1. Introduction
Leishmaniases are a complex of infectious diseases, including visceral and cutaneous forms, caused by different species of
Leishmania protozoa and transmitted to mammals by the bite of a phlebotomine sandfly vector. Those are associated with poor housing, lack of financial resources, population displacement, malnutrition, and a weak immune system, leading to their classification as neglected tropical diseases [
1]. Chemotherapy is essential to treat and control visceral (VL) and cutaneous (CL) leishmaniases, as there is no vaccine approved for human use [
1]. Pentavalent antimony (Sb(V)) complexes, despite their use for more than 70 years, are still used as first-choice drugs in several developing countries for treatment of all forms of leishmaniases. These include sodium stibogluconate (Pentostam
®) and meglumine antimoniate (Glucantime
®, Sanofi-Aventis Farmacêutica Ltda, São Paulo, Brazil). Pentavalent antimonials are prodrugs in which the metal is reduced to the active and toxic trivalent form [
2]. Unfortunately, the use of conventional antimonial drugs is limited by the need for parenteral administration through repeated doses for a long period of time, by severe toxicities, and the development of drug resistance [
2]. The few other drugs available, including amphotericin B (AmB) and miltefosine (Milt), also present drawbacks, mainly toxicities and emergence of resistance. Thus, there is a great need for new drugs and novel delivery strategies to improve existing drugs [
3].
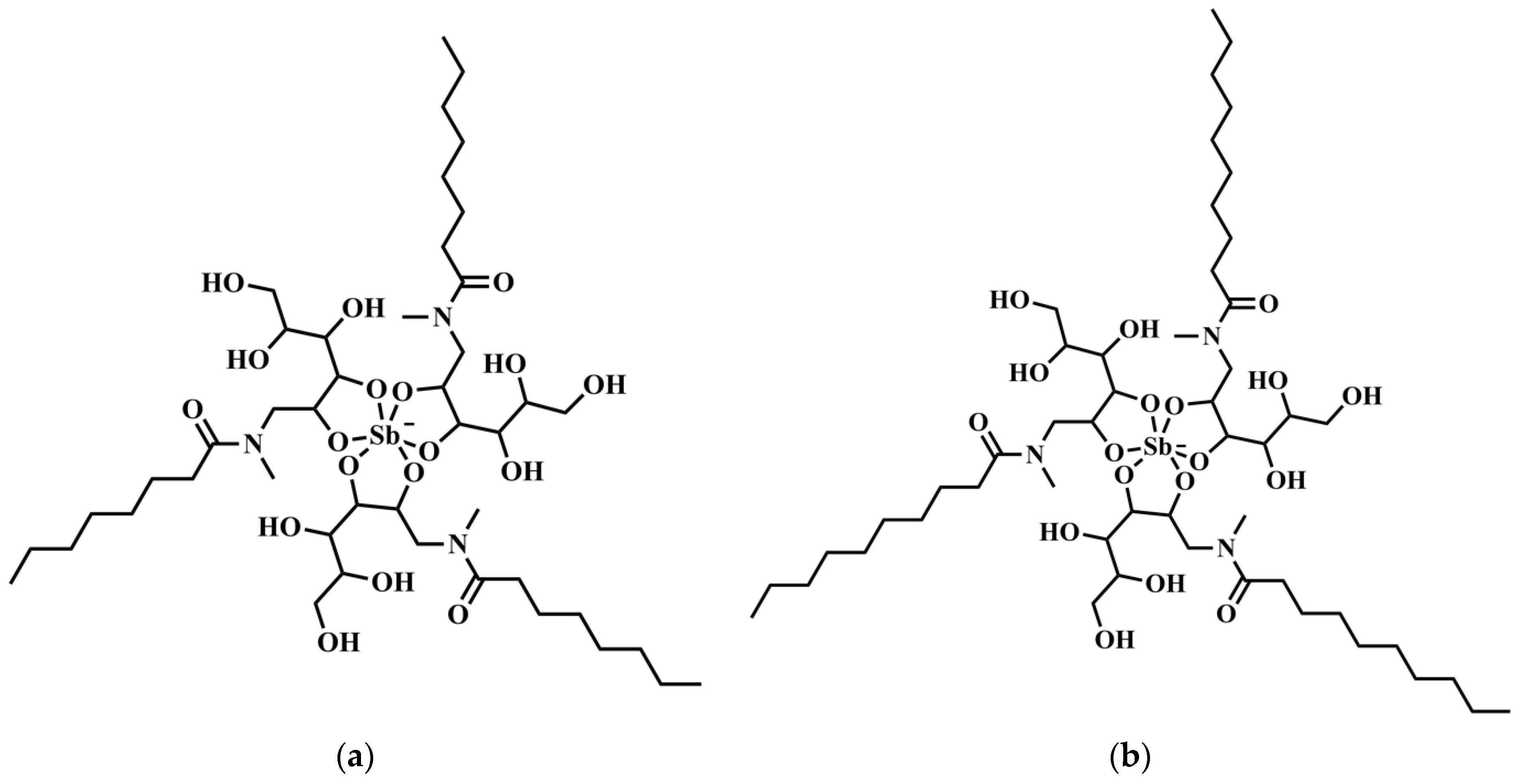
Amphiphilic Sb(V) complexes were first introduced by our group to achieve orally effective pentavalent antimonials [
4]. Two complexes, named SbL8 and SbL10, were obtained through reaction of KSb(OH)
6 with the non-ionic surfactants,
N-octanoyl-
N-methylglucamide (L8) and
N-decanoyl-
N-methylglucamine (L10), at a molar ratio of 1:3, respectively. A pharmacokinetic study of SbL8 by oral route in mice showed greater and more sustained levels of Sb in serum and liver when compared with Glucantime
®, resulting in area under the curve (AUC) and mean residence time (MRT) about 4-fold greater [
4,
5]. SbL8 further demonstrated antileishmanial activities by the oral route in murine models of VL and CL caused by New World
Leishmania species [
4,
5,
6]. SbL8 self-assembles in aqueous solution, forming micelle-like nanoparticles capable of incorporating other lipophilic substances [
5,
6]. Nevertheless, the therapeutic potential of these nanosystems for carrying and delivering Sb(V) and other co-incorporated substances has not been fully investigated, mainly regarding their use by parenteral and topical routes. Indeed, nanoparticles have been extensively studied as drug carrier for treatment of VL by parenteral route, exploiting their natural tendencies to accumulate in the macrophages of the liver, spleen and bone marrow, which are the main infection sites [
3,
7]. Nanoassemblies are also promising in topical formulations for CL, due to improved drug solubility and penetration into the skin, when compared to conventional formulation [
8,
9].
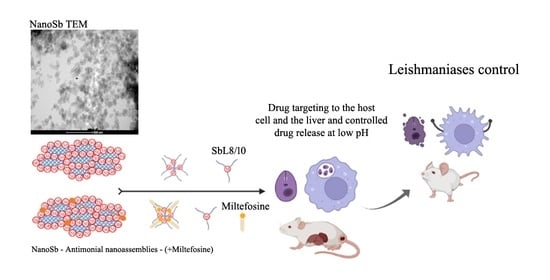
In the present work, we tested the hypothesis as to whether SbL8 and SbL10 nanoassemblies may favor drug targeting to the infection sites after parenteral and topical administrations. To achieve this goal, we evaluated the thermodynamic and kinetic stabilities of these nanoassemblies at different pHs and investigated their ability to promote Sb uptake by macrophages and the liver, as well as their in vitro antileishmanial activities and therapeutic efficacies in murine models of VL and CL.
