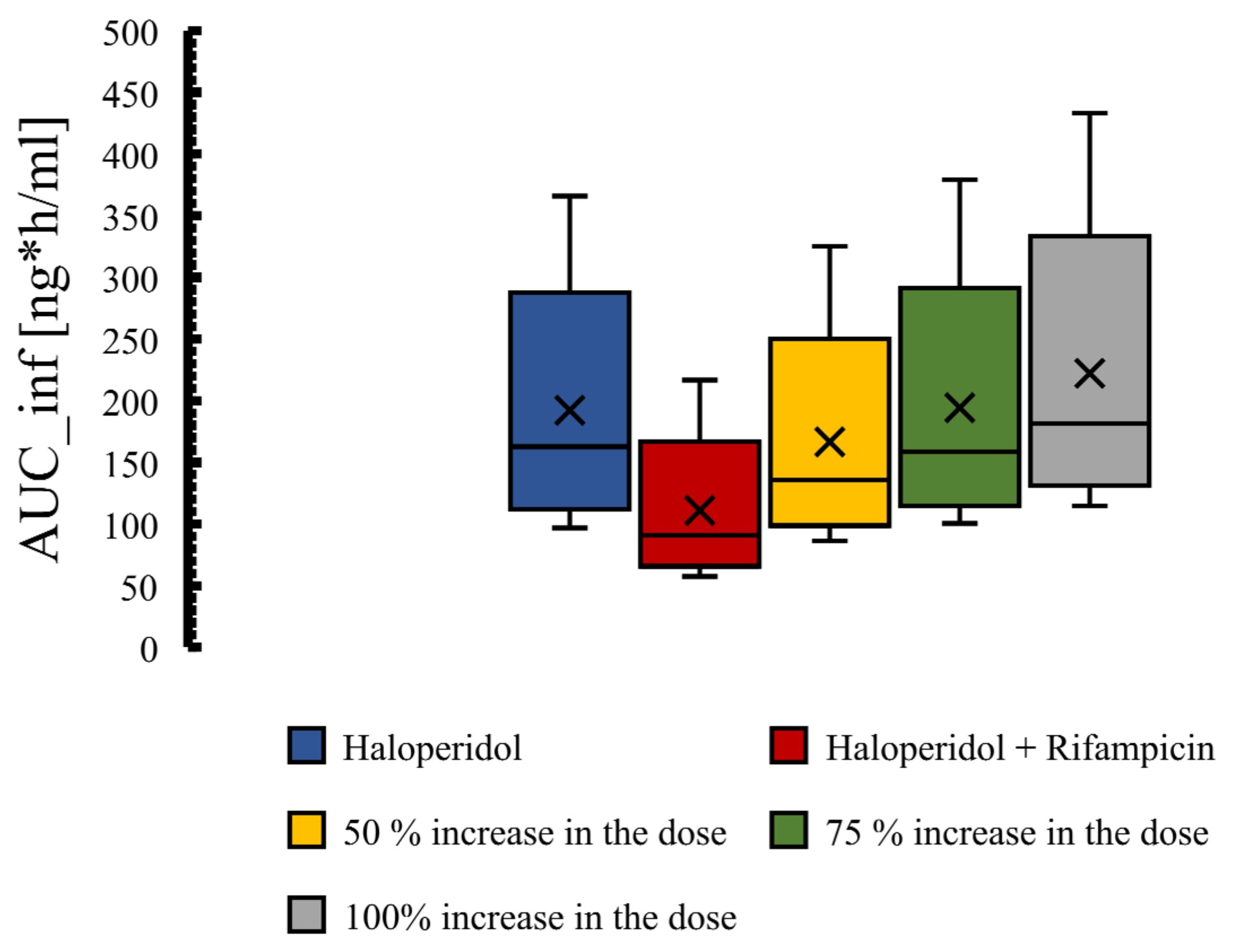
3.4. The PBPK Model for Tuberculotic Population and Evaluation of Its Performance to Predict Drug Interaction with the Anti-Tuberculotic Agent and Dose Optimization
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tandon, R.; Nasrallah, H.A.; Keshavan, M.S. Schizophrenia,“just the facts” 4. Clinical features and conceptualization. Schizophr. Res. 2009, 110, 1–23. [Google Scholar] [PubMed]
- Stefansson, H.; Ophoff, R.A.; Steinberg, S.; Andreassen, O.A.; Cichon, S.; Rujescu, D.; Werge, T.; Pietiläinen, O.P.; Mors, O.; Mortensen, P.B. Common variants conferring risk of schizophrenia. Nature 2009, 460, 744–747. [Google Scholar] [PubMed]
- Tandon, R.; Gaebel, W.; Barch, D.M.; Bustillo, J.; Gur, R.E.; Heckers, S.; Malaspina, D.; Owen, M.J.; Schultz, S.; Tsuang, M. Definition and description of schizophrenia in the DSM-5. Schizophr. Res. 2013, 150, 3–10. [Google Scholar] [PubMed]
- Cloutier, M.; Aigbogun, M.S.; Guerin, A.; Nitulescu, R.; Ramanakumar, A.V.; Kamat, S.A.; DeLucia, M.; Duffy, R.; Legacy, S.N.; Henderson, C. The economic burden of schizophrenia in the United States in 2013. J. Clin. Psychiatry 2016, 77, 5379. [Google Scholar]
- Hatfield, A.B. Psychological costs of schizophrenia to the family. Soc. Work 1978, 23, 355–359. [Google Scholar]
- Murray, C.J.; Lopez, A.D. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997, 349, 1436–1442. [Google Scholar]
- Hasan, A.; Falkai, P.; Wobrock, T.; Lieberman, J.; Glenthoj, B.; Gattaz, W.F.; Thibaut, F.; Möller, H.; WFSBP Task force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: Update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J. Biol. Psychiatry 2013, 14, 2–44. [Google Scholar]
- Hasan, A.; Falkai, P.; Wobrock, T.; Lieberman, J.; Glenthoj, B.; Gattaz, W.F.; Thibaut, F.; Möller, H.; WFSBP Task force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) Guidelines for Biological Treatment of Schizophrenia, part 1: Update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J. Biol. Psychiatry 2012, 13, 318–378. [Google Scholar]
