
PKCeta Promotes Stress-Induced Autophagy and Senescence in Breast Cancer Cells, Presenting a Target for Therapy


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Antibodies and Reagents
2.3. Cell Lysis and Western Blot Analysis
2.4. SA-β-Galactosidase Staining
2.5. Statistical Analysis
3. Results
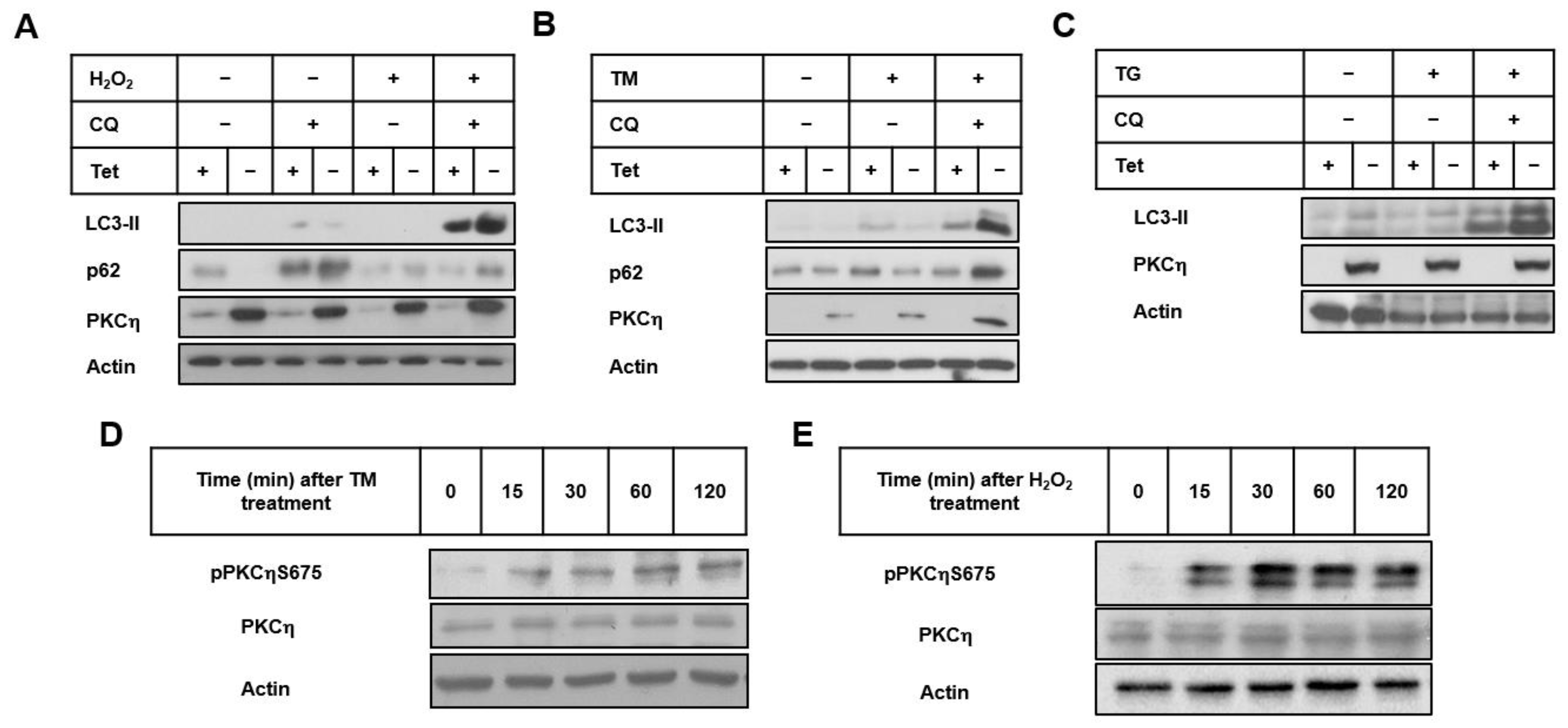
3.1. PKCη Enhances Autophagy Induced by ER and Oxidative Stress
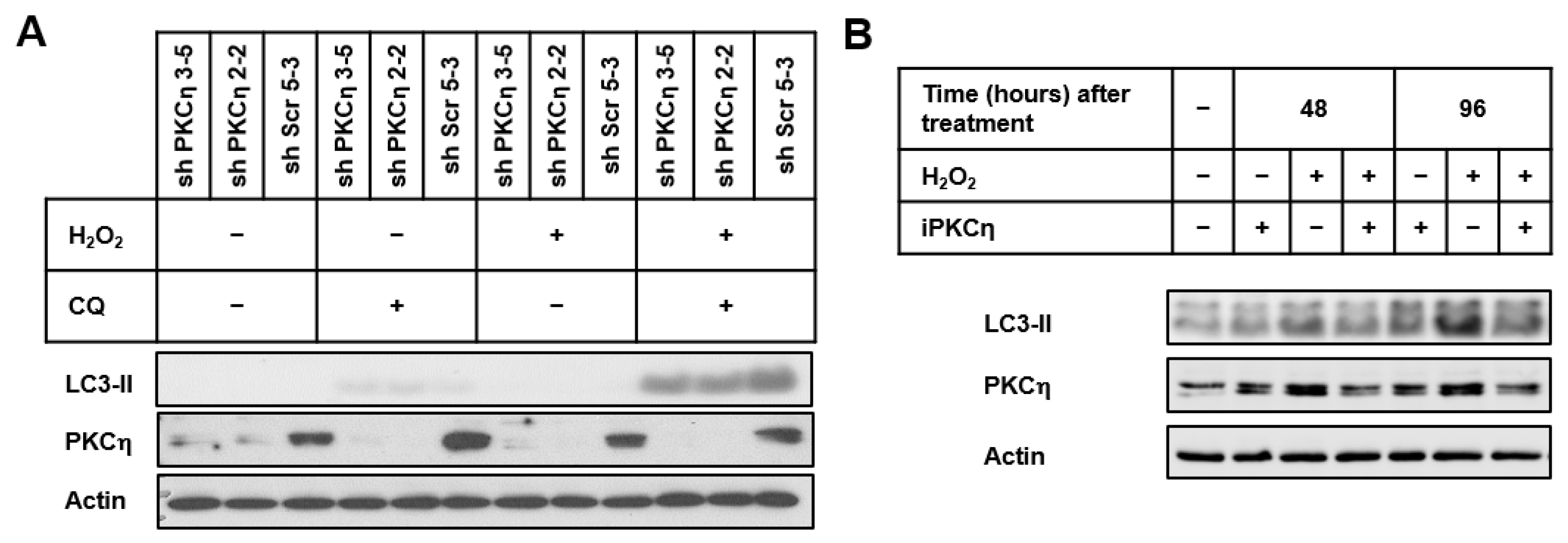
3.2. PKCη Knockdown Reduces Autophagy
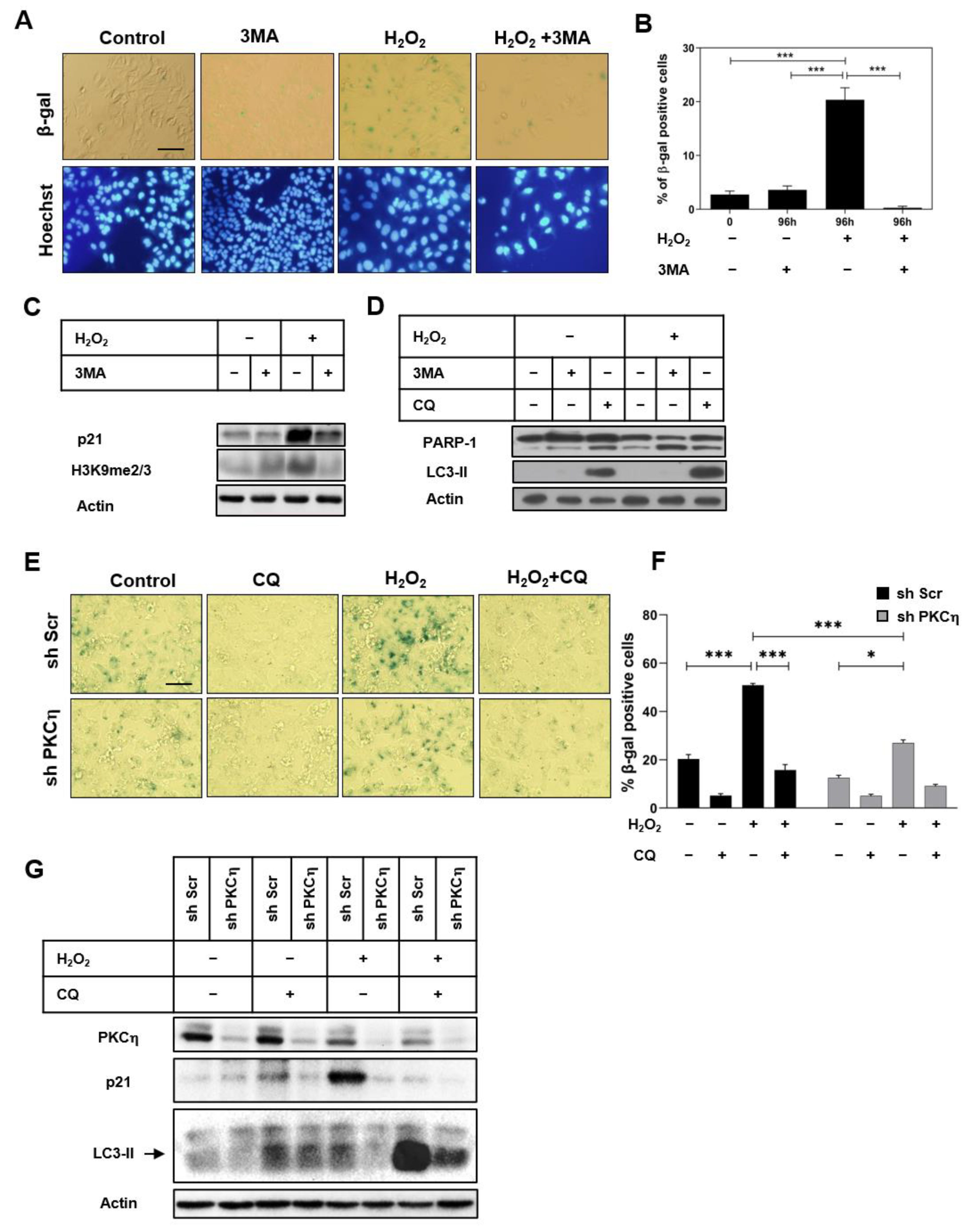
3.3. Inhibition of Autophagy Attenuates the Induction of Senescence by PKCη
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Perez-Hernandez, M.; Arias, A.; Martinez-Garcia, D.; Perez-Tomas, R.; Quesada, R.; Soto-Cerrato, V. Targeting Autophagy for Cancer Treatment and Tumor Chemosensitization. Cancers 2019, 11, 1599. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.R.; Neves, S.P.; Santos, L.S.; Dias, R.B.; Bezerra, D.P. Challenges and Therapeutic Opportunities of Autophagy in Cancer Therapy. Cancers 2020, 12, 3461. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. Autophagy and senescence in cancer therapy. J. Cell. Physiol. 2014, 229, 6–9. [Google Scholar] [CrossRef]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Goehe, R.W.; Di, X.; Sharma, K.; Bristol, M.L.; Henderson, S.C.; Valerie, K.; Rodier, F.; Davalos, A.R.; Gewirtz, D.A. The autophagy-senescence connection in chemotherapy: Must tumor cells (self) eat before they sleep? J. Pharmacol. Exp. Ther. 2012, 343, 763–778. [Google Scholar] [CrossRef]