2. Materials and Methods
2.1. Reagents, Culture Media and Drugs
M199, αMEM, RPMI-1640 culture media, adenosine, NaHCO
3, NaOH, HEPES, penicillin-streptomycin solution, dimethyl sulfoxide (DMSO) and phorbol 12-myristate 13-acetate (PMA) reagent grade, Giemsa stain,
N-octanoyl-
N-methylglucamide (L8, 98%),
N-decanoyl-
N-methylglucamide (L10, 98%), potassium hexahydroxoantimonate (KSb(OH)
6), potassium antimony(III) tartrate, 1,6-diphenylhexatriene (DPH), miltefosine (Milt) powder (≥98%) and amphotericin B (AmB) (European Pharmacopoeia (EP) Reference Standard) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Trypan blue was from Merck. Hemin was obtained from Honeywell International Inc. (Charlotte, North Carolina, USA) and fetal calf serum (FCS) was supplied by Invitrogen or Cultilab (Brazil) and inactivated by heating at 56 °C for 1 h (HIFCS). Propylene glycol (PG) was obtained from Labsynth (Diadema, Brazil). Double-distilled deionized water was used throughout all the experiments. Glucantime
® was from Sanofi-Aventis Farmacêutica Ltda (São Paulo, Brazil). The fluorescent analog of miltefosine (MT-11-BDP) (11-(4′,4′-difluoro-1′,3′,5′,7′-tetramethyl-4′-bora-3′a,4′a-diaza-s-indacen-2′-yl)undecylphosphocholine) was synthesized as described previously [
10].
2.2. Synthesis of Amphiphilic Antimony Complexes
The amphiphilic complexes SbL8 and SbL10 were synthesized, as previously described [
4]. Briefly, KSb(OH)
6 and L8 or L10 surfactant were co-dissolved in water at a 1:3 Sb/surfactant molar ratio and a final surfactant concentration of 0.08 M. The mixture was heated at 60 °C under agitation until complete solvent evaporation. The resulting film was dissolved in water at 25 °C, and the dispersion was finally freeze-dried.
2.3. Preparation of the Formulations for Biological Assays
To prepare the formulations for in vitro assays and parenteral administration, the freeze-dried compounds were first dispersed in 0.15 M NaCl solution at 175 mM Sb concentration. Incorporation of Milt into SbL10 suspension was performed as described previously, through simple addition of Milt to the SbL10 suspension and incubation for 1 h at room temperature [
6].
To prepare formulations for topical use, the freeze-dried compounds were dispersed in a 1:1 (v/v) water:PG mixture at 12% Sb (w/v) and hydroxyethyl cellulose (Natrosol™, Ashland, Catlettsburg, KY, USA) was dispersed in the resulting solution at 1% (w/v) final concentration.
2.4. Evaluation of Incorporation of Miltefosine Fluorescent Analog
MT-11-BDP was used as a fluorescent indicator of the incorporation of Milt in SbL10 nanoassemblies, as previously described [
5]. An aliquot of 10 mM ethanolic solution of the MT-11-BDP was added to the bottom of a tube and the solvent was evaporated. The dispersion of SbL10 or L10 in saline was then added to reach an Sb/MT-11-BDP molar ratio of 130:1, followed by incubation for 1 h at 25 °C. A saline solution of MT-11-BDP without surfactant was used as control. For fluorescence measurement, the solutions were diluted in PBS at an L8 concentration of 1 mM. Fluorescence measurements were carried out using the Cary Eclipse™ spectrofluorometer (Varian Inc., Australia) and a 1-cm cuvette compartment with temperature control and magnetic stirring. Fluorescence emission spectra were recorded at 25 °C, with excitation wavelength (λ) set at 500 nm.
2.5. Physicochemical Characterization of SbL8 and SbL10 Nanoassemblies
The formulations were characterized for particle size distribution (mean hydrodynamic diameter and polydispersity index) by dynamic light scattering (DLS) and zeta-potential, using the Zetasizer equipment (Nano ZS90; Malvern Instruments, Malvern, UK) and measurements at a fixed angle of 90°. Dispersion Technology Software, version 6.12, was used to collect and analyze the data obtained. SbL8 and SbL10 dispersions were diluted in 0.15 M NaCl solution at final concentration of 30 mM of Sb, and kept at 25 °C during the entire experiment. The particle size was also investigated using nanoparticle tracking analysis (NTA; Malvern Instruments, UK) and NTA 3.44 software (Malvern Panalytical, Malvern, UK) to collect and analyze data. Measurements were performed at a final Sb concentration of 1 mM in phosphate buffer saline (PBS, 0.15 M NaCl, 10 mM phosphate) at pH 7.2.
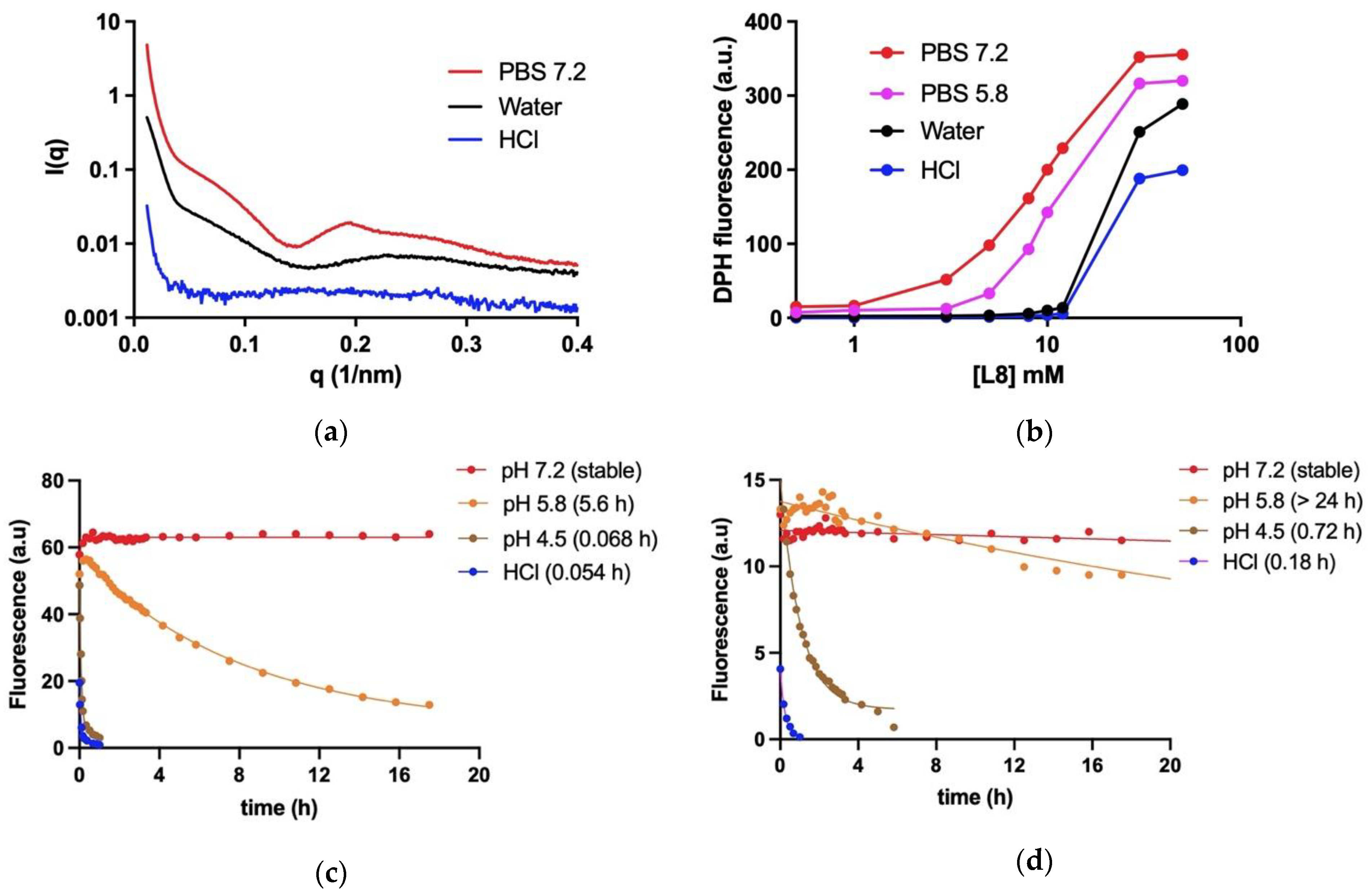
The formation of hydrophobic environment in aqueous dispersions of L8 and SbL8 was investigated using the lipophilic fluorescent probe DPH, as described previously [
4], exploiting the increase in DPH fluorescence upon incorporation into hydrophobic environment. To evaluate the dependence of DPH fluorescence on SbL8 concentration, DPH was added from a tetrahydrofuran stock solution at a final concentration of 5 × 10
−7 M to dispersions of SbL8 at various concentrations in either water (pH 5.5), HCl 0.05 M or PBS at pH 5.8 or 7.2. After 24-h incubation at 25 °C under light protection, the fluorescence intensity was measured at excitation and emission wavelengths of 360 and 428 nm, respectively. Fluorescence measurements were carried out using a Cary Eclipse™ fluorescence spectrometer. Temperature was controlled at 25 °C through a jacketed cuvette holder from a refrigerated circulating water bath.