- Wilson, M.P.; Pepper, D.; Currier, G.W.; Holloman, G.H., Jr.; Feifel, D. The psychopharmacology of agitation: Consensus statement of the American Association for Emergency Psychiatry Project BETA Psychopharmacology Workgroup. West. J. Emerg. Med. 2012, 13, 26. [Google Scholar] [CrossRef]
- Agar, M.R.; Lawlor, P.G.; Quinn, S.; Draper, B.; Caplan, G.A.; Rowett, D.; Sanderson, C.; Hardy, J.; Le, B.; Eckermann, S. Efficacy of oral risperidone, haloperidol, or placebo for symptoms of delirium among patients in palliative care: A randomized clinical trial. JAMA Intern. Med. 2017, 177, 34–42. [Google Scholar]
- Hardy, J.R.; O’Shea, A.; White, C.; Gilshenan, K.; Welch, L.; Douglas, C. The efficacy of haloperidol in the management of nausea and vomiting in patients with cancer. J. Pain Symptom Manage. 2010, 40, 111–116. [Google Scholar] [PubMed]
- Boettger, S.; Breitbart, W.; Passik, S. Haloperidol and risperidone in the treatment of delirium and its subtypes. Eur. J. Psychiatry 2011, 25, 59–67. [Google Scholar]
- McCue, R.E.; Waheed, R.; Urcuyo, L.; Orendain, G.; Joseph, M.D.; Charles, R.; Hasan, S.M. Comparative effectiveness of second-generation antipsychotics and haloperidol in acute schizophrenia. Br. J. Psychiatry 2006, 189, 433–440. [Google Scholar] [PubMed]
- Kapur, S.; Mamo, D. Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2003, 27, 1081–1090. [Google Scholar]
- Meltzer, H.Y. Update on typical and atypical antipsychotic drugs. Annu. Rev. Med. 2013, 64, 393–406. [Google Scholar]
- Richelson, E. Receptor pharmacology of neuroleptics: Relation to clinical effects. J. Clin. Psychiatry 1999, 60, 5–14. [Google Scholar]
- Moore, G.; Pfaff, J.A. Assessment and emergency management of the acutely agitated or violent adult. In UpToDate; UpToDate Inc.: Waltham, MA, USA; Available online: https://www.uptodate.com (accessed on 15 February 2021).