- Zhang, J.W.; Zhang, S.S.; Song, J.R.; Sun, K.; Zong, C.; Zhao, Q.D.; Liu, W.T.; Li, R.; Wu, M.C.; Wei, L.X. Autophagy inhibition switches low-dose camptothecin-induced premature senescence to apoptosis in human colorectal cancer cells. Biochem. Pharmacol. 2014, 90, 265–275. [Google Scholar] [CrossRef]
- Chen, J.L.; Lin, H.H.; Kim, K.J.; Lin, A.; Forman, H.J.; Ann, D.K. Novel roles for protein kinase Cdelta-dependent signaling pathways in acute hypoxic stress-induced autophagy. J. Biol. Chem. 2008, 283, 34432–34444. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Lin, H.H.; Kim, K.J.; Lin, A.; Ou, J.H.; Ann, D.K. PKC delta signaling: A dual role in regulating hypoxic stress-induced autophagy and apoptosis. Autophagy 2009, 5, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Coward, J.; Ambrosini, G.; Musi, E.; Truman, J.; Haimovitz-Friedman, A.; Allegood, J.; Wang, E.; Merrill, A.J.; Schwartz, G. Safingol (L-threo-sphinganine) induces autophagy in solid tumor cells through inhibition of PKC and the PI3-kinase pathway. Autophagy 2009, 5, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, Y.; Tashiro, S.; Onodera, S.; Ikejima, T. Involvement of PKC signal pathways in oridonin-induced autophagy in HeLa cells: A protective mechanism against apoptosis. Biochem. Biophys. Res. Commun. 2009, 378, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, K.; Wu, J.; Kaufman, R.J. Protein kinase Cθ is required for autophagy in response to stress in the endoplasmic reticulum. J. Biol. Chem. 2008, 283, 15370–15380. [Google Scholar] [CrossRef]
- Zurgil, U.; Ben-Ari, A.; Atias, K.; Isakov, N.; Apte, R.; Livneh, E. PKCη promotes senescence induced by oxidative stress and chemotherapy. Cell Death Dis. 2014, 5, e1531. [Google Scholar] [CrossRef]