To evaluate the kinetic stability of SbL8 nanoassemblies, a dispersion of SbL8 was prepared at 50 mM L8 in deionized water and incubated overnight with DPH at 10−5 M final concentration. The suspension was diluted 50 times in either HCl 0.05 M, PBS pH 7.2, 5.8 or 4.5 in quartz cuvette maintained at 37 °C under magnetic stirring. To evaluate the kinetic stability of SbL10 nanoassemblies, a dispersion of SbL10 was prepared at 50 mM of L10 in deionized water and further incubated overnight at 25 °C with 10−4 M DPH final concentration. The SbL10 suspension was then diluted at 0.1 mM L10 in either HCl 0.05 M, PBS 7.2, 5.8 or 4.5 in a quartz cuvette maintained at 37 °C under magnetic stirring. DPH fluorescence was then registered continuously as a function of time at excitation and emission wavelengths of 360 and 428 nm, respectively. The half-times of dissociation of the nanoassemblies were determined through non-linear regression fit according to mono-exponential decay model, using the GraphPad Prism software©, Version 9, GraphPad Software, LLC (San Diego, CA, USA).
Small-angle X-ray scattering (SAXS) studies were conducted in line 2 of LNLS—Brazilian Synchrotron Light Laboratory/MCT (Campinas, Sao Paulo, Brazil). This line is equipped with a monochromator (λ = 1.54 Å), an ionization chamber, and a 300 k Pilatus detector placed at 1 m from the sample to record the intensity of the scattering. The scattering from the samples was subtracted from that of the system without the sample. The intensities of all samples were measured in relative units, but for the purpose of comparison, the measurements were standardized under the same experimental conditions. A volume of 50 µL of SbL8 dispersions in either water, HCl 0.05 M or PBS at pH 7.2 was injected into the sample compartment maintained at 25 °C. The I(q) vs. q curves were obtained after subtraction of the corresponding blank from the signal of each sample.
Transmission electron microscopy (TEM) was also used to characterize the morphology of SbL8 nanoassemblies. A solution of SbL8 at 25 mM was prepared in PBS 7.2. The sample was deposited on formvar film (previously deionized copper grid) and treated with osmium tetroxide (OsO4) as contrast agent. The images were obtained using a Tecnai G2-Spirit-FEI-2006 (120 kV) microscope (FEI Company, Hillsboro, OR, USA), equipped with high resolution (11 megapixels) and high-speed digital cameras, located at the Electron Microscopy Center of UFMG.
2.6. Antimony Intracellular Accumulation
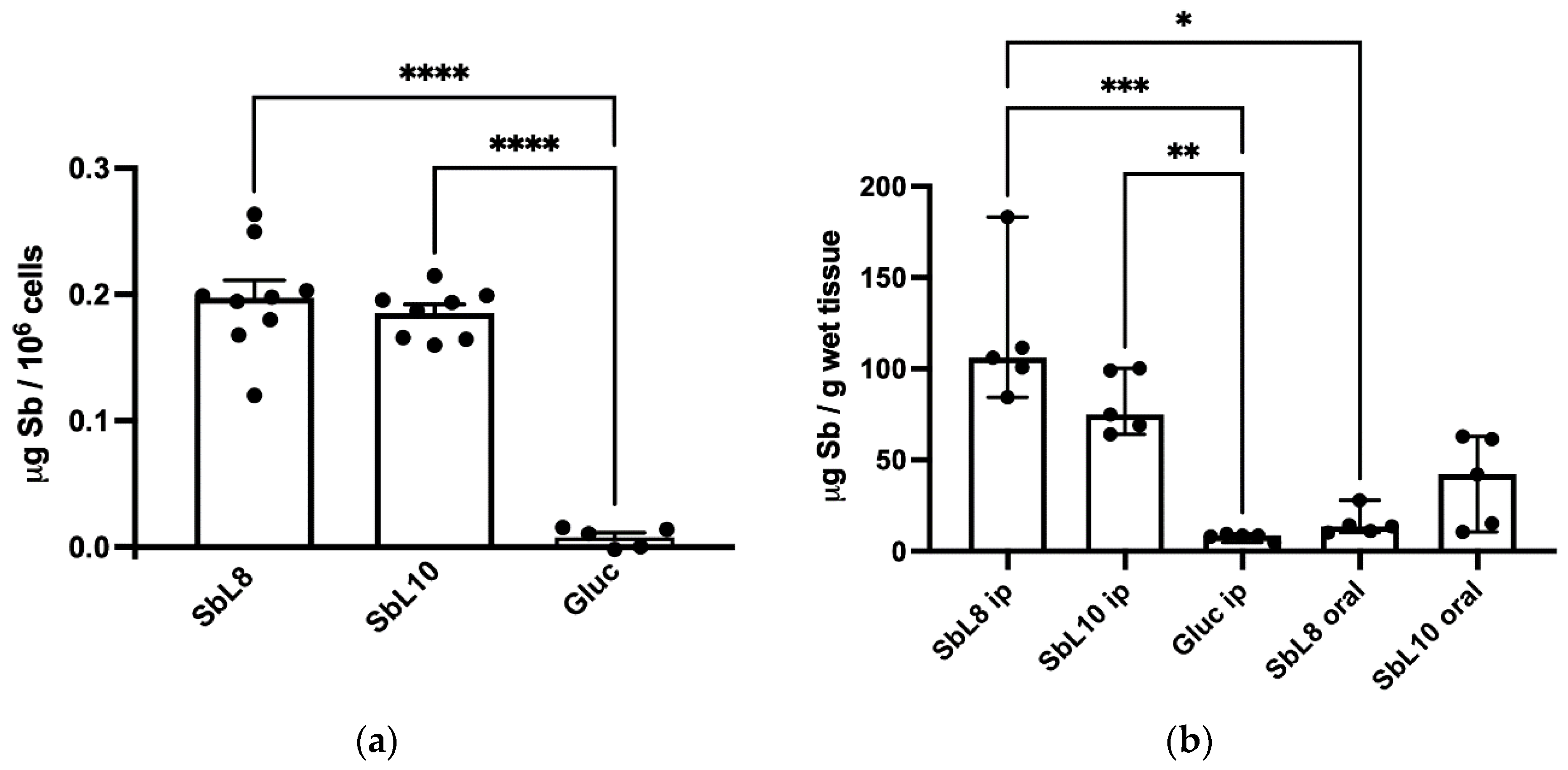
The intracellular accumulation of Sb was evaluated in THP-1 acute monocytic human leukemia cell line (ATCC TIB-202) using 6-wells plates. In each well, 2 × 10
6 THP-1 cells were differentiated into macrophage-like, adherent and non-dividing phenotype, via treatment with 100 ng/mL PMA for 72 h in complete RPMI-1640 medium. Cells were then exposed for 4 h at 37 °C to the antimonial drugs (Glucantime
®, SbL8 and SbL10) at 50 µg Sb/mL in RPMI-1640 medium complemented with 10% HIFCS (
n = 6–9, for each antimonial drug). Adherent cells were then washed 4 times with 2 mL of ice-cold PBS and digested overnight with 65% nitric acid at room temperature. The digested samples were then diluted 50-fold with 0.2% nitric acid and Sb concentration was determined by graphite furnace atomic absorption spectrometry (GFAAS), using a Perkin–Elmer AA600 graphite furnace atomic absorption spectrometer, as described previously [
11]. Cells without drug exposure were used for blank subtraction.