- Beach, S.R.; Gross, A.F.; Hartney, K.E.; Taylor, J.B.; Rundell, J.R. Intravenous haloperidol: A systematic review of side effects and recommendations for clinical use. Gen. Hosp. Psychiatry 2020, 67, 42–50. [Google Scholar]
- Kudo, S.; Ishizaki, T. Pharmacokinetics of haloperidol. Clin. Pharmacokinet. 1999, 37, 435–456. [Google Scholar]
- Rostami-Hodjegan, A.; Toon, S. Physiologically Based Pharmacokinetics as a Component of Model-Informed Drug Development: Where We Were, Where We Are, and Where We Are Heading. J. Clin. Pharmacol. 2020, 60, S12–S16. [Google Scholar] [CrossRef]
- Hartmanshenn, C.; Scherholz, M.; Androulakis, I.P. Physiologically-based pharmacokinetic models: Approaches for enabling personalized medicine. J. Pharmacokinet. Pharmacodyn. 2016, 43, 481–504. [Google Scholar]
- Maeng, H.; Chow, E.C.; Fan, J.; Pang, K.S. Physiologically based pharmacokinetic (PBPK) modeling: Usefulness and applications. Encycl. Drug Metab. Interact. 2011, 3, 1–48. [Google Scholar]
- Arya, V.; Venkatakrishnan, K. Role of Physiologically Based Pharmacokinetic Modeling and Simulation in Enabling Model-Informed Development of Drugs and Biotherapeutics. J. Clin. Pharmacol. 2020, 60, S7–S11. [Google Scholar] [CrossRef] [PubMed]
- Kuepfer, L.; Niederalt, C.; Wendl, T.; Schlender, J.; Willmann, S.; Lippert, J.; Block, M.; Eissing, T.; Teutonico, D. Applied concepts in PBPK modeling: How to build a PBPK/PD model. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 516–531. [Google Scholar] [CrossRef]
- Kuepfer, L.; Lippert, J.; Eissing, T. Multiscale mechanistic modeling in pharmaceutical research and development. Adv. Syst. Biol. 2012, 736, 543–561. [Google Scholar]
- Hanke, N.; Frechen, S.; Moj, D.; Britz, H.; Eissing, T.; Wendl, T.; Lehr, T. PBPK models for CYP3A4 and P-gp DDI prediction: A modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 647–659. [Google Scholar] [CrossRef] [Green Version]
- Kanacher, T.; Lindauer, A.; Mezzalana, E.; Michon, I.; Veau, C.; Mantilla, J.D.G.; Nock, V.; Fleury, A. A Physiologically-Based Pharmacokinetic (PBPK) Model Network for the Prediction of CYP1A2 and CYP2C19 Drug–Drug–Gene Interactions with Fluvoxamine, Omeprazole, S-mephenytoin, Moclobemide, Tizanidine, Mexiletine, Ethinylestradiol, and Caffeine. Pharmaceutics 2020, 12, 1191. [Google Scholar] [CrossRef]
- Horiuchi, K.; Ohnishi, S.; Matsuzaki, T.; Funaki, S.; Watanabe, A.; Mizutare, T.; Matsumoto, S.; Nezasa, K.; Hasegawa, H. Improved human pharmacokinetic prediction of hepatically metabolized drugs with species-specific systemic clearance. J. Pharm. Sci. 2018, 107, 1443–1453. [Google Scholar] [CrossRef]
- Kato, Y.; Nakajima, M.; Oda, S.; Fukami, T.; Yokoi, T. Human UDP-glucuronosyltransferase isoforms involved in haloperidol glucuronidation and quantitative estimation of their contribution. Drug Metab. Dispos. 2012, 40, 240–248. [Google Scholar] [CrossRef]
- Chang, W. Reduced haloperidol: A factor in determining the therapeutic benefit of haloperidol treatment? Psychopharmacology 1992, 106, 289–296. [Google Scholar] [CrossRef]
- Ye, M.; Nagar, S.; Korzekwa, K. A physiologically based pharmacokinetic model to predict the pharmacokinetics of highly protein-bound drugs and the impact of errors in plasma protein binding. Biopharm. Drug Dispos. 2016, 37, 123–141. [Google Scholar] [CrossRef]
- Jemikalajah, J.D.; Okogun, G.; Adu, M.E.; Okolie, G.C. Evaluation of serum proteins in pulmonary tuberculosis. Afr. J. Cell. Pathol. 2014, 3, 20–24. [Google Scholar] [CrossRef]