- Fima, E.; Shtutman, M.; Libros, P.; Missel, A.; Shahaf, G.; Kahana, G.; Livneh, E. PKCη enhances cell cycle progression, the expression of G1 cyclins and p21 in MCF-7 cells. Oncogene 2001, 20, 6794–6804. [Google Scholar] [CrossRef]
- Basu, A. The Enigmatic Protein Kinase C-eta. Cancers 2019, 11, 214. [Google Scholar] [CrossRef]
- Rotem-Dai, N.; Oberkovitz, G.; Abu-Ghanem, S.; Livneh, E. PKCη confers protection against apoptosis by inhibiting the pro-apoptotic JNK activity in MCF-7 cells. Exp. Cell Res. 2009, 315, 2616–2623. [Google Scholar] [CrossRef]
- Maissel, A.; Marom, M.; Shtutman, M.; Shahaf, G.; Livneh, E. PKCη is localized in the Golgi, ER and nuclear envelope and translocates to the nuclear envelope upon PMA activation and serum-starvation: C1b domain and the pseudosubstrate containing fragment target PKCη to the Golgi and the nuclear envelope. Cell. Signal. 2006, 18, 1127–1139. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Harhaji-Trajkovic, L.; Arsikin, K.; Kravic-Stevovic, T.; Petricevic, S.; Tovilovic, G.; Pantovic, A.; Zogovic, N.; Ristic, B.; Janjetovic, K.; Bumbasirevic, V.; et al. Chloroquine-mediated lysosomal dysfunction enhances the anticancer effect of nutrient deprivation. Pharm. Res. 2012, 29, 2249–2263. [Google Scholar] [CrossRef] [PubMed]