2.7. Hepatic Accumulation of Antimony
BALB/c mice (female, 6 to 8 weeks old, 18 to 20 g) were obtained from the Animal Facility Center of the Federal University of Minas Gerais (UFMG). Free access to a standard diet was allowed, and tap water was supplied ad libitum. The animals were handled according to the protocol approved by the Ethical Committee for Animal Experimentation of the UFMG (protocol no. 163/2019). Five groups of BALB/c mice (
n = 5) were used for the biodistribution study of Sb. They received a single dose of each compound either by i.p. or oral route and antimony level was determined in the liver 24 h after administration. The compounds were Glucantime
® (200 mg Sb/kg, i.p. route), SbL8 or SbL10 (20 mg Sb/kg, i.p. route), or SbL8 and SbL10 (200 mg Sb/kg, oral route). Animal euthanasia was undertaken by cervical dislocation after ketamine-xylazine anesthesia. The livers were recovered, homogenized, and subjected to digestion with nitric acid in a dry block (MA 4004; Marconi, São Paulo, Brazil). Antimony concentration was determined in digested liver by GFAAS using a Perkin–Elmer AA600, as described previously [
11]. The analytical method for determination of Sb in the liver was validated and showed suitable levels of precision (coefficient of variation [CV] < 5%), accuracy (80 to 120% analyte recovery), and linearity (range of 10 to 180 µg of Sb/L). The quantification limit of the analytical method was 0.93 µg of Sb/g for the liver.
2.8. In Vitro Cytotoxicity and Antileishmanial Activity
RAW264.7 (mouse leukemic monocyte macrophage) cell line from the European Collection of Cell Cultures (UK) was used for the cytotoxicity assay and for Leishmania infection to determine activity against intracellular amastigotes. RAW264.7 cell were kept in 75-cm2 sterile flasks in a total volume of 20 mL of RPMI-1640 medium complemented with 10% HIFCS, at 37 °C in 5% CO2 atmosphere incubator. Cells were passed every 4 days (log phase) at 100,000 cells per mL of inoculum.
Promastigotes forms of
Leishmania (L.)
donovani (MHOM/ET/67/HU3, also called LV9) were cultivated in M199 medium plus 10 mM of HEPES pH 7 supplemented with adenosine 100 µM and hemin at 0.5 mg/L, 10% FCS, 100 I.U./mL penicillin and 100 µg/mL streptomycin, pH 7, as adapted from a previous study [
12]. Cells were passed in fresh medium every 4 days (log phase) at 200,000 parasites per mL in a final volume of 5 mL kept into 25 cm
2 bottles at 27 °C (+/− 2 °C) incubator. A maximum number of 10 passages from primary culture of promastigotes isolated from spleen of experimentally infected rodents were used for all experiments.
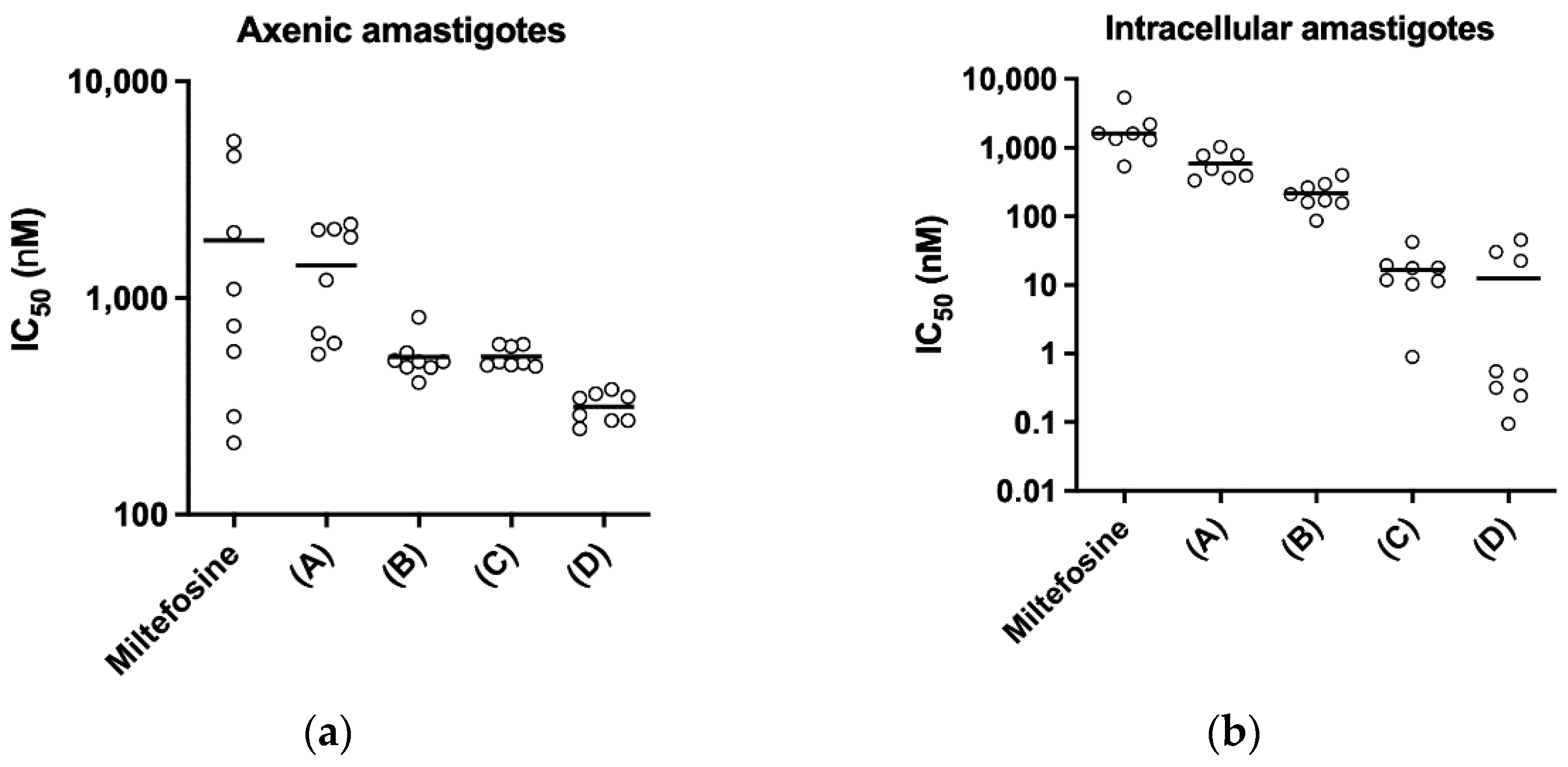
For cytotoxicity test, RAW264.7 cells were resuspended in 20 mL of fresh medium and counted with Trypan blue at 0.4% (w/v) in Neubauer chamber. Cell suspension was adjusted to 100,000 cells/mL and aliquoted into a sterile 96-well plate (200 µL/well). Plates were incubated for 24 h at 37 °C, 5% CO2. Afterwards, supernatants were discarded and 100 µL of new medium containing serial dilutions of SbL8, SbL10, free ligands L8 and L10, Glucantime®, AmB, Milt, potassium antimony(III) tartrate were distributed in triplicate for each concentration. Plates were incubated for 24 h in the same conditions as described above. After this period, plates were centrifuged, supernatants were removed and 100 µL of new medium containing 100 µM resazurin, were added to each well. Plates were further incubated for 4 h and fluorescence intensities (FI) were read with Spark® Tecan Spectrometer (Männedorf, Switzerland, EU) at 530–570 nm excitation and 585–590 nm emission wavelength. CC50 (cytotoxic concentration for 50% of cells) were calculated for each drug, taken DMSO at 10% (v/v) and RPMI-1640 medium supplemented with 10% HIFCS as negative (0%) and positive (100%) controls for viability, respectively.
Axenic amastigote-like forms were obtained and used as previously described [
13]. Briefly, stationary phase cultures of LV9 were centrifuged at 500×
g for 10 min, and parasite pellet was resuspended into fresh complete M199 containing 200 µM of Ca
2+ and Mg
2+ at pH 6. Cells were incubated at 37 °C in 5% CO
2 atmosphere for 72 h before drug screening. After incubation, plates were centrifuged, and supernatant was discarded and substituted by serial dilutions of aforementioned drugs (100 µL/well). After 24 h of incubation in the same above conditions, resazurin solution was added at final concentration of 100 µM and plates were incubated for additional 24 h. Fluorescence was read as above and IC