- Frechen, S.; Solodenko, J.; Wendl, T.; Dallmann, A.; Ince, I.; Lehr, T.; Lippert, J.; Burghaus, R. A generic framework for the physiologically-based pharmacokinetic platform qualification of PK-Sim and its application to predicting cytochrome P450 3A4–mediated drug–drug interactions. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Willmann, S.; Lippert, J.; Sevestre, M.; Solodenko, J.; Fois, F.; Schmitt, W. PK-Sim (R): A physiologically based pharmacokinetic ‘whole-body’ model. Biosilico 2003, 4, 121–124. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- Wojtyniak, J.; Britz, H.; Selzer, D.; Schwab, M.; Lehr, T. Data digitizing: Accurate and precise data extraction for quantitative systems pharmacology and physiologically-based pharmacokinetic modeling. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Holley, F.O.; Magliozzi, J.R.; Stanski, D.R.; Lombrozo, L.; Hollister, L.E. Haloperidol kinetics after oral and intravenous doses. Clin. Pharmacol. Ther. 1983, 33, 477–484. [Google Scholar] [CrossRef]
- Magliozzi, J.R.; Gillespie, H.; Lombrozo, L.; Hollister, L.E. Mood alteration following oral and intravenous haloperidol and relationship to drug concentration in normal subjects. J. Clin. Pharmacol. 1985, 25, 285–290. [Google Scholar] [CrossRef]
- Chang, W.; Francis Lam, Y.W.; Jann, M.W.; Chen, H. Pharmacokinetics of haloperidol and reduced haloperidol in Chinese schizophrenic patients after intravenous and oral administration of haloperidol. Psychopharmacology 1992, 106, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Forsman, A.; Öhman, R. On the pharmacokinetics of haloperidol. Nord. Psykiatr. Tidsskr. 1974, 28, 441–448. [Google Scholar] [CrossRef]
- Forsman, A.; Mårtensson, E.; Nyberg, G.; Öhman, R. A gas chromatographic method for determining haloperidol. Naunyn Schmiedebergs Arch. Pharm. 1974, 286, 113–124. [Google Scholar] [CrossRef]
- Takeda, M.; Nishinuma, K.; Yamashita, S.; Matsubayashi, T.; Tanino, S.; Nishimura, T. Serum haloperidol levels of schizophrenics receiving treatment for tuberculosis. Clin. Neuropharmacol. 1986, 9, 386–397. [Google Scholar] [CrossRef]
- Cheng, Y.F.; Paalzow, L.K.; Bondesson, U.; Ekblom, B.; Eriksson, K.; Eriksson, S.O.; Lindberg, A.; Lindström, L. Pharmacokinetics of haloperidol in psychotic patients. Psychopharmacology 1987, 91, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Ashford, J.W.; Archer, S.M.; Rudy, A.C.; Wermeling, D.P. Comparison of intranasal administration of haloperidol with intravenous and intramuscular administration: A pilot pharmacokinetic study. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2008, 28, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Jann, M.W.; Lam, Y.F.; Chang, W. Reversible metabolism of haloperidol and reduced haloperidol in Chinese schizophrenic patients. Psychopharmacology 1990, 101, 107–111. [Google Scholar] [CrossRef]
- Midha, K.K.; Chakraborty, B.S.; Ganes, D.A.; Hawes, E.M.; Hubbard, J.W.; Keegan, D.L.; Korchinski, E.D.; McKay, G. Intersubject variation in the pharmacokinetics of haloperidol and reduced haloperidol. J. Clin. Psychopharmacol. 1989, 9, 98–104. [Google Scholar] [CrossRef]
- Midha, K.K.; Chakraborty, B.S.; Schwede, R.; Hawes, E.M.; Hubbard, J.W.; McKay, G. Comparative bioavailability of a new commercial tablet formulation and two lots of a reference formulation of haloperidol. J. Pharm. Sci. 1989, 78, 443–447. [Google Scholar] [CrossRef]