- Zurgil, U.; Ben-Ari, A.; Rotem-Dai, N.; Karp, G.; Krasnitsky, E.; Frost, S.A.; Livneh, E. PKCη is an anti-apoptotic kinase that predicts poor prognosis in breast and lung cancer. Biochem. Soc. Trans. 2014, 42, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Bjorkoy, G.; Lamark, T.; Pankiv, S.; Overvatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Antal, C.E.; Newton, A.C. Tuning the signalling output of protein kinase C. Biochem. Soc. Trans. 2014, 42, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Newton, A. Protein kinase C: Structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev. 2001, 101, 2353–2364. [Google Scholar] [CrossRef]
- Tamarkin, A.; Zurgil, U.; Braiman, A.; Hai, N.; Krasnitsky, E.; Maissel, A.; Ben-Ari, A.; Yankelovich, L.; Livneh, E. DNA damage targets PKCη to the nuclear membrane via its C1b domain. Exp. Cell Res. 2011, 317, 1465–1475. [Google Scholar] [CrossRef]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.; Meijer, A.J.; Codogno, P. Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar] [CrossRef]
- Mele, L.; Del Vecchio, V.; Liccardo, D.; Prisco, C.; Schwerdtfeger, M.; Robinson, N.; Desiderio, V.; Tirino, V.; Papaccio, G.; La Noce, M. The role of autophagy in resistance to targeted therapies. Cancer Treat. Rev. 2020, 88, 102043. [Google Scholar] [CrossRef]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef]
- Guo, L.; Xie, B.; Mao, Z. Autophagy in premature senescent cells is activated via AMPK pathway. Int. J. Mol. Sci. 2012, 13, 3563–3582. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zou, P.; Zou, J.; Wang, J.; Zhou, D.; Liu, L. Autophagy regulates ROS-induced cellular senescence via p21 in a p38 MAPKalpha dependent manner. Exp. Gerontol. 2011, 46, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Slobodnyuk, K.; Radic, N.; Ivanova, S.; Llado, A.; Trempolec, N.; Zorzano, A.; Nebreda, A.R. Autophagy-induced senescence is regulated by p38α signaling. Cell Death Dis. 2019, 10, 376. [Google Scholar] [CrossRef] [PubMed]