50 (inhibitory concentration of 50%) were determined for each drug, taken as 100% and 0% viability the values measured for untreated and 10% DMSO-treated amastigote-like, respectively.
The assay on intracellular amastigotes was performed as described previously [
13,
14]. Briefly, a total of 20.000 RAW264.7 cells in 200 µL of fresh complete RPMI plus 10% HIFCS, cultivated as described above, were added to each well in a Nunc™ Lab-Tek™ Chamber Slide System (Thermo Fisher Scientific™, Waltham, MA, USA) and incubated at 37 °C in 5% CO
2 for 24 h before
Leishmania infection. Axenic LV9 amastigote-like forms (obtained as described above) were added at 10:1 parasites/RAW264.7 cell ratio in fresh complete RPMI 10% HIFCS and incubated for 24 h in the same conditions to allow parasite infection. After this period, wells were washed twice and fresh medium containing serial dilutions of test drugs was added (100 µL/well). Plates were incubated for additional 24 h. Wells were then washed three times with PBS, fixed with ice-cold methanol (>99%) for 2 min, stained for 5–10 min with 10% aqueous solution of Giemsa stain, washed in water and air-dried for further microscope analysis. At least 300 macrophages were counted per well, regardless of whether those were infected or not. Only assays in which the percentage of infected macrophages was higher than 80% were considered. The infection index was defined as II = (% infection rate × amastigotes/infected macrophage)/100. The IC
50 in this case is the drug concentration in which the II corresponds to 50%, using the non-treated control as 100% [
14].
Both IC50 and CC50 values in all experiments were calculated in GraphPad Prism® 9.2.0 software through non-linear regression of log(inhibitor) vs. response (variable slope). Only results in each Hillslope was around −3 and R Square was >0.95 were considered. Results are from a representative experiment out of at least three independent experiments.
2.9. Chequerboard Assay
In order to explore possible pharmacological interactions (synergism, additive effect or antagonism) between the clinically used leishmanicidal compounds Milt and the novel amphiphilic Sb complex SbL10 described in this work, a chequerboard assay was performed. After determining the IC
50 of each compound itself in LV9-infected macrophages, as described above the top concentration for each compound, set as 4 × IC
50 (Milt or SbL10)/5 were calculated and different combinations of Milt in SbL10 aqueous solution were prepared based on subsequent variations of each top concentration at: (A) 0.2:0.8; (B) 0.4:0.6; (C) 0.6:0.4 and (D) 0.8:0.2 ratios before serial dilutions in a two-dimensional concentration array, as described and adapted from previous studies [
12,
15,
16,
17]. Serial dilutions of each compound alone were also tested. RAW264.7 were plated in a 96-well plate and infected with amastigote-like forms of LV9, as described in the previous topic. A total of 24 h after infection, washing and cell-resting, 100 µL of drug solutions were distributed per well and incubated 48 h at 37 °C under 5% CO
2 atmosphere. At the end of the incubation, cells were lysed and DNA was extracted by adding 50 µL of DirectPCR
® Lysis Reagent (Viagen Biotech Inc, Eurogentec, France) with Proteinase K (1 mg/mL), and plates were further incubated for 2 h at 4 °C, according to manufacturers’ information. Parasite load in cells was determined using SYBR Green I (Invitrogen, France) assay, as described previously [
12] in which the fluorescence dye is shown to be enhanced upon parasite DNA coupling. A total of 40 µL of lysis buffer containing 0.05% of SYBR
® Green (5 μL of SYBR Green I/10 mL probe) and 10 µL of cell lysate were used per reaction. Untreated, non-infected and infected macrophages were used as controls. The Mastercycler
® ep realplex real-time PCR machine (Eppendorf, France) was used to directly evaluate the fluorescence, after the application of the following program: 90 °C for 1 min and ramp time of 5 min until 10 °C, fluorescence obtained in continue and in a hold step at 10 °C. The fluorescence values at 10 °C were analyzed to determine the IC
50 for each drug and its combination. The IC
50 for each association was then used to calculate the fractional inhibitory concentration (FICs) which corresponds to IC
50 of molecule A when mixed with molecule B/IC
50 of molecule A alone [
15].