- Zhao, P.; Rowland, M.; Huang, S. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin. Pharmacol. Ther. 2012, 92, 17–20. [Google Scholar] [CrossRef]
- Ke, A.; Barter, Z.; Rowland-Yeo, K.; Almond, L. Towards a best practice approach in PBPK modeling: Case example of developing a unified efavirenz model accounting for induction of CYPs 3A4 and 2B6. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.A.; Dolgos, H. Requirements to establishing confidence in physiologically based pharmacokinetic (PBPK) models and overcoming some of the challenges to meeting them. Clin. Pharmacokinet. 2019, 58, 1355–1371. [Google Scholar] [CrossRef] [PubMed]
- Kalam, M.N.; Rasool, M.F.; Alqahtani, F.; Imran, I.; Rehman, A.U.; Ahmed, N. Development and Evaluation of a Physiologically Based Pharmacokinetic Drug-Disease Model of Propranolol for Suggesting Model Informed Dosing in Liver Cirrhosis Patients. Drug Des. Dev. Ther. 2021, 15, 1195. [Google Scholar] [CrossRef]
- Kovar, L.; Schräpel, C.; Selzer, D.; Kohl, Y.; Bals, R.; Schwab, M.; Lehr, T. Physiologically-based pharmacokinetic (PBPK) modeling of buprenorphine in adults, children and preterm neonates. Pharmaceutics 2020, 12, 578. [Google Scholar] [CrossRef] [PubMed]
- Inaba, T.; Kovacs, J. Haloperidol reductase in human and guinea pig livers. Drug Metab. Dispos. 1989, 17, 330–333. [Google Scholar]
- Shi, S.M.; Di, L. The role of carbonyl reductase 1 in drug discovery and development. Expert Opin. Drug Metab. Toxicol. 2017, 13, 859–870. [Google Scholar] [CrossRef]
- Hua, W.; Zhang, H.; Ryu, S.; Yang, X.; Di, L. Human tissue distribution of carbonyl reductase 1 using proteomic approach with liquid chromatography-tandem mass spectrometry. J. Pharm. Sci. 2017, 106, 1405–1411. [Google Scholar] [CrossRef]
- El-Khateeb, E.; Achour, B.; Scotcher, D.; Al-Majdoub, Z.M.; Athwal, V.; Barber, J.; Rostami-Hodjegan, A. Scaling factors for clearance in adult liver cirrhosis. Drug Metab. Dispos. 2020, 48, 1271–1282. [Google Scholar] [CrossRef]
- Usuki, E.; Pearce, R.; Parkinson, A.; Castagnoli, N. Studies on the conversion of haloperidol and its tetrahydropyridine dehydration product to potentially neurotoxic pyridinium metabolites by human liver microsomes. Chem. Res. Toxicol. 1996, 9, 800–806. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, L.; Nguyen, K.; Fretland, A.J. Utility of intersystem extrapolation factors in early reaction phenotyping and the quantitative extrapolation of human liver microsomal intrinsic clearance using recombinant cytochromes P450. Drug Metab. Dispos. 2011, 39, 373–382. [Google Scholar] [CrossRef]
- Thelen, K.; Coboeken, K.; Willmann, S.; Burghaus, R.; Dressman, J.B.; Lippert, J. Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part 1: Oral solutions. J. Pharm. Sci. 2011, 100, 5324–5345. [Google Scholar] [CrossRef] [PubMed]
- Thelen, K.; Coboeken, K.; Willmann, S.; Dressman, J.B.; Lippert, J. Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part II: Extension to describe performance of solid dosage forms. J. Pharm. Sci. 2012, 101, 1267–1280. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.T.; Lade, J.M.; Tran, T.B.; Dahal, U.P. Fraction unbound for liver microsome and hepatocyte incubations for all major species can be approximated using a single-species surrogate. Drug Metab. Dispos. 2019, 47, 419–423. [Google Scholar] [CrossRef]
