
Tailoring of Rosuvastatin Calcium and Atenolol Bilayer Tablets for the Management of Hyperlipidemia Associated with Hypertension: A Preclinical Study

 ,
,  , , , ,
, , , ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of ROS Solid Dispersions (ROS-SDs)
2.3. Dissolution Study of the Prepared ROS-SDs
2.4. Differential Scanning Calorimetry Characterizations (DSC)
2.5. Analysis of Powder X-ray Diffraction (PXRD)
2.6. Fourier-Transform Infrared Spectroscopy (FT-IR) Characterizations
2.7. Preparation of Immediate-Release Layer (IRL) of ROS (ROS-IRL)
2.8. Preparation of AT Floating Sustained-Release Layer (SRL) (AT-SRL)
2.9. Regression Analysis
2.10. Pre-Compression Characterization of Different Prepared Tablets
2.10.1. Angle of Repose Study
2.10.2. Bulk Density Study
2.10.3. Tapped Density Study
2.10.4. Carr’s Index (CI) and Hausner’s Ratio (HR) Study
2.11. Post-Compression Tablet Evaluation
2.11.1. Drug Content Study
2.11.2. Tablet Weight Variation Study
2.11.3. Tablet Thickness Study
2.11.4. Tablet Friability and Hardness Study
2.11.5. In Vitro Buoyancy Study
2.11.6. In Vitro Drug Release Study
In Vitro Release of ROS-IRL Mono Tablets
In Vitro Release of AT-SRL Mono Tablets
In Vitro Drug Release Kinetics Study
2.12. Bilayer Tablets (BLTs) Compression Study
2.13. Accelerated Stability Study
2.14. In Vivo Assessment Study