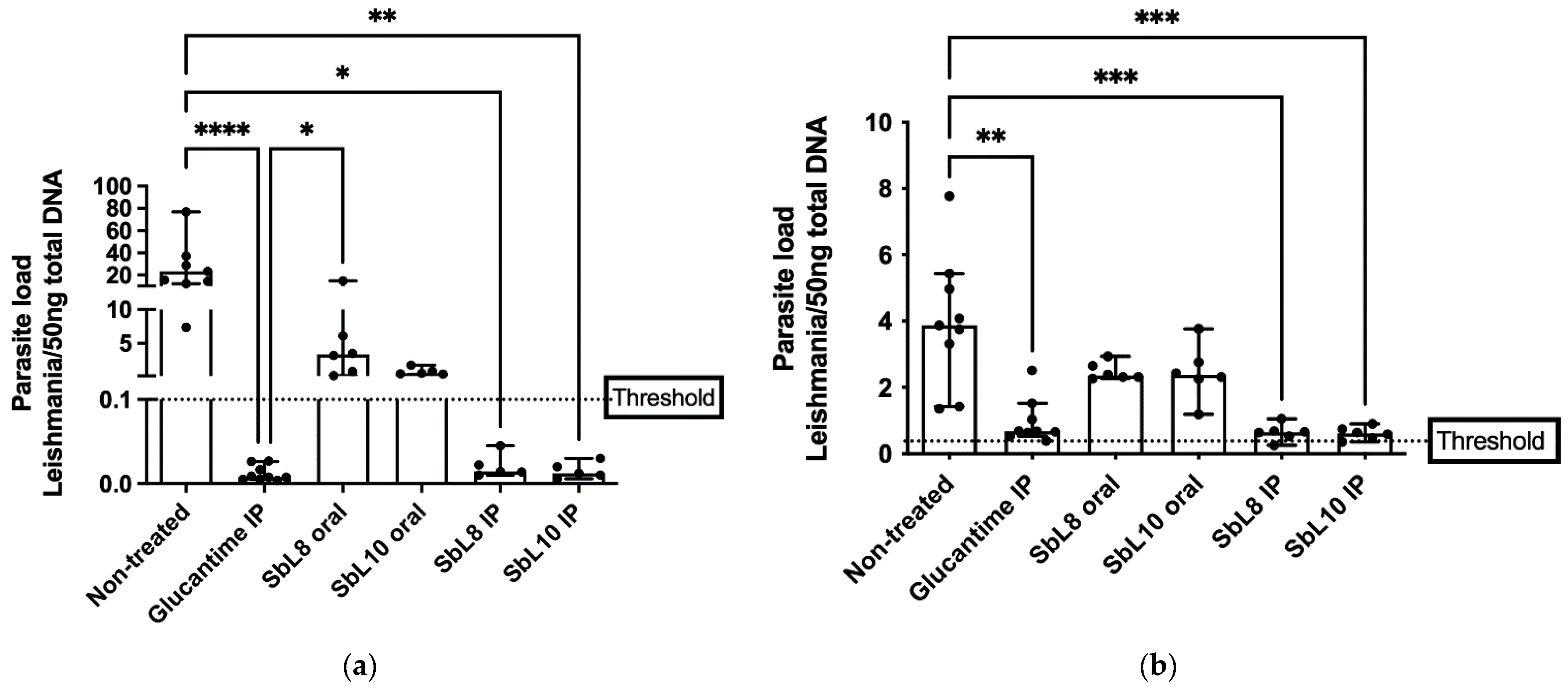
2.10. Antileishmanial Chemotherapy in Murine VL Model
The in vivo experiment and animal handle described here were carried out according to European Directive 2010/63/EU, the article R214.89 of the French Animal Welfare Law n˚2013–1118. The specific protocols for in vivo testing of antileishmanial potential drugs were approved by the Paris-Saclay University’s institutional ethic committee for animals handling, license CEEA 26-063/2013 and followed previously described studies [
18,
19]. Golden hamsters (
Mesocricetus auratus) previously infected with
L. donovani (LV9) were euthanized and spleens were processed to obtain viable amastigotes for mice infection. BALB/c mice (Janvier Labs, France), females, aged from 6 to 8 weeks were then infected with an inoculum of 5 × 10
6 parasites, by retro-orbital intravenous injection. One week after infection, mice received different treatments per group (
n = 6–9) once a day for 30 days, in the following schemes: a. Untreated control; b. Glucantime
® (200 mg Sb/kg/day; i.p. route; c. SbL8 (20 mg Sb/kg/day; i.p.); d. SbL10 (20 mg Sb/kg/day; i.p.); e. SbL8 at 200 mg Sb/kg/day, by oral route, and f. SbL10 (200 mg Sb/kg/day; oral route). After the 30-days treatment, mice were euthanized, and their liver and spleen were analyzed for determination of parasite load by qPCR as described below (
Section 2.12).
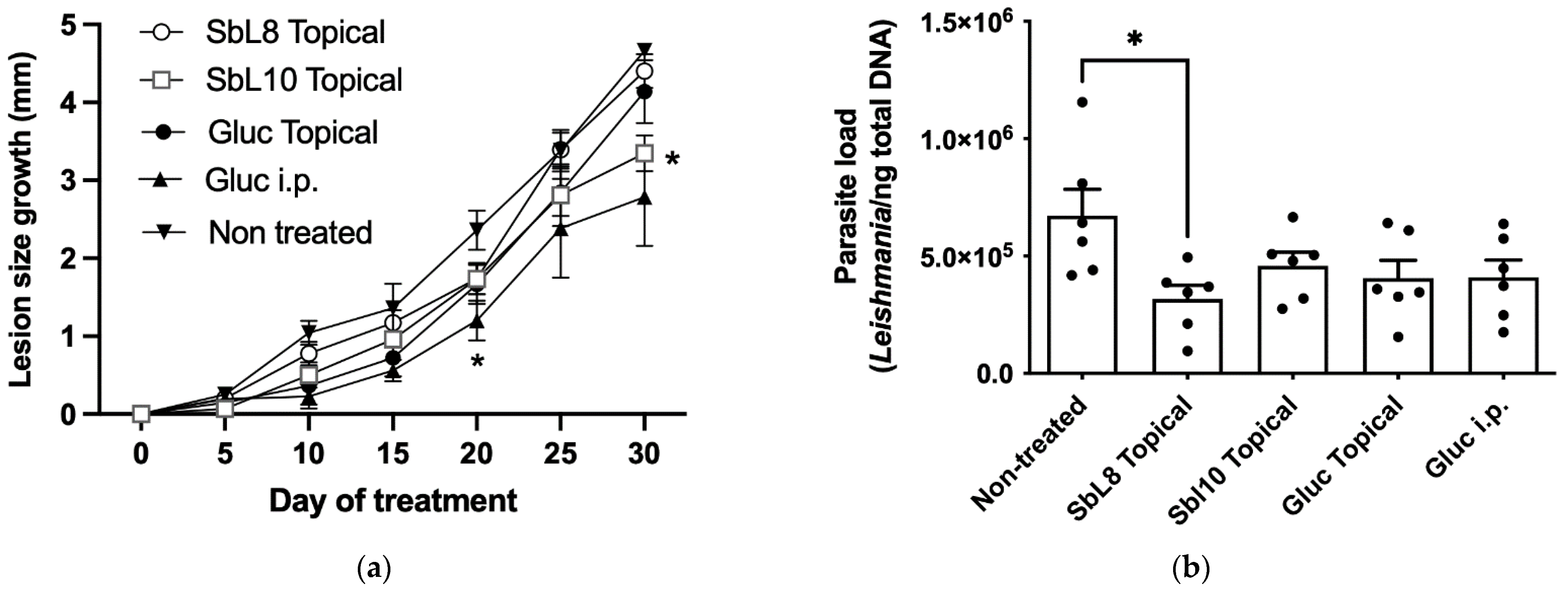
2.11. Antileishmanial Chemotherapy in Murine CL Model
The Leishmania strain used for cutaneous infection Leishmania (L.) amazonensis (MHOM/BR/1989/BA199) was obtained from the cryopreservation bank of the Leishmania Biology Laboratory at ICB, UFMG. Cells were maintained in vitro as promastigotes at 24 °C, pH 7.0, in αMEM supplemented with 10% HIFCS, 100 μg/mL kanamycin, and 50 μg/mL, ampicillin in BOD incubator. The promastigotes were grown in cell culture flasks of 25-mL with initial inoculum of 1 × 106 cells/mL and transferred to fresh medium, twice a week, once they reached the stationary growth phase.