- Ishizaki, J.; Yokogawa, K.; Nakashima, E.; Ichimura, F. Prediction of changes in the clinical pharmacokinetics of basic drugs on the basis of octanol-water partition coefficients. J. Pharm. Pharmacol. 1997, 49, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Settimo, L.; Bellman, K.; Knegtel, R. Comparison of the accuracy of experimental and predicted pKa values of basic and acidic compounds. Pharm. Res. 2014, 31, 1082–1095. [Google Scholar] [CrossRef]
- Margaillan, G.; Rouleau, M.; Klein, K.; Fallon, J.K.; Caron, P.; Villeneuve, L.; Smith, P.C.; Zanger, U.M.; Guillemette, C. Multiplexed targeted quantitative proteomics predicts hepatic glucuronidation potential. Drug Metab. Dispos. 2015, 43, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Eyles, D.W.; Stedman, T.J.; Pond, S.M. Nonlinear relationship between circulating concentrations of reduced haloperidol and haloperidol: Evaluation of possible mechanisms. Psychopharmacology 1994, 116, 161–166. [Google Scholar] [CrossRef]
- Zheng, L.; Xu, M.; Tang, S.; Song, H.; Jiang, X.; Wang, L. Physiologically based pharmacokinetic modeling of oxycodone in children to support pediatric dosing optimization. Pharm. Res. 2019, 36, 171. [Google Scholar] [CrossRef]
- Biesdorf, C.; Martins, F.S.; Sy, S.K.; Diniz, A. Physiologically-based pharmacokinetics of ziprasidone in pregnant women. Br. J. Clin. Pharmacol. 2019, 85, 914–923. [Google Scholar] [CrossRef]
- Yamazaki, S.; Evers, R.; De Zwart, L. Physiologically-based pharmacokinetic modeling to evaluate in vitro-to-in vivo extrapolation for intestinal P-glycoprotein inhibition. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 55–67. [Google Scholar] [CrossRef]
- Magliozzi, J.R.; Hollister, L.E. Elimination half-life and bioavailability of haloperidol in schizophrenic patients. J. Clin. Psychiatry 1985, 46, 20–21. [Google Scholar] [PubMed]
- Yun, M.; Kwon, J.; Kwon, K. Pharmacokinetics and bioequivalence of haloperidol tablet by liquid chromatographic mass spectrometry with electrospray ionization. Arch. Pharm. Res. 2005, 28, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Franken, L.G.; Mathot, R.; Masman, A.D.; Baar, F.; Tibboel, D.; van Gelder, T.; Koch, B.; de Winter, B. Population pharmacokinetics of haloperidol in terminally ill adult patients. Eur. J. Clin. Pharmacol. 2017, 73, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.P.; Kozielska, M.; Johnson, M.; Mafirakureva, N.; Vermeulen, A.; Liu, J.; de Greef, R.; Rujescu, D.; Groothuis, G.M.; Danhof, M. Population pharmacokinetic-pharmacodynamic modeling of haloperidol in patients with schizophrenia using positive and negative syndrome rating scale. J. Clin. Psychopharmacol. 2013, 33, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sassen, S.D.; van der Jagt, M.; Endeman, H.; Koch, B.C.; Hunfeld, N.G. Pharmacokinetics of Haloperidol in Critically Ill Patients: Is There an Association with Inflammation? Pharmaceutics 2022, 14, 549. [Google Scholar] [CrossRef]
- Yukawa, E.; Hokazono, T.; Yukawa, M.; Ichimaru, R.; Maki, T.; Matsunaga, K.; Ohdo, S.; Anai, M.; Higuchi, S.; Goto, Y. Population pharmacokinetics of haloperidol using routine clinical pharmacokinetic data in Japanese patients. Clin. Pharmacokinet. 2002, 41, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.J.; Holford, N.H. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 303–332. [Google Scholar] [CrossRef] [PubMed]