- Raveh-Amit, H.; Hai, N.; Rotem-Dai, N.; Shahaf, G.; Gopas, J.; Livneh, E. Protein kinase Cη activates NF-κB in response to camptothecin-induced DNA damage. Biochem. Biophys. Res. Commun. 2011, 412, 313–317. [Google Scholar] [CrossRef]
- Chang, H.; Zou, Z. Targeting autophagy to overcome drug resistance: Further developments. J. Hematol. Oncol. 2020, 13, 159. [Google Scholar] [CrossRef]
- Jayaram, D.R.; Frost, S.; Argov, C.; Liju, V.B.; Anto, N.P.; Muraleedharan, A.; Ben-Ari, A.; Sinay, R.; Smoly, I.; Novoplansky, O.; et al. Unraveling the hidden role of a uORF-encoded peptide as a kinase inhibitor of PKCs. Proc. Natl. Acad. Sci. USA 2021, 118, e2018899118. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rotem-Dai, N.; Muraleedharan, A.; Livneh, E. PKCeta Promotes Stress-Induced Autophagy and Senescence in Breast Cancer Cells, Presenting a Target for Therapy. Pharmaceutics 2022, 14, 1704. https://doi.org/10.3390/pharmaceutics14081704
Rotem-Dai N, Muraleedharan A, Livneh E. PKCeta Promotes Stress-Induced Autophagy and Senescence in Breast Cancer Cells, Presenting a Target for Therapy. Pharmaceutics. 2022; 14(8):1704. https://doi.org/10.3390/pharmaceutics14081704
Chicago/Turabian StyleRotem-Dai, Noa, Amitha Muraleedharan, and Etta Livneh. 2022. "PKCeta Promotes Stress-Induced Autophagy and Senescence in Breast Cancer Cells, Presenting a Target for Therapy" Pharmaceutics 14, no. 8: 1704. https://doi.org/10.3390/pharmaceutics14081704