BALB/c mice (female, 6 to 8 weeks old, 18 to 20 g) were obtained from the Animal Facility Center of the Federal University of Minas Gerais (UFMG). Free access to a standard diet was allowed, and tap water was supplied ad libitum. The animals were handled according to the protocol approved by the Ethical Committee for Animal Experimentation of the UFMG (protocol no. 54/2020). Mice were first inoculated intradermally at the tail base with 1 × 10
6 stationary phase promastigotes of
L. amazonensis. Chemotherapy was initiated 35 days post-infection, corresponding to the first ulceration sign of the infection papule, with daily doses for 30 days. Animals were divided in groups of 6 individuals receiving the following treatments: a. topical application of 50 µL of 1:1 water:PG hydrogel containing SbL8 at 12% Sb (
w/
v); b. topical application of 50 µL of 1:1 water:PG hydrogel containing SbL10 at 12% Sb (
w/
v); c. topical application on the lesion of 50 µL of 1:1 water:PG hydrogel containing Glucantime
® at 12% Sb (
w/
v); i.p. administration of Glucantime
® at 200 mg Sb/Kg/day; d. non-treated control group. After chemotherapy (65 days post-infection), animals were euthanized and the lesion was removed for evaluation of the parasitic load by qPCR, as described below (
Section 2.12).
2.12. Evaluation of Parasite Load by qPCR
Evaluation of anti-leishmanial activity in the VL and CL murine models were performed at the end of each chemotherapeutic protocol, as described previously [
5,
6]. Animals were submitted to euthanasia to collect and analyze organs (liver and spleen in VL; excision of the whole lesion in CL). Organs were weighed and homogenized in cold PBS (1 mL per 100 mg of tissue) with the aid of a tissue dissociator. One hundred microliters of each organ homogenate were added in a microtube containing lysis buffer and Proteinase K, vortexed and incubated at 56 °C overnight to extract genomic DNA according to manufacturers’ instructions, with the “Illustra tissue & cells genomic Prep Mini Spin Kit” (GE, Healthcare Lifesciences) or NucleoSpin
® Tissue Kit (MN, Macherey-Nagel GmbH & Co. KG, Dürin, Germany). Likewise, genomic DNA was extracted from 1 million axenic amastigote-like of
L. donovani (LV9) or
L. amazonensis promastigotes, and used for serial dilutions defining the standard curve as absolute quantitative control. The DNA concentrations were measured by spectrophotometry (Abs at 280/260 nm) and adjusted to 50 ng/μL per reaction. The final volume was 20 μL per reaction that included ultrapure water, SYBR Green PCR Master Mix (Warrington, UK), 10 pmol of each oligonucleotide, as sense (forward, 5′-CCTATTTTACACCAACCCCCAGT-3′) and antisense primers (reverse, 5′-GGGTAGGGGCGTTC TGCGAAA-3′) constructed for amplification of the mini-circle region present in the kinetoplast DNA (kDNA) of approximately 120 bp. Real-time qPCR was performed as described previously [
6,
18,
19]. The amplification protocol included an annealing temperature and extension of 60 °C, with melting curve construction, on the Applied Biosystems™7500 Fast Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA) and the analysis was made using the 7500 System Software. Parasite load was determined by absolute quantification based on the standard curve Ct vs. gDNA mass in pg. Results are shown as the parasite load that represents the number of
Leishmania per ng of total DNA.
Additionally, in the cutaneous infection model, lesion development was followed during the treatment and plot of the change in lesion size with respect to time zero was obtained as a function of time. As the lesions developed into a circular shape, the size was calculated from the means of the coronal and sagittal lesion diameters, using a universal calliper, 150 mm, Digimess (São Paulo, Brazil), every 5 days from the onset of the treatment.
2.13. Statistical Analyses
One-way ANOVA with Tukey’s post-test (for normally distributed data) or Kruskal–Wallis non-parametric test with Dunn’s post-test were used for statistical analyses of parasite load, with significance level p < 0.05. The normal distribution was checked with the following tests: Anderson-Darling test, D’Agostino & Pearson test and Shapiro–Wilk test. Two-way ANOVA (repeated measures) was used to compare the variation in lesion size between the experimental groups, followed by Dunnett’s post-test. p < 0.05, p < 0.01, p < 0.001 and p < 0.0001 were marked with *, **, *** and ****, respectively. The graphics and statistical analyses were performed using GraphPad Prism® (version 9) software (GraphPad Software LLC, San Diego, CA, USA).
4. Discussion
The present study provides new insights into the physicochemical properties and the therapeutic potential of SbL8 and SbL10 nanoassemblies for VL and CL. Previous studies addressed the potential of amphiphilic antimony complexes essentially for oral delivery of Sb [
4,
5,
6]. Here, we moved a step forward by investigating the potential of these nanosystems to carry and deliver Sb(V) and other associated lipophilic substance for parenteral and topical applications.
Fluorescence probing of hydrophobic environment and SAXS measurements demonstrated for the first time the stability of nanoassemblies at neutral pH in aqueous solution, but also the conformational change and release of incorporated lipophilic marker upon medium acidification at pH values close to that of gastric fluid and parasitophorous vacuole. The conformational change as a function of pH is supported by modifications in the SAXS curve profile and DPH partitioning. However, the precise morphological changes in the nanoparticle require further characterization, as we were unable to detect significant difference by TEM between PBS (pH 7.2) (
Figure S6) and water [
5]. The pH dependence of the stability of the nanoassemblies may be attributed to the acid–base properties of Sb(V). Acidification of the medium is expected to favor protonation of the hydroxyl group of Sb atom, resulting in conformational change of the complexes and their nanoassemblies. Such a protonation is supported by the reduction of the nanoparticle zeta-potential after acidification of the medium (
Table S1). These data are also consistent with previous reports that 1:2 Sb(V)-GMP complex is stable at neutral pH but rapidly dissociates at pH 5 [
21]. Interestingly, SbL10 nanoassemblies were found to be more stable than SbL8 ones. This may be attributed to the longer acyl chain of L10 with an enhancement of hydrophobic interactions.