- Huisinga, W.; Solms, A.; Fronton, L.; Pilari, S. Modeling interindividual variability in physiologically based pharmacokinetics and its link to mechanistic covariate modeling. CPT Pharmacomet. Syst. Pharmacol. 2012, 1, 1–10. [Google Scholar] [CrossRef]
- Jamei, M.; Dickinson, G.L.; Rostami-Hodjegan, A. A framework for assessing inter-individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: A tale of ‘bottom-up’vs ‘top-down’recognition of covariates. Drug Metab. Pharmacokinet. 2009, 24, 53–75. [Google Scholar]
- Willmann, S.; Höhn, K.; Edginton, A.; Sevestre, M.; Solodenko, J.; Weiss, W.; Lippert, J.; Schmitt, W. Development of a physiology-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J. Pharmacokinet. Pharmacodyn. 2007, 34, 401–431. [Google Scholar] [CrossRef]












| Dose | N | Female (n) | Age (Year) | Weight (kg) | Population | Reference |
|---|---|---|---|---|---|---|
| Haloperidol Administered Intravenously | ||||||
| 0.125 mg/kg | 6 | 0 | 21–37 | Mean: 70.5 | Healthy | [40] |
| 0.125 mg/kg | 12 | 0 | 19–37 | 56–92 | Healthy | [41] |
| 0.125 mg/kg | 8 | 0 | 21–48 | Mean: 63.3 | Psychotic otherwise healthy | [42] |
| 10 mg | 12 | 0 | Healthy | [43] | ||
| 5 mg | 1 | 1 | Mean: 55 | 74 | Psychotic otherwise healthy | [44] |
| 5 mg | 2 | 20 s–60 s | Tuberculotic (control) | [45] | ||
| 5 mg | 3 | 20 s–60 s | Tuberculotic (intervention) | [45] | ||
| 3.5 mg | 6 | 3 | 20–43 | Mean: 67 | Psychotic otherwise healthy | [46] |
| 2.5 mg | 4 | 2 | 24–37 | 63–82 | Healthy | [47] |
| Reduced Haloperidol after Haloperidol Administered Intravenously | ||||||
| 0.125 mg/kg | 8 | 0 | 21–48 | Mean: 63.3 | Psychotic otherwise healthy | [42] |
| Haloperidol Administered Orally | ||||||
| 0.503 mg/kg | 8 | 0 | 19–37 | Mean: 70.8 | Healthy | [40] |
| 0.500 mg/kg | 9 | 0 | 19–37 | 56–92 | Healthy | [41] |
| 0.500 mg/kg | 6 | 0 | 21–48 | Mean: 63.3 | Psychotic otherwise healthy | [42] |
| 10 mg | 6 | 0 | 32–57 | 43–66 | Psychotic otherwise healthy | [48] |
| 5 mg | 28 | 0 | 18–50 | Mean: 71.5 | Healthy | [49,50] |
| 2 mg | 8 | 5 | Mean: 32 | Mean: 67 | Psychotic otherwise healthy | [46] |
| Reduced Haloperidol after Haloperidol Administered Orally | ||||||
| 0.500 mg/kg | 6 | 0 | 21–48 | 63.3 ± 6.7 | Psychotic otherwise healthy | [42] |
| Reduced haloperidol administered orally | ||||||
| 10 mg | 6 | 0 | 32–57 | 43–66 | Psychotic otherwise healthy | [48] |
| PK Parameter | Predicted | Observed | Pre/Obs Ratio | PE (%) |
|---|---|---|---|---|
| 0.125 mg/kg IV in adult healthy [40] | ||||
| AUC_inf [ng·h/mL] | 168 | 202 | 0.83 | 17% |
| C_max [ng/mL] | 28 | 27 * | 1.04 | 4% |
| CL [mL/min/kg] | 12.3 | 11.1 | 1.11 | 9.3% |
| 0.125 mg/kg IV in adult healthy [41] | ||||
| AUC_inf [ng·h/mL] | 171.3 | 173.3 * | 1.01 | 1.2% |
| C_max [ng/mL] | 32.5 | 32.5 * | 1.00 | 0.00% |
| CL [mL/min/kg] | 12.0 | 12.02 * | 0.99 | 0.00% |
| 0.125 mg/kg IV in adult schizophrenic otherwise healthy [42] | ||||
| AUC_inf [ng·h/mL] | 271 | 383 | 0.71 | 6.3% |
| C_max [ng/mL] | 77.22 | 63.10 * | 1.22 | 22.4% |
| CL [mL/min/kg] | 7.56 | 6.20 | 1.22 | 21.9% |
| 10 mg IV in adult healthy [43] | ||||
| AUC_inf [ng·h/mL] | 193 | 202 * | 0.96 | 4.5% |
| C_max [ng/mL] | 31.50 | 25.70 * | 1.21 | 23% |
| CL [mL/min/kg] | 12.20 | 11.90 * | 1.03 | 2.5% |
| 5 mg IV in adult psychotic otherwise healthy [44] | ||||
| AUC_inf [ng·h/mL] | 97.56 | 86.20 * | 1.13 | 13.2% |
| C_max [ng/mL] | 108.70 | 115 * | 0.95 | 5.5% |
| CL [mL/min/kg] | 10 | 13 * | 0.77 | 23% |