2.14.1. Experimental Animals
2.14.2. Experimental Design
2.15. Statistical Analysis
3. Results and Discussion
3.1. Characterization of ROS-SDs
3.1.1. In Vitro Dissolution Study
3.1.2. Differential Scanning Calorimetry Characterizations
3.1.3. FT-IR Spectroscopy Characterizations
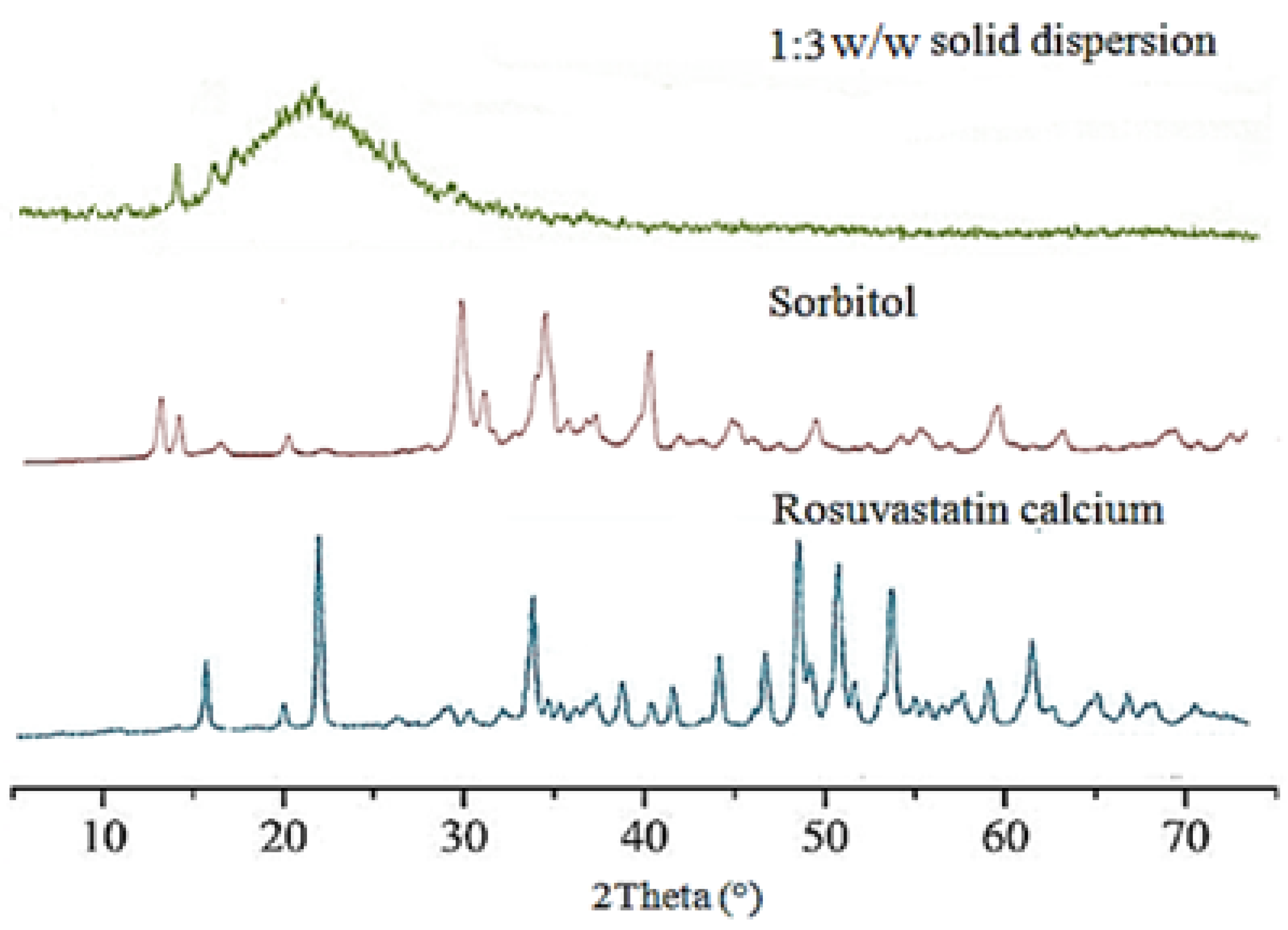
3.1.4. Powder X-ray Diffraction Analysis
3.2. Pre-Compression Characterization of Tablets
3.3. Characterization of the Prepared Mono Tablets
3.4. In Vitro Buoyancy Study
3.5. In Vitro Drug Release Studies
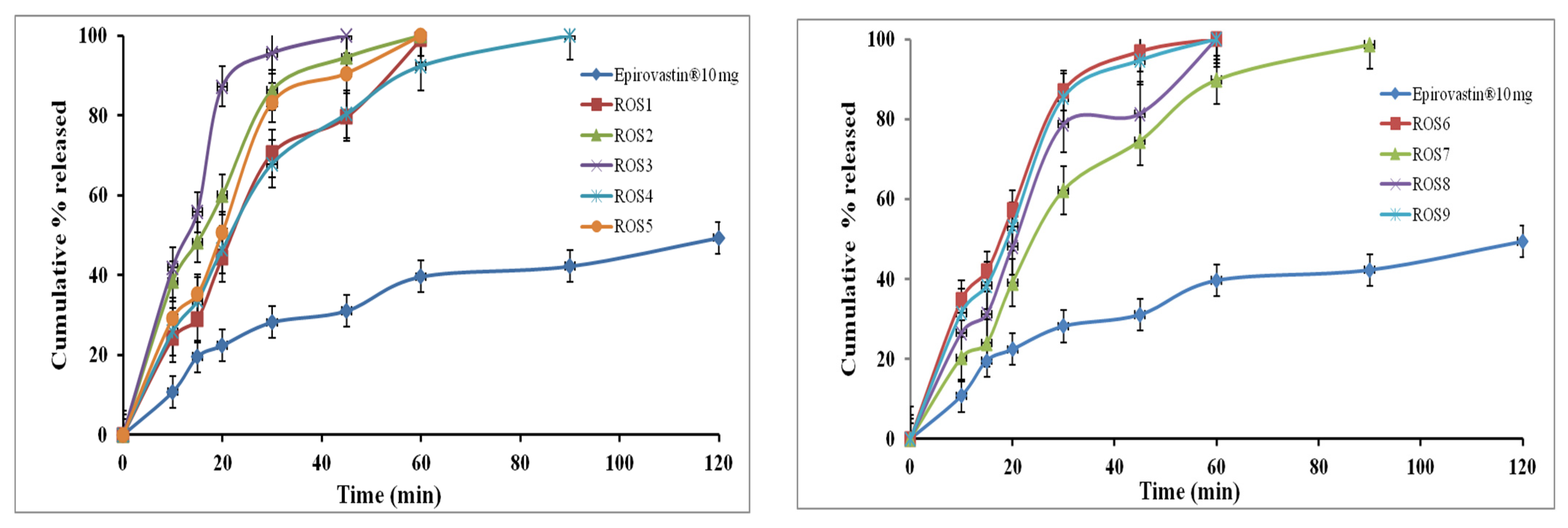
3.5.1. In Vitro ROS Release from IRL Mono Tablets
3.5.2. In Vitro AT Release from Floating SR Mono Tablets
3.6. In Vitro Drug Release Kinetic Modeling
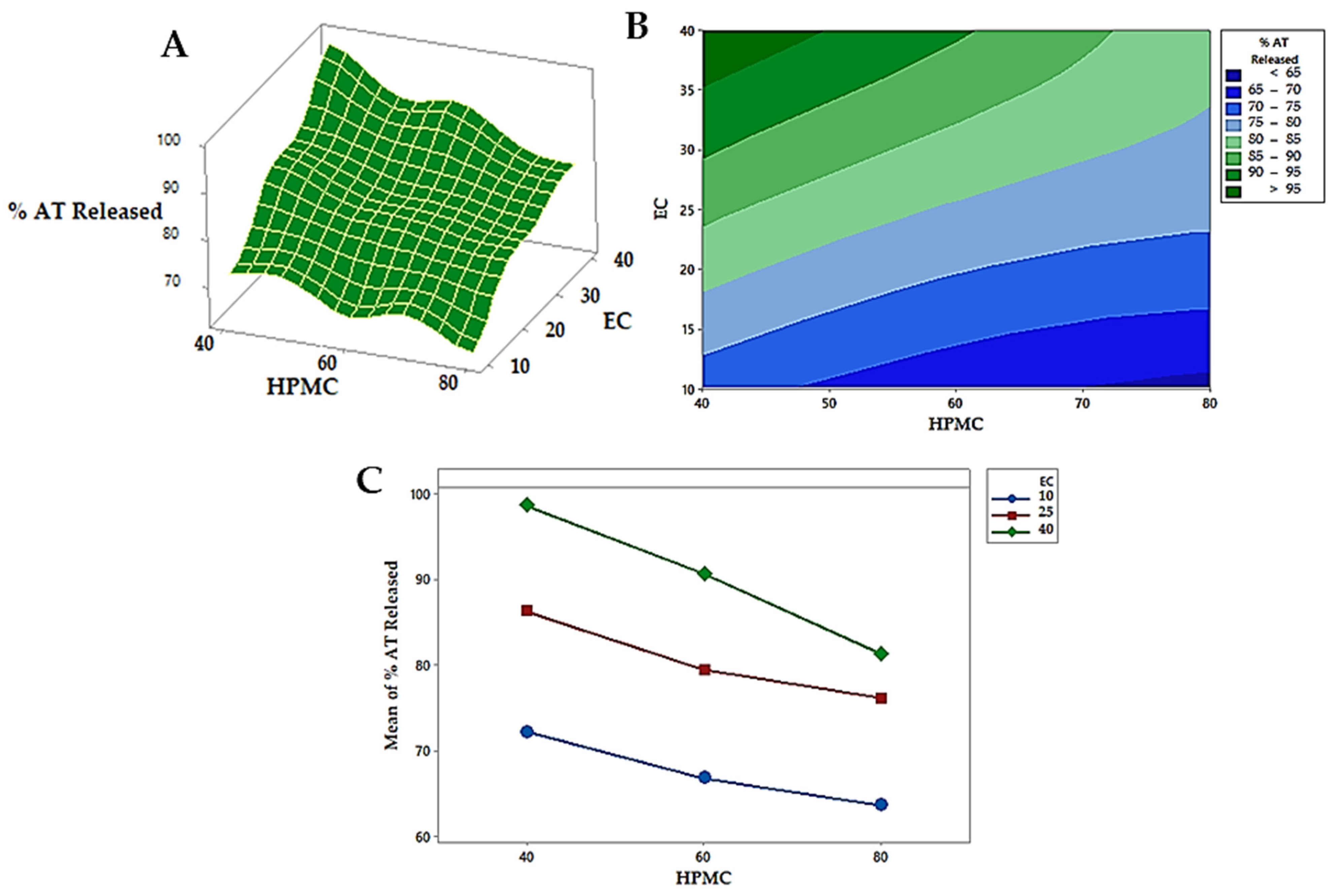
3.7. Formulation and Evaluation of BLTs
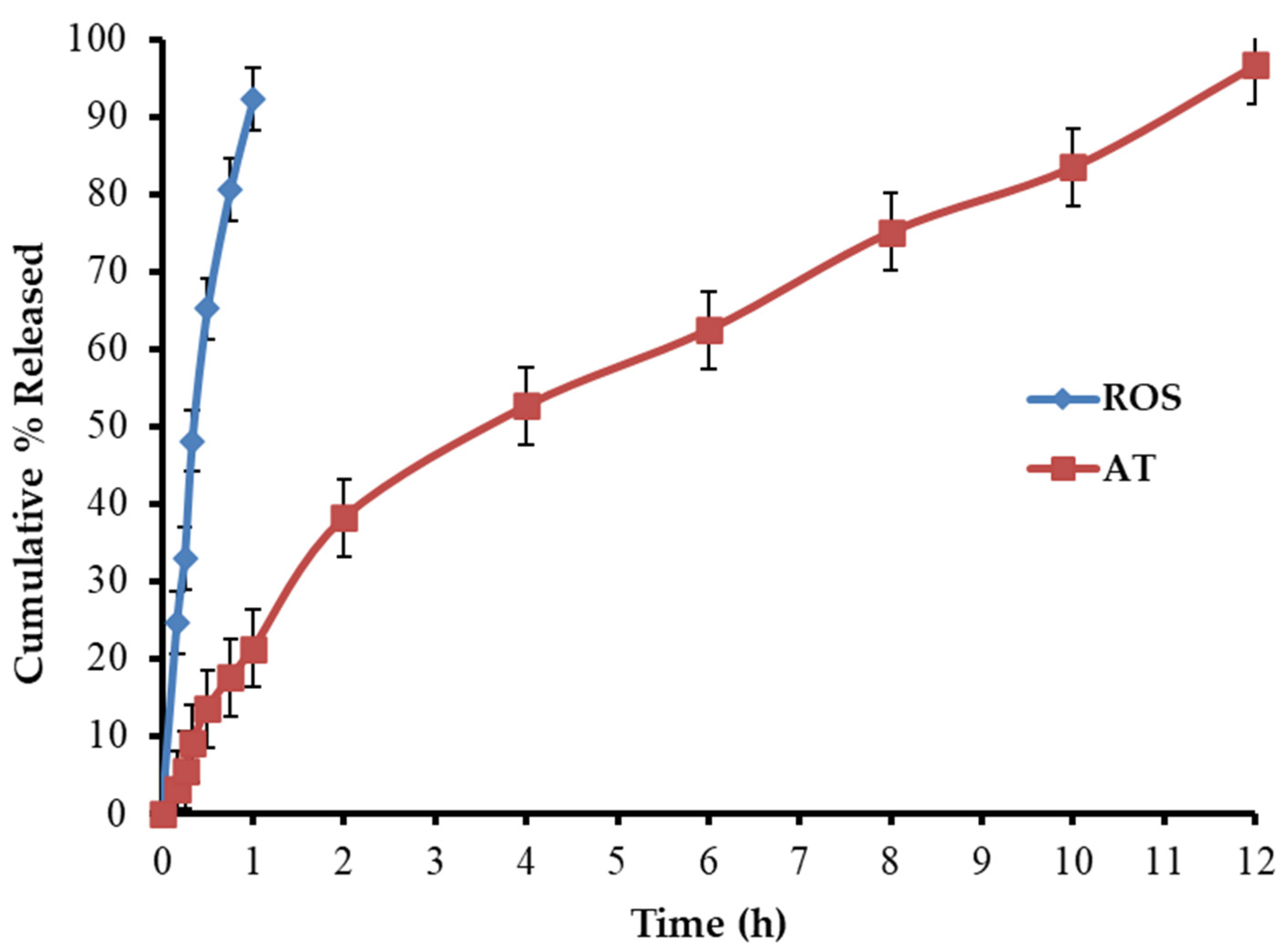
In Vitro Dissolution Investigation of BLTs
3.8. Stability Studies
3.9. In Vivo Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alqahtani, M.S.; Kazi, M.; Alsenaidy, M.A.; Ahmad, M.Z. Advances in oral drug delivery. Front. Pharm. 2021, 12, 618411. [Google Scholar] [CrossRef] [PubMed]
- Ansel, H.C.; Popovich, N.G.; Allen, L.V., Jr. Ansel’s Pharmaceutical Dosage Forms Drug Delivery Systems. Available online: https://thepoint.lww.com/allen9e (accessed on 20 April 2020).
- Abebe, A.; Akseli, I.; Sprockel, O.; Kottala, N.; Cuitiño, A.M. Review of bilayer tablet technology. Int. J. Pharm. 2014, 461, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Syan, N.; Mathur, P. Bi-layer tablet technology—Opening new ways in drug delivery systems: An overview. Int. J. Res. Pharm. Biomed. Sci. 2013, 4, 8–16. [Google Scholar]
- Jha, M.K.; Rahman, M.H.; Rahman, M.M. Biphasic oral solid drug delivery system: A review. Int. J. Pharm. Sci. Res. 2011, 2, 1108. [Google Scholar]
- Elsayed, M.; Aboelez, M.O.; Elsadek, B.E.; Sarhan, H.A.; Khaled, K.A.; Belal, A.; Khames, A.; Hassan, Y.A.; Abdel-Rheem, A.A.; Elkaeed, E.B.J.P. Tolmetin Sodium Fast Dissolving Tablets for Rheumatoid Arthritis Treatment: Preparation and Optimization Using Box-Behnken Design and Response Surface Methodology. Pharmaceutics 2022, 14, 880. [Google Scholar] [CrossRef] [PubMed]
- Hesham, M.; Jelan, A.; Mahmoud, A. Development and optimization of itopride hydrochloride fast disintegrating tablets using factorial design and response surface methodology. Int. J. Pharm. Sci. Res. 2015, 6, 1661–1672. [Google Scholar]
- Belal, A.; Elanany, M.A.; Santali, E.Y.; Al-Karmalawy, A.A.; Aboelez, M.O.; Amin, A.H.; Abdellattif, M.H.; Mehany, A.B.; Elkady, H.J.M. Screening a Panel of Topical Ophthalmic Medications against MMP-2 and MMP-9 to Investigate Their Potential in Keratoconus Management. Molecules 2022, 27, 3584. [Google Scholar] [CrossRef]
- Abdel-Rahman, L.H.; Abu-Dief, A.M.; Aboelez, M.O.; Abdel-Mawgoud, A.A.H.; Biology, P.B. DNA interaction, antimicrobial, anticancer activities and molecular docking study of some new VO (II), Cr (III), Mn (II) and Ni (II) mononuclear chelates encompassing quaridentate imine ligand. J. Photochem. 2017, 170, 271–285. [Google Scholar] [CrossRef]
- Shirse, P. Formulation and evaluation of bilayer tablets of diclofrenac sodium with ranitidine HCl for sustained and immediate release. J. Appl. Pharm. Sci. 2012, 2, 136. [Google Scholar] [CrossRef] [Green Version]
- Khodairy, A.; Ali, A.M.; Aboelez, M.O.; El-Wassimy, M.T. One-Pot Multicomponent Synthesis of Novel 2-Tosyloxyphenylpyrans under Green and Conventional Condition with Anti-inflammatory Activity. J. Heterocycl. Chem. 2017, 54, 1442–1449. [Google Scholar] [CrossRef]
- Ghattas, A.-E.-B.A.; Khodairy, A.; Moustafa, H.M.; Hussein, B.R.; Farghaly, M.M.; Aboelez, M.O. Synthesis, in vitro antibacterial and in vivo anti-inflammatory activity of some new pyridines. Pharm. Chem. J. 2017, 51, 652–660. [Google Scholar] [CrossRef]
- Morris, M.J.; Velkoska, E.; Cole, T.J. Central and peripheral contributions to obesity-associated hypertension: Impact of early overnourishment. Exp. Phys. 2005, 90, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Kamel, M.S.; Belal, A.; Aboelez, M.O.; Shokr, E.K.; Abdel-Ghany, H.; Mansour, H.S.; Shawky, A.M.; El-Remaily, M.A.E.A.A.A. Microwave-Assisted Synthesis, Biological Activity Evaluation, Molecular Docking, and ADMET Studies of Some Novel Pyrrolo [2, 3-b] Pyrrole Derivatives. Molecules 2022, 27, 2061. [Google Scholar] [CrossRef] [PubMed]
- Aboelez, M.O.; Elhady, O.M.; Shaykoon, M.S.A.; Shoman, M.E.; Ahmed, S.A.; Abuo-Rahma, G.E.-D.A.; Chemistry, A. Design, Synthesis and Hypolipidemic Activity of Novel Hydrazones of Nicotinic acid Hydrazide. J. Pharm. Appl. Chem. 2016, 2, 43–49. [Google Scholar] [CrossRef]
- Khule, N.R.; Mahale, N.; Chaudhari, S. Formulation development and evaluation of once a day regioselective dual component gastroretentive tablet of rosuvastatin calcium and metoprolol succinate. Blood 2012, 15, 18. [Google Scholar]
- Shoman, M.E.; Aboelez, M.O.; Shaykhon, M.S.; Ahmed, S.A.; Abuo-Rahma, G.E.-D.A.; Elhady, O.M. New nicotinic acid-based 3, 5-diphenylpyrazoles: Design, synthesis and antihyperlipidemic activity with potential NPC1L1 inhibitory activity. Mol. Divers. 2021, 25, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.D.; Warwick, M.J.; Dane, A.L.; Hill, S.J.; Giles, P.B.; Phillips, P.J.; Lenz, E. Metabolism, excretion, and pharmacokinetics of rosuvastatin in healthy adult male volunteers. Clin. Ther. 2003, 25, 2822–2835. [Google Scholar] [CrossRef]
- Sanjana, A.; Ahmed, M.G. Development and in vitro evaluation of rosuvastatin tablets by floating drug delivery system. Asian. J. Pharm. 2017, 11, S260–S264. [Google Scholar]
- Sarfraz, R.M.; Ahmad, M.; Mahmood, A.; Minhas, M.U.; Yaqoob, A. Development and evaluation of rosuvastatin calcium based microparticles for solubility enhancement: An in vitro study. Adv. Polym. Technol. 2017, 36, 433–441. [Google Scholar] [CrossRef]
- Martin, P.D.; Mitchell, P.D.; Schneck, D.W. Pharmacodynamic effects and pharmacokinetics of a new HMG-CoA reductase inhibitor, rosuvastatin, after morning or evening administration in healthy volunteers. Br. J. Clin. Pharm. 2002, 54, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.D.; Warwick, M.J.; Dane, A.L.; Brindley, C.; Short, T. Absolute oral bioavailability of rosuvastatin in healthy white adult male volunteers. Clin. Ther. 2003, 25, 2553–2563. [Google Scholar] [CrossRef]
- Yang, Y.; Faustino, P.J.; Volpe, D.A.; Ellison, C.D.; Lyon, R.C.; Yu, L.X. Biopharmaceutics classification of selected β-blockers: Solubility and permeability class membership. Mol. Pharm. 2007, 4, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.; Dangi, R.; Patidar, R.; Rathi, J.; Asati, V. Formulation and evaluation of sustain released matrix tablet of atenolol. J. Drug Deliv Ther. 2019, 9, 183–189. [Google Scholar] [CrossRef] [Green Version]
- HV, G.; Balamuralidhara, V.; Kumar, P. Formulation and in vitro evaluation of gastric floating tablets of atenolol. J. Pharm. Res. 2010, 3, 1450–1455. [Google Scholar]
- Waqar, A.B.; Koike, T.; Yu, Y.; Inoue, T.; Aoki, T.; Liu, E.; Fan, J. High-fat diet without excess calories induces metabolic disorders and enhances atherosclerosis in rabbits. Atherosclerosis 2010, 213, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, L.; You, J.; Cao, J.; Fu, X. Effect of 7-difluoromethyl-5, 4′-dimethoxygenistein on aorta atherosclerosis in hyperlipidemia ApoE−/− mice induced by a cholesterol-rich diet. Drug Des. Dev. Ther. 2013, 7, 233. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, M.S.; Abdelhafez, W.A.; Zayed, G.; Samy, A.M. In vitro and in vivo characterization of fast dissolving tablets containing gliquidone–pluronic solid dispersion. Drug Dev. Ind. Pharm. 2019, 45, 1973–1981. [Google Scholar] [CrossRef]
- Afifi, S. Solid Dispersion Approach Improving Dissolution Rate of Stiripentol: A Novel Antiepileptic Drug. Iran. J. Pharm. Res. 2015, 14, 1001–1014. [Google Scholar]
- Mohamed, M.S.; Abdelhafez, W.A.; Zayed, G.; Samy, A.M. Optimization, in-vitro release and in-vivo evaluation of gliquidone nanoparticles. AAPS PharmSciTech 2020, 21, 35. [Google Scholar] [CrossRef]
- Shaker, M.A. Dissolution and bioavailability enhancement of Atorvastatin: Gelucire semi-solid binary system. J. Drug Deliv. Sci. Technol. 2018, 43, 178–184. [Google Scholar] [CrossRef]
- Gupta, K.; Singhvi, I. Formulation and characterization of mouth dissolving tablets of ezetimibe by frosta technique using ezetimibe: Hydroxypropyl-β-cyclodextrin solid dispersion. Int. J. Pharm. Erud. 2015, 5, 18–28. [Google Scholar]
- de Oliveira Eloy, J.; Saraiva, J.; de Albuquerque, S.; Marchetti, J.M. Solid dispersion of ursolic acid in Gelucire 50/13: A strategy to enhance drug release and trypanocidal activity. Aaps PharmsciTech 2012, 13, 1436–1445. [Google Scholar] [CrossRef] [Green Version]
- Elsayed, M.M.; Mostafa, M.E.; Alaaeldin, E.; Sarhan, H.A.; Shaykoon, M.S.; Allam, S.; Ahmed, A.R.; Elsadek, B.E. Design and characterisation of novel Sorafenib-loaded carbon nanotubes with distinct tumour-suppressive activity in hepatocellular carcinoma. Int. J. Nanomed. 2019, 14, 8445. [Google Scholar] [CrossRef] [Green Version]
- Elsayed, M.M.A.; Okda, T.M.; Atwa, G.M.K.; Omran, G.A.; Abd Elbaky, A.E.; Ramadan, A.E.h. Design and Optimization of Orally Administered Luteolin Nanoethosomes to Enhance Its Anti-Tumor Activity against Hepatocellular Carcinoma. Pharmaceutics 2021, 13, 648. [Google Scholar] [CrossRef] [PubMed]
- Refaat, H.; Naguib, Y.W.; Elsayed, M.; Sarhan, H.A.; Alaaeldin, E. Modified spraying technique and response surface methodology for the preparation and optimization of propolis liposomes of enhanced anti-proliferative activity against human melanoma cell line A375. Pharmaceutics 2019, 11, 558. [Google Scholar] [CrossRef] [Green Version]
- Saddik, M.S.; Elsayed, M.; Abdelkader, M.S.A.; El-Mokhtar, M.A.; Abdel-Aleem, J.A.; Abu-Dief, A.M.; Al-Hakkani, M.F.; Farghaly, H.S.; Abou-Taleb, H.A. Novel Green Biosynthesis of 5-Fluorouracil Chromium Nanoparticles Using Harpullia pendula Extract for Treatment of Colorectal Cancer. Pharmaceutics 2021, 13, 226. [Google Scholar] [CrossRef]
- El-Shenawy, A.; Ahmed, M.; Mansour, H.; El Rasoul, S. Torsemide Fast Dissolving Tablets: Development, Optimization Using Box–Bhenken Design and Response Surface Methodology, In Vitro Characterization, and Pharmacokinetic. AAPS PharmSciTech 2017, 2, 2168–2179. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.M.; Abd El-Rasoul, S.; Auda, S.H.; Ibrahim, M.A. Emulsification/internal gelation as a method for preparation of diclofenac sodium–sodium alginate microparticles. Saudi Pharm. J. 2013, 21, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auda, S.H.; Ahmed, M.M.; El-Rasoul, A.; Saleh, K.I. Formulation and physicochemical characterization of piroxicam containing polymer films. Bull. Pharm. Sci. Assiut 2010, 33, 33–42. [Google Scholar] [CrossRef]
- Kate, V.; Payghan, S. Development of Directly Compressible Mucoadehsive Fast Disintegrating Sublingual Tablet System of Piroxicam Using 3 factor, 3 Level Box Behnken Design. Asian J. Biomed. Pharm. Sci. 2013, 3, 19–29. [Google Scholar]
- Ahmed, M.M. Effect of different formulation variables on release characteristics of gastro-floating microspheres of ethyl cellulose/carbopol 934P encapsulating sorafenib. Int. J. Pharm. Pharm. Sci. 2019, 11, 64–70. [Google Scholar] [CrossRef]
- Elsayed, M. Controlled release alginate-chitosan microspheres of tolmetin sodium prepared by internal gelation technique and characterized by response surface modeling. Braz. J. Pharm. Sci. 2021, 56, 1–15. [Google Scholar] [CrossRef]
- Hariprasanna, R.; Dewalkar, H.; Patil, B.S.; Yachwad, R.; More, M. Design and developmnent of ziprazidone fast dissolving tablets by vacuum drying technique. J. Appl. Pharm. 2012, 4, 567–576. [Google Scholar]
- Sree, P.K.; Martha, S.; Sharma, J.; Prasad, B.D. Formulation and evaluation of oral fast disintegrating tablets of Delafloxacin. Int. J. Pharm. Biol. Sci. 2019, 9, 1035–1044. [Google Scholar]
- Singh, I.; Kumar, P. Preformulation studies for direct compression suitability of cefuroxime axetil and paracetamol: A graphical representation using SeDeM diagram. Acta. Pol. Pharm. 2012, 69, 87–93. [Google Scholar] [PubMed]
- Zaid, A.N.; Rowa’J, A.-R.; Ghoush, A.A.; Qaddumi, A.; Zaaror, Y.A. Weight and content uniformity of lorazepam half-tablets: A study of correlation of a low drug content product. Saudi Pharm. J. 2013, 21, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Rasoul, A.; Ahmed, M.M. Chitosan polymer as a coat of calcium alginate microcapsules loaded by non-steroidal antiinflammatory drug. Bull. Pharm. Sci. Assiut 2010, 33, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Haritha, B. A review on evaluation of tablets. J. Form. Sci. Bioavailab. 2017, 1, 1–5. [Google Scholar]
- Ibrahim, M.A.; Amal El Sayeh, F. Optimized furosemide taste masked orally disintegrating tablets. Saudi Pharm. J. 2017, 25, 1055–1062. [Google Scholar] [CrossRef]
- Patel, V.F.; Patel, N.M. Intragastric floating drug delivery system of cefuroxime axetil: In vitro evaluation. AAPS PharmSciTech. 2006, 7, E118–E124. [Google Scholar] [CrossRef]
- Li, L.; Li, J.; Si, S.; Wang, L.; Shi, C.; Sun, Y.; Liang, Z.; Mao, S. Effect of formulation variables on in vitro release of a water-soluble drug from chitosan–sodium alginate matrix tablets. Asian J. Pharm. Sci. 2015, 10, 314–321. [Google Scholar] [CrossRef] [Green Version]
- Shah, K.U.; Khan, G.M. Regulating drug release behavior and kinetics from matrix tablets based on fine particle-sized ethyl cellulose ether derivatives: An in vitro and in vivo evaluation. Sci. World J. 2012, 2012, 842348. [Google Scholar] [CrossRef] [Green Version]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar]
- Higuchi, T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Abdul Althaf, S.; Sailaja, P.; Ashwin Kumar, M. Formulation, evaluation and mathematical modelling of clopidogrel bisulphate & aspirin immediate release bilayer tablets. Pharm. Anal. Acta 2012, 3, 194. [Google Scholar]
- Elkot, M.; Elsayed, M.; Alaaeldin, E.; Sarhan, H.; Shaykoon, M.S.A.; Elsadek, B. Accelerated stability testing of microcapsulated sorafenib-loaded carbon nanotubes prepared by emulsification/internal gelation method. Int. J. Pharm. Pharm. Res. 2019, 16, 126–139. [Google Scholar]
- Bellini, L.; Veladiano, I.A.; Schrank, M.; Candaten, M.; Mollo, A. Prospective clinical study to evaluate an oscillometric blood pressure monitor in pet rabbits. BMC Vet. Res. 2018, 14, 1–8. [Google Scholar] [CrossRef]
- Baskaran, G.; Salvamani, S.; Azlan, A.; Ahmad, S.A.; Yeap, S.K.; Shukor, M.Y. Hypocholesterolemic and antiatherosclerotic potential of Basella alba leaf extract in hypercholesterolemia-induced rabbits. Evid.-Based Complement. Altern. Med. 2015, 2015, 751714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saharan, V.; Kukkar, V.; Kataria, M.; Gera, M.; Choudhury, P.K. Dissolution enhancement of drugs. Part I: Technologies and effect of carriers. Int. J. Health Res. 2009, 2, 107–124. [Google Scholar] [CrossRef]
- El Maghraby, G.M.; Elsergany, R.N. Fast disintegrating tablets of nisoldipine for intra-oral administration. Pharm. Dev. Technol. 2014, 19, 641–650. [Google Scholar] [CrossRef]
- Hasan, A.; Abd Elghany, M.; Sabry, S. Design and characterization of intra-oral fast dissolving tablets containing diacerein-solid dispersion. J. Appl. Pharm. Sci. 2020, 10, 44–53. [Google Scholar]
- Moustafa, A.H.; Ahmed, W.W.; Awad, M.F.; Aboelez, M.O.; Khodairy, A.; Amer, A.A. Eco-friendly and regiospecific intramolecular cyclization reactions of cyano and carbonyl groups in N, N-disubstituted cyanamide. Mol. Divers. 2022, 26, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Elkhodairy, K.A.; Hassan, M.A.; Afifi, S.A. Formulation and optimization of orodispersible tablets of flutamide. Saudi Pharm. J. 2014, 22, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Chambin, O.; Champion, D.; Debray, C.; Rochat-Gonthier, M.; Le Meste, M.; Pourcelot, Y. Effects of different cellulose derivatives on drug release mechanism studied at a preformulation stage. J. Control. Release 2004, 95, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, J.; Barry, K.; Cerqueira, P.; Iqbal, T.; O’neill, J.; Roos, Y. Effect of composition and storage conditions on the flowability of dairy powders. Int. Dairy J. 2007, 17, 383–392. [Google Scholar] [CrossRef]
- Ahuja, S.; Scypinski, S. Handbook of Modern Pharmaceutical Analysis; Academic Press: Cambridge, MA, USA, 2001; Volume 3. [Google Scholar]
- Durgapal, S.; Mukhopadhyay, S.; Goswami, L. Preparation, characterization and evaluation of floating microparticles of ciprofloxacin. Int. J. Appl. Pharm. 2017, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Su’udiyah, U.F. The Effect of Ethyl Cellulose 10 Cps on the floating Characteristic and Ranitidine HCL release from floating Tablet (using HPMC K15M Matrix). Res. J. Pharm. Technol. 2021, 14, 735–740. [Google Scholar] [CrossRef]
- Fentie, M.; Belete, A.; Gebre-Mariam, T. Formulation and optimization of controlled release floating microspheres of Furosemide from ethylcellulose and hydroxypropyl methylcellulose polymer blends. Int. J. Pharm. Sci. Res. 2014, 5, 70. [Google Scholar]
- Setty, C.M.; Prasad, D.; Gupta, V.; Sa, B. Development of fast dispersible aceclofenac tablets: Effect of functionality of superdisintegrants. Indian J. Pharm. Sci. 2008, 70, 180. [Google Scholar] [CrossRef] [Green Version]
- Battu, S.K.; Repka, M.A.; Majumdar, S.; Rao, Y.M. Formulation and evaluation of rapidly disintegrating fenoverine tablets: Effect of superdisintegrants. Drug Dev. Ind. Pharm. 2007, 33, 1225–1232. [Google Scholar] [CrossRef]
- Zhao, N.; Augsburger, L.L. Functionality comparison of 3 classes of superdisintegrants in promoting aspirin tablet disintegration and dissolution. AAPS PharmsciTech 2005, 6, E634–E640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolhuis, G.; Zuurman, K.; Te Wierik, G. Improvement of dissolution of poorly soluble drugs by solid deposition on a super disintegrant. II. The choice of super disintegrants and effect of granulation. Eur. J. Pharm. Sci. 1997, 5, 63–69. [Google Scholar] [CrossRef]
- Gharti, K.; Budhathoki, U.; Thapa, P.; Bhargava, A. Formulation in vitro evaluation of floating tablets of hydroxypropyl methylcellulose and polyethylene oxide using ranitidine hydrochloride as a model drug. J. Young Pharm. 2012, 4, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Elsayed, M.; El Rasoul, S.A.; Hussein, A.K. Response surface methodology as a useful tool for development and optimization of sustained release ketorolac tromethamine niosomal organogels. J. Pharm. Innov. 2020, 15, 664–677. [Google Scholar] [CrossRef]
- Pham, A.T.; Lee, P.I. Probing the mechanisms of drug release from hydroxypropylmethyl cellulose matrices. Pharm. Res. 1994, 11, 1379–1384. [Google Scholar] [CrossRef]
- Sreenivas, S.; Dandagi, P.; Gadad, A.; Godbole, A.; Hiremath, S.; Mastiholimath, V.; Bhagawati, S. Orodispersible tablets: New-fangled drug delivery system-A review. Indian J. Pharm. Educ. 2005, 39, 177–185. [Google Scholar]
- Gupta, M.; Pandey, S.; Chauhan, B.S.; Gupta, N. Design, development and evaluation of rosuvastatin calcium and diltiazem hydrochloride bilayer tablet using combination concept of sustained layer with conventional layer. Turk. J. Pharm. Sci. 2014, 11, 269–284. [Google Scholar]
- El-Shenawy, A.A.; Mahmoud, R.A.; Mahmoud, E.A.; Mohamed, M.S. Intranasal In Situ Gel of Apixaban-Loaded Nanoethosomes: Preparation, Optimization, and In Vivo Evaluation. AAPS PharmSciTech 2021, 22, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Gouda, R.; Baishya, H.; Qing, Z. Application of mathematical models in drug release kinetics of carbidopa and levodopa ER tablets. J. Dev. Drugs 2017, 6, 1–8. [Google Scholar]
- Gierszewska-Drużyńska, M.; Ostrowska-Czubenko, J. Mechanism of water diffusion into noncrosslinked and ionically crosslinked chitosan membranes. Prog. Chem. Appl. Chitin Its Deriv. 2012, 17, 63–70. [Google Scholar]
- Bouin, A.-S.; Wierer, M. Quality standards of the European Pharmacopoeia. J. Ethnopharmacol. 2014, 158, 454–457. [Google Scholar] [CrossRef]
- Sharma, U.; Kishore, J.; Garg, A.; Anand, T.; Chakraborty, M.; Lali, P. Dyslipidemia and associated risk factors in a resettlement colony of Delhi. J. Clin. Lipidol. 2013, 7, 653–660. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F. Code | CCS | CP | SSG | Lactose Monohydrate |
|---|---|---|---|---|
| ROS1 | 9 | *** | *** | 97.5 |
| ROS2 | 12 | *** | *** | 94.5 |
| ROS3 | 15 | *** | *** | 91.5 |
| ROS4 | *** | 9 | *** | 97.5 |
| ROS5 | *** | 12 | *** | 94.5 |
| ROS6 | *** | 15 | *** | 91.5 |
| ROS7 | *** | *** | 9 | 97.5 |
| ROS8 | *** | *** | 12 | 94.5 |
| ROS9 | *** | *** | 15 | 91.5 |
| Independent Variables | Levels | ||
|---|---|---|---|
| High (+1) | Medium (0) | Low (−1) | |
| HPMC (% w/w), (X1) | 40 | 30 | 20 |
| EC (% w/w), (X2) | 20 | 12.5 | 5 |
| Dependent response | Aim | ||
| % released at 12 h (Y1) | Maximize | ||
| AT/SR | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ingredients (mg) | AT1 | AT2 | AT3 | AT4 | AT5 | AT6 | AT7 | AT8 | AT9 |
| AT | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| HPMC K100 | 40 | 40 | 60 | 60 | 80 | 60 | 80 | 80 | 40 |
| EC | 10 | 25 | 25 | 10 | 10 | 40 | 25 | 40 | 40 |
| Sodium bicarbonate | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Mg Stearate | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Lactose monohydrate | 78 | 63 | 43 | 58 | 38 | 28 | 23 | 8 | 48 |
| Formula | % DE30 min *, n = 3 | % DE60 min *, n = 3 | % DE120 min *, n = 3 | RDR30 min **, n = 3 | RDR60 min **, n = 3 | RDR120 min **, n = 3 |
|---|---|---|---|---|---|---|
| Pure ROS | 5.45 ± 0.708 | 12.50 ± 1.11 | 23.98 ± 1.55 | 1.00 ± 0.152 | 1.00 ± 0.108 | 1.00 ± 0.116 |
| SD *** 1:1 | 16.67 ± 1.75 | 27.49 ± 1.13 | 41.07 ± 2.09 | 2.52 ± 0.426 | 1.60 ± 0.243 | 1.55 ± 0.145 |
| SD 1:2 | 22.57 ± 1.69 | 39.88 ± 2.15 | 60.30 ± 2.56 | 3.43 ± 0.611 | 2.36 ± 0.780 | 2.22 ± 0.207 |
| SD 1:3 | 29.21 ± 2.16 | 50.31 ± 2.06 | 70.97 ± 3.23 | 5.10 ± 0.351 | 2.82 ± 0.565 | 2.49 ± 0.233 |
| SD 1:4 | 29.14 ± 1.84 | 50.05 ± 1.78 | 69.99 ± 1.94 | 5.25 ± 0.444 | 2.73 ± 0.621 | 2.42 ± 1.78 |
| F. Code | The Angle of Repose (θ) | Carr’s Index (%) | Hausner’s Ratio |
|---|---|---|---|
| ROS tablets powder blends | |||
| ROS1 | 29.23 ± 1.55 | 16.30 ± 2.23 | 1.32 ± 0.023 |
| ROS2 | 24.66 ± 1.29 | 19.25 ± 1.89 | 1.21 ± 0.012 |
| ROS3 | 18.59 ± 1.06 | 14.84 ± 1.25 | 1.20 ± 0.007 |
| ROS4 | 25.83 ± 1.32 | 16.90 ± 2.08 | 1.27 ± 0.071 |
| ROS5 | 22.18 ± 1.66 | 13.78 ± 1.33 | 1.30 ± 0.037 |
| ROS6 | 16.59 ± 1.10 | 11.72 ± 1.94 | 1.10 ± 0.055 |
| ROS7 | 17.82 ± 1.41 | 12.45 ± 2.61 | 1.19 ± 0.072 |
| ROS8 | 27.74 ± 1.20 | 18.61 ± 1.73 | 1.28 ± 0.026 |
| ROS9 | 20.65 ± 1.32 | 19.04 ± 1.40 | 1.12 ± 0.047 |
| AT tablets powder blends | |||
| AT1 | 22.74 ± 1.30 | 17.42 ± 1.91 | 1.09 ±0.046 |
| AT2 | 25.81 ± 1.05 | 12.84 ± 2.04 | 1.13 ± 0.109 |
| AT3 | 19.25 ± 1.22 | 16.51 ± 2.80 | 1.22 ± 0.047 |
| AT4 | 26.49 ± 1.09 | 10.30 ± 2.87 | 1.20 ± 0.040 |
| AT5 | 21.20 ± 1.07 | 16.33 ± 1.26 | 1.23 ± 0.030 |
| AT6 | 28.70 ± 1.32 | 14.94 ± 1.84 | 1.11 ± 0.037 |
| AT7 | 25.23 ± 1.63 | 11.70 ± 2.84 | 1.18 ± 0.015 |
| AT8 | 27.77 ± 1.27 | 11.94 ± 1.22 | 1.24 ± 0.052 |
| AT9 | 20.35 ± 2.14 | 12.40 ± 1.87 | 1.19 ± 0.017 |
| F. Code | Drug Content (%) | Weight (mg) | Thickness (mm) | Friability (%) | Hardness (Kg/cm2) |
|---|---|---|---|---|---|
| ROS Immediate-Release Mono Tablets | |||||
| ROS1 | 96.12 ± 4.34 | 140.85 ± 3.82 | 1.87 ± 0.34 | 0.672 ± 0.004 | 3.85 ± 0.52 |
| ROS2 | 99.54 ± 2.87 | 144.43 ± 2.94 | 1.91 ± 0.56 | 0.580 ± 0.001 | 3.25 ± 0.40 |
| ROS3 | 100.82 ± 3.40 | 151.24 ± 5.43 | 2.08 ± 0.81 | 0.532 ± 0.007 | 4.60 ± 0.71 |
| ROS4 | 97.09 ± 1.28 | 144.56 ± 3.30 | 1.90 ± 0.24 | 0.693 ± 0.003 | 4.09 ± 0.55 |
| ROS5 | 95.70 ± 4.20 | 150.00 ± 2.75 | 1.99 ± 0.62 | 0.802 ± 0.006 | 3.95 ± 0.84 |
| ROS6 | 95.29 ± 2.06 | 144.14 ± 3.08 | 1.81 ± 0.11 | 0.596 ± 0.003 | 4.58 ± 0.22 |
| ROS7 | 98.95 ± 3.09 | 141.74 ± 2.79 | 1.83 ± 0.26 | 0.895 ± 0.009 | 4.45 ± 0.95 |
| ROS8 | 97.18 ± 1.25 | 149.10 ± 3.42 | 1.95 ± 0.61 | 0.711 ± 0.004 | 3.54 ± 0.66 |
| ROS9 | 100.04 ± 2.31 | 140.72 ± 2.83 | 1.90 ± 0.56 | 0.655 ± 0.006 | 4.11 ± 0.38 |
| AT Sustained-Release Mono Tablets | |||||
| AT1 | 98.60 ± 2.34 | 193.56 ± 4.10 | 2.99 ± 0.37 | 0.734 ± 0.004 | 5.60 ± 0.18 |
| AT2 | 97.50 ± 2.67 | 195.94 ± 3.81 | 2.90 ± 0.62 | 0.631 ± 0.002 | 5.84 ± 0.23 |
| AT3 | 99.21 ± 1.90 | 197.78 ± 2.55 | 3.09 ± 0.11 | 0.540 ± 0.005 | 7.69 ± 0.54 |
| AT4 | 95.33 ± 2.33 | 201.40 ± 4.26 | 2.92 ± 0.20 | 0.628 ± 0002 | 6.52 ± 0.12 |
| AT5 | 98.10 ± 2.84 | 189.67 ± 2.38 | 3.01 ± 0.73 | 0.583 ± 0.005 | 7.72 ± 0.40 |
| AT6 | 100.42 ± 1.25 | 202.62 ± 5.73 | 3.11 ± 0.74 | 0.555 ± 0.008 | 6.66 ± 0.31 |
| AT7 | 100.11 ± 2.69 | 199.50 ± 2.85 | 2.95 ± 0.26 | 0.773 ± 0.003 | 7.15 ± 0.27 |
| AT8 | 96.85 ± 2.55 | 204.93 ± 5.20 | 3.00 ± 0.18 | 0.730 ± 0.004 | 6.50 ± 0.16 |
| AT9 | 96.35 ± 1.62 | 200.21 ± 5.83 | 3.05 ± 0.34 | 0.749 ± 0.001 | 5.98 ± 0.21 |
| F. Code | Buoyancy Lag Time (min) | Total Floating Time (h) |
|---|---|---|
| AT1 | 4.42 ± 0.21 | 22.41 ± 1.20 |
| AT2 | 5.53 ± 0.32 | 23.12 ± 0.80 |
| AT3 | 2.82 ± 0.08 | 18.91 ± 1.14 |
| AT4 | 2.10 ± 0.11 | 17.54 ± 1.31 |
| AT5 | 1.20 ± 0.15 | 13.83 ± 0.74 |
| AT6 | 3.75 ± 0.41 | 20.10 ± 0.89 |
| AT7 | 1.53 ± 0.16 | 14.95 ± 1.08 |
| AT8 | 2.08 ± 0.24 | 15.18 ± 1.14 |
| AT9 | 6.35 ± 0.83 | 24.01 ± 1.91 |
| Source | Sum of Squares | Df | Mean Square | F-Ratio | p-Value |
|---|---|---|---|---|---|
| A:HPMC | 230.64 | 1 | 230.64 | 167.94 | 0.0010 |
| B:EC | 772.935 | 1 | 772.935 | 562.82 | 0.0002 |
| AA | 1.62 | 1 | 1.62 | 1.18 | 0.3569 |
| AB | 19.36 | 1 | 19.36 | 14.10 | 0.0330 |
| BB | 8.405 | 1 | 8.405 | 6.12 | 0.0898 |
| Total error | 4.12 | 3 | 1.37333 | ||
| Total correction | 1037.08 | 8 |
| F. Code | Zero-Order | First-Order | Higuchi-Diffusion | n | |||
|---|---|---|---|---|---|---|---|
| r2 | K0 | r2 | K1 | r2 | KH | ||
| Kinetics of ROS-Ca2+-IRT | |||||||
| ROS1 | 0.958 | 0.593 | 0.981 | 0.010 | 0.979 | 7.136 | ….. |
| ROS2 | 0.307 | 0.284 | 0.636 | 0.048 | 0.114 | 1.247 | ….. |
| ROS3 | 0.459 | 0.443 | 0.526 | 0.038 | 0.307 | 3.496 | ….. |
| ROS4 | 0.970 | 0.534 | 0.990 | 0.008 | 0.982 | 6.376 | ….. |
| ROS5 | 0.956 | 0.851 | 0.983 | 0.038 | 0.976 | 0.239 | ….. |
| ROS6 | 0.099 | 0.087 | 0.811 | 0.049 | 0.112 | 1.159 | ….. |
| ROS7 | 0.969 | 0.459 | 0.983 | 0.006 | 0.978 | 5.460 | ….. |
| ROS8 | 0.969 | 0.755 | 0.991 | 0.019 | 0.980 | 9.001 | ….. |
| ROS9 | 0.312 | 0.280 | 0.896 | 0.051 | 0.480 | 5.081 | ….. |
| Kinetics of AT-SRT | |||||||
| AT1 | 0.964 | 5.769 | 0.982 | 0.103 | 0.995 | 21.65 | 0.703 |
| AT2 | 0.958 | 6.593 | 0.960 | 0.150 | 0.980 | 24.76 | 0.621 |
| AT3 | 0.960 | 6.167 | 0.981 | 0.122 | 0.992 | 23.14 | 0.6719 |
| AT4 | 0.994 | 5.36 | 0.977 | 0.086 | 0.941 | 19.39 | ….. |
| AT5 | 0.988 | 5.08 | 0.948 | 0.077 | 0.898 | 18.01 | ….. |
| AT6 | 0.941 | 7.01 | 0.966 | 0.183 | 0.985 | 26.6 | 0.602 |
| AT7 | 0.966 | 5.71 | 0.987 | 0.112 | 0.991 | 22.22 | 0.619 |
| AT8 | 0.989 | 6.47 | 0.958 | 0.129 | 0.956 | 23.64 | ….. |
| AT9 | 0.949 | 7.59 | 0.842 | 0.297 | 0.992 | 28.81 | 0.606 |
| Parameters/Time | Initial Time | 1 Month | 2 Months | 4 Months | 6 Months | |
|---|---|---|---|---|---|---|
| Drug content (%) | AT | 99.65 ± 1.6 | 99.20 ± 2.3 | 98.48 ± 1.0 | 97.77 ± 1.7 | 96.80 ± 2.4 |
| ROS | 98.54 ± 2.3 | 98.11 ± 1.8 | 97.61 ± 1.4 | 96.33 ± 3.2 | 95.16 ± 1.7 | |
| Average weight (mg) | 347.51 ± 4.1 | 347.44 ± 3.7 | 347.02 ± 4.2 | 346.79 ± 4.3 | 346.54 ± 2.6 | |
| Hardness (kg/cm2) | 6.43 ± 0.2 | 6.47 ± 0.3 | 6.25 ± 0.2 | 6.17 ± 0.8 | 6.20 ± 0.2 | |
| Cumulative % drug released | ROS at 60 min | 92.34 ± 2.2 | 92.49 ± 1.4 | 92.33 ± 2.0 | 91.55 ± 1.5 | 91.70 ± 1.2 |
| AT at 12 h | 96.65 ± 3.3 | 96.16 ± 1.3 | 96.27 ± 2.0 | 96.10 ± 1.0 | 96.39 ± 2.1 | |
| Time (Week) | Groups | Cholesterol (mg/dL) | TG (mg/dL) | HDL-c (mg/dL) | LDL-c (mg/dL) |
|---|---|---|---|---|---|
| Week 0 | A (control) | 74.38 ± 0.61 d | 51.37 ± 0.92 c | 22.37 ± 0.80 a | 41.72 ± 0.29 c |
| B (HFD) | 71.14 ± 0.84 d | 48.14 ± 0.20 c | 19.14 ± 0.97 a | 42.37 ± 0.47 c | |
| C (HFD) | 75.15 ± 0.39 d | 52.15 ± 0.19 c | 23.15 ± 0.82 a | 41.57 ± 0.51 c | |
| Week 4 | A (control) | 76.11 ± 0.15 d | 53.11 ± 0.48 c | 24.11 ± 0.41 a | 41.37 ± 0.62 c |
| B (HFD) | 82.54 ± 0.48 cd | 59.54 ± 0.39 c | 19.27 ± 0.73 a | 51.36 ± 0.54 b | |
| C (HFD) | 83.78 ± 0.67 cd | 60.78 ± 0.71 c | 18.74 ± 0.27 b | 52.88 ± 0.67 b | |
| Week 8 | A (control) | 70.72 ± 0.87 d | 47.72 ± 0.52 c | 25.49 ± 0.19 a | 35.68 ± 0.27 c |
| B (HFD) | 93.87 ± 0.59 c | 70.87 ± 0.97 b | 20.58 ± 0.30 b | 59.11 ± 0.19 b | |
| C (HFD) | 95.73 ± 0.74 c | 72.73 ± 0.74 b | 19.24 ± 0.43 b | 61.94 ± 0.24 b | |
| Week 10 | A (control) | 76.81 ± 1.25 d | 53.81 ± 0.29 c | 26.28 ± 0.81 a | 39.76 ± 0.35 c |
| B (HFD) | 101.27 ± 0.91 b | 78.27 ± 0.63 b | 18.25 ± 0.37 b | 67.36 ± 0.64 b | |
| C (HFD) | 107.67 ± 0.28 b | 84.67 ± 0.18 b | 17.23 ± 0.23 b | 73.50 ± 0.74 b | |
| Week 12 | A (control) | 74.34 ± 1.23 d | 51.34 ± 0.34 c | 26.37 ± 0.12 a | 37.70 ± 0.67 c |
| B (HFD) | 125.74 ± 0.28 a | 102.74 ± 0.41 a | 15.27 ± 0.28 c | 89.92 ± 0.42 b | |
| C (HFD) | 128.22 ± 0.36 a | 105.22 ± 0.59 a | 14.79 ± 0.71 c | 92.38 ± 0.18 b | |
| Week 14 | A (control) | 78.59 ± 0.71 d | 55.59 ± 0.37 c | 26.59 ± 0.60 a | 40.88 ± 0.17 c |
| B (HFD) | 138.45 ± 0.29 a | 115.44 ± 0.82 a | 14.23 ± 0.37 c | 101.13 ± 0.28 a | |
| C (HFD) + bilayer | 119.91 ± 0.20 b | 96.91 ± 0.24 ab | 17.29 ± 0.81 b | 83.23 ± 0.39 b | |
| Week 15 | A (control) | 77.18 ± 0.89 d | 54.18 ± 0.37 c | 25.33 ± 0.39 a | 41.01 ± 0.71 c |
| B (HFD) | 151.67 ± 0.87 a | 128.67 ± 0.42 a | 13.28 ± 0.19 c | 112.65 ± 0.80 a | |
| C (HFD) + bilayer | 105.29 ± 0.19 b | 82.29 ± 0.57 b | 18.29 ± 0.47 b | 70.54 ± 0.76 b | |
| Week 16 | A (control) | 71.25 ± 0.38 d | 58.81 ± 0.38 c | 27.29 ± 0.38 a | 38.19 ± 0.89 c |
| B (HFD) | 174.48 ± 0.34 a | 151.48 ± 0.41 a | 11.17 ± 0.67 c | 133.01 ± 0.27 a | |
| C (HFD) + bilayer | 94.89 ± 0.49 cd | 71.89 ± 0.08 bc | 21.23 ± 0.89 a | 59.28 ± 0.63 bc |
| Time (Week) | Groups | Blood Pressure (mmHg) | Heart Rate (Number/Minute) | Body Weight (gm) |
|---|---|---|---|---|
| Week 0 | A (control) | 75.83 ± 1.31 d | 112.14 ± 0.87 c | 1081.80 ± 0.91 d |
| B (HFD) | 78.41 ± 2.24 d | 118.36 ± 0.97 c | 1117.13 ± 1.51 d | |
| C (HFD) | 77.15 ± 1.69 d | 112.87 ± 0.71 c | 1095.48 ± 1.09 d | |
| Week 4 | A (control) | 80.31 ± 2.27 d | 115.21 ± 1.91 c | 1321.47 ± 1.38 d |
| B (HFD) | 95.79 ± 1.94 cd | 135.54 ± 1.47 c | 1647.21 ± 0.56 c | |
| C (HFD) | 95.96 ± 2.37 cd | 137.78 ± 1.38 c | 1660.49 ± 2.17 c | |
| Week 8 | A (control) | 74.87 ± 1.12 d | 121.58 ± 1.74 c | 1551.29 ± 2.35 c |
| B (HFD) | 93.87 ± 0.59 c | 142.31 ± 2.63 b | 2259.28 ± 3.18 b | |
| C (HFD) | 95.73 ± 0.74 c | 151.87 ± 3.17 b | 2191.37 ± 2.97 b | |
| Week 10 | A (control) | 81.24 ± 2.19 d | 118.38 ± 1.78 c | 1628.36 ± 3.46 c |
| B (HFD) | 111.57 ± 2.35 b | 156.38 ± 2.17 b | 2561.92 ± 3.48 a | |
| C (HFD) | 113.17 ± 1.63 b | 161.31 ± 3.29 b | 2449.39 ± 2.37 a | |
| Week 12 | A (control) | 82.91 ± 2.47 d | 122.11 ± 2.64 c | 1711.14 ± 2.19 c |
| B (HFD) | 123.29 ± 3.12 a | 172.25 ± 3.57 a | 2778.32 ± 3.08 a | |
| C (HFD) | 132.46 ± 2.48 a | 166.39 ± 2.78 a | 1760.27 ± 3.91 a | |
| Week 14 | A (control) | 81.58 ± 2.84 d | 115.71 ± 3.26 c | 1817.07 ± 2.49 c |
| B (HFD) | 141.12 ± 3.38 a | 185.68 ± 4.15 a | 2892.33 ± 4.17 a | |
| C (HFD) + bilayer | 122.17 ± 2.67 b | 159.81 ± 3.38 ab | 2423.01 ± 2.93 b | |
| Week 15 | A (control) | 84.55 ± 3.19 d | 117.27 ± 3.84 c | 1895.33 ± 3.61 c |
| B (HFD) | 148.34 ± 2.49 a | 188.38 ± 4.31 a | 3012.14 ± 4.74 a | |
| C (HFD) + bilayer | 109.37 ± 3.28 b | 145.37 ± 3.42 b | 2101.15 ± 1.78 b | |
| Week 16 | A (control) | 81.67 ± 3.19 d | 114.32 ± 2.78 c | 1937.92 ± 2.48 c |
| B (HFD) | 165.34 ± 4.27 a | 192.67 ± 4.59 a | 3130.39 ± 4.37 a | |
| C (HFD) + bilayer | 91.27 ± 2.38 cd | 136.47 ± 3.24 bc | 2071.36 ± 2.84 bc |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsayed, M.M.A.; Aboelez, M.O.; Mohamed, M.S.; Mahmoud, R.A.; El-Shenawy, A