The pH dependence of the stability of these nanoassemblies has important implications for therapeutics. First, it strongly suggests that SbL8 and SbL10 nanosystems release incorporated lipophilic substance in the stomach and are therefore unable to act as a drug carrier by the oral route. On the other hand, these nanoassemblies may be able to carry a lipophilic drug in the bloodstream after parenteral administration, which further release Sb(V) and the co-incorporated drug into the acidic parasitophorous vacuole following phagocytosis. The smaller size of SbL10 nanosystems and their greater stability may afford a better accumulation into the parasite through endocytosis via flagellar pocket. The higher activity of SbL10 against the intramacrophage amastigotes in comparison to axenic amastigotes and the increased activity of Milt after incorporation in SbL10 nanoassemblies (at 2:3 ratio and below) support the specific drug release in the parasitophorous vacuole, rather than nanoparticle endocytosis by the parasite.
The improved targeting of Sb to the host cell (in vitro) and the liver (in vivo) from SbL8 and SbL10 are major findings of the present study. The high intracellular accumulation of Sb can be explained by the amphiphilic character of the complexes (
Figure 1) and their self-assembling, in contrast to the marked hydrophilicity of meglumine antimoniate, which may facilitate binding of the nanoassemblies onto the cell surface and their subsequent internalization via phagocytosis. The hydrophobic interaction of the acyl tails of amphiphilic complexes with the lipid membrane may contribute to cell surface binding. Alternatively, the negatively charged polar head of the complexes may interact with receptors on the macrophage surface leading to phagocytosis of the nanosystem. Scavenger receptors are candidates for interaction with amphiphilic Sb complexes as they are known to bind to a range of polyanionic molecules such as phosphatidylserine [
22]. Our results are also consistent with previous reports that incorporation of meglumine antimoniate into chitosan-based nanoparticles or liposomes resulted in significantly higher Sb uptake by macrophages and antileishmanial activity, in comparison to the free drug [
23,
24]. Opsonization is another well-known process that contributes in vivo to rapid clearance of nanoparticulate carriers by macrophages of the mononuclear phagocytic system (MPS) [
25]. It consists of the adsorption onto the surface of nanoparticles of opsonins, such as immunoglobulins and complement proteins such as C3, C4, and C5 [
26]. Such phenomenon is also expected to contribute to the ability of SbL8 and SbL10 nanosystems to target the liver. Thus, our data supports a scenario in which these Sb nanoassemblies passively target macrophages and organs of the MPS, resulting in higher antileishmanial activity achieved at lower Sb dose in the VL model.
The current work uncovers for the first time the therapeutic potential of SbL8/SbL10 nanoassemblies administered by parenteral route leading to the enhancement of their leishmanicidal effectiveness in comparison to the oral route. Interestingly, it is also the first report on efficacy of amphiphilic Sb(V) complexes on the murine VL model caused by L. donovani. The lower cytotoxicity and higher selectivity index of SbL10, regarding the SbL8, suggest a greater potential of SbL10 for treatment of VL. Therapeutic effectiveness was also achieved at lower Sb dose, as compared with the conventional pentavalent antimonials in humans (20 mg Sb/kg/day) equivalent to 240 mg Sb/kg/day in mice. The higher in vitro antileishmanial activity of SbL10 compared to SbL8 in the intramacrophage amastigote model did not translate into significant difference in in vivo activity. As a possible explanation for this apparent discrepancy, the interaction of nanoassemblies with serum components may alter their surface or structural organization and affect their intracellular processing, resulting in either increased delivery of Sb to the parasite from SbL8 or decreased drug delivery from SbL10.
As a main drawback, conventional antimonial therapy is often accompanied by local pain during intramuscular injection and by severe side effects that include cardiotoxicity, pancreatitis, hepatotoxicity and nephrotoxicity [
2]. Although toxicity was not addressed in depth in our study, the high drug targeting to the liver and the lower Sb dose required for amphiphilic complexes by i.p. route may result in lower drug accumulation into the heart, pancreas and kidneys and reduced metal-related toxicities in these organs.
Another limitation of conventional antimonial therapy is the high risk of emergence of parasite resistance to antimony [
27]. Typical changes observed in Sb-resistant
Leishmania strains refer to the overexpression of ATP-binding cassette (ABC) transporters responsible for either Sb sequestration inside intracellular vesicles or active extrusion of Sb out of the cells [
28]. Down-regulation of the aquaglyceroporin (AQP1) responsible for Sb entry into the parasite has also been reported. As a potential therapeutic benefit of amphiphilic Sb complexes, to be investigated in future studies, the delivery of Sb into the parasite through endocytosis of nanoassemblies may bypass the transporters involved in Sb resistance and keep the cell sensitive to the drug.
Evidence was also obtained for the effectiveness of the topical application of SbL8/SbL10 according to the decrease of the size of the lesion for SbL10 and of the parasite load for SbL8. Nevertheless, the BALB/c mice have an extreme susceptibility to the
L. amazonensis species used in this study, with the development of increasing, ulcerative, metastatic and difficult-to-treat lesions, which may differ from the course of human CL. In humans, overexuberant cutaneous inflammation is observed in mucosal leishmaniasis which is often refractory to drug treatment [
29]. Thus, our experimental CL model may contribute to the moderate activity of the antimonial drugs either applied topically (SbL8, SbL10) or administered parenterally (Glucantime
®). The activity of the amphiphilic Sb complexes may also be limited by their high affinity for lipid membranes resulting in low skin penetration. Further improvement of the efficacy of topical treatment with these nanoformulations may be achieved by increasing the daily regimen into more usual twice-a-day application and by co-incorporating another active agent in the formulation.
The ability of SbL8 nanoassemblies to incorporate another amphiphilic drug, such as Milt or AmB, and the proposal of combination therapy have been described previously [
6]. The present study addresses a gap of knowledge by identifying the most appropriate routes of administration for the use of Sb nanoassemblies as carriers of other lipophilic drugs. Indeed, our work strongly supports the use of parenteral or topical route to guarantee the stability of the nanocarrier and enjoy the full benefit of its targeting properties. Here, we also show that incorporation of Milt in SbL10 suspension at 2:3 ratio (and below) results in highly active formulation in vitro, in the sub-micromolar range. Thus, it would be worth evaluating in future studies the in vivo antileishmanial activity of this specific drug combination by parenteral or topical route. The marked dependence of the pharmacological interaction between SbL10 and Milt upon the drug ratio can be attributed to a drug carrier effect, e.g., the incorporation of Milt in SbL10 micelles, which may predominate at low Milt/SbL10 ratio. Our proposal is that SbL10 nanoassemblies may act as carrier for Milt, therefore enhancing the drug uptake by the host cell or even the parasite. Future studies using the fluorescent Milt analog may help to confirm this model.
Macrophages can be activated to kill intracellular parasites, through increased pro-inflammatory mediator expression (IL-12, IFN-γ and TNF-α) and down-regulation of disease-promoting cytokines (IL-10 and TGF-β). Thus, the progression of the disease, its physiopathology and the effectiveness of drugs including antimonials strongly depend on the host immunologic status [
29]. In this context, immunomodulators have been investigated for their efficacy in the treatment of leishmaniasis, with the idea that they may also be used in combination with leishmanicidal drugs [
29,
30]. Previous studies highlighted therapeutic benefits in CL from the combination of pentavalent antimonials with CpG ODN D35, imiquimod or GM-CSF [
31,
32,
33]. Therefore, one can expect a more effective treatment of CL from the incorporation an immunomodulator in the amphiphilic Sb topical formulation.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}