| 3.5 mg IV in adult psychotic otherwise healthy [46] | ||||
| AUC_inf [ng·h/mL] | 144.30 | 175.72 * | 0.82 | 18% |
| C_max [ng/mL] | 15.13 | 19.2 * | 0.79 | 21.2% |
| CL [mL/min/kg] | 6.30 | 6.50 | 0.97 | 3.1% |
| 2.5 mg IV in adult healthy [47] | ||||
| AUC_inf [ng·h/mL] | 56 | 60.40 | 0.93 | 7.3% |
| C_max [ng/mL] | 22.00 | 27.43 * | 0.80 | 19.8% |
| CL [mL/min/kg] | 10.62 | 11.55 * | 0.92 | 8.2% |
| Predictability assessment | ||||
| AUC_inf | C_max | CL | ||
| MFE | 0.91 | 1.00 | 1.01 | |
| RMSE | 46.2 | 6.76 | 1.38 | |
| PK Parameter | Predicted | Observed | Pre/Obs Ratio | PE (%) |
|---|---|---|---|---|
| 0.503 mg/kg Oral in Adult Healthy [40] | ||||
| AUC_inf [ng·h/mL] | 537.4 | 566 | 0.95 | 5.1% |
| C_max [ng/mL] | 33.60 | 37.4 * | 0.90 | 10.2% |
| CL [mL/min/kg] | 15.61 | 15.41 * | 1.01 | 1.3% |
| 0.500 mg/kg Oral in Adult Healthy [41] | ||||
| AUC_inf [ng·h/mL] | 544 | 500.1 * | 1.10 | 8.8% |
| C_max [ng/mL] | 35.34 | 35.83 * | 0.99 | 1.3% |
| CL [mL/min/kg] | 15.20 | 16.64 * | 0.91 | 8.7% |
| 0.500 mg/kg Oral in Adult Schizophrenic Otherwise Healthy [42] | ||||
| AUC_inf [ng·h/mL] | 550 | 512.6 | 1.07 | 7.3% |
| C_max [ng/mL] | 20.81 | 19.76 * | 1.05 | 5.3% |
| CL [mL/min/kg] | 15.12 | 15.73 * | 0.96 | 3.9% |
| 10 mg Oral in Adult Psychotic Otherwise Healthy [48] | ||||
| AUC_inf [ng·h/mL] | 212.8 | 200.5 | 1.06 | 6.1% |
| C_max [ng/mL] | 9.13 | 8.34 * | 1.08 | 9.5% |
| CL [mL/min/kg] | 15 | 18.2 | 0.82 | 17.6% |
| 5 mg Oral in Adult Healthy [49] | ||||
| AUC_inf [ng·h/mL] | 40.10 | 44 * | 0.91 | 8.9% |
| C_max [ng/mL] | 1.28 | 1.27 * | 1.01 | 0.8% |
| CL [mL/min/kg] | 29.0 | 25.85 * | 1.12 | 12.2% |
| 2 mg Oral in Adult Psychotic Otherwise Healthy [46] | ||||
| AUC_inf [ng·h/mL] | 90.73 | 103 * | 0.88 | 12% |
| C_max [ng/mL] | 3.95 | 3.54 * | 1.12 | 11.6% |
| CL [mL/min/kg] | 5.50 | 4.93 * | 1.12 | 11.7% |
| Predictability Assessment | ||||
| AUC_inf | C_max | CL | ||
| MFE | 0.99 | 1.03 | 0.99 | |
| RMSE | 27.27 | 1.66 | 1.96 | |
| Group | Data | AUC_inf [ng·h/mL] | Half-Life [h] |
|---|---|---|---|
| Control group | Predicted | 174 | 11.5 |
| Observed | 186.1 | 10.1 | |
| Fold error | 0.93 | 1.22 | |
| Rifampicin 600 mg | Predicted | 103.85 | 4.76 |
| Observed | 105.03 | 4.93 | |
| Fold error | 0.99 | 0.97 | |
| Pred. AUC ratio rifampicin/control | 0.60 | ||
| Pred. T1/2 ratio rifampicin/control | 0.41 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alasmari, M.S.; Alasmari, F.; Alasmari, A.F.; Alshamsan, A.; Alsanea, S.; Rasool, M.F.; Alqahtani, F. Development and Evaluation of a Physiologically Based Pharmacokinetic Model for Predicting Haloperidol Exposure in Healthy and Disease Populations. Pharmaceutics 2022, 14, 1795. https://doi.org/10.3390/pharmaceutics14091795
Alasmari MS, Alasmari F, Alasmari AF, Alshamsan A, Alsanea S, Rasool MF, Alqahtani F. Development and Evaluation of a Physiologically Based Pharmacokinetic Model for Predicting Haloperidol Exposure in Healthy and Disease Populations. Pharmaceutics. 2022; 14(9):1795. https://doi.org/10.3390/pharmaceutics14091795
Chicago/Turabian StyleAlasmari, Mohammed S., Fawaz Alasmari, Abdullah F. Alasmari, Aws Alshamsan, Sary Alsanea, Muhammad F. Rasool, and Faleh Alqahtani. 2022. "Development and Evaluation of a Physiologically Based Pharmacokinetic Model for Predicting Haloperidol Exposure in Healthy and Disease Populations" Pharmaceutics 14, no. 9: 1795. https://doi.org/10.3390/pharmaceutics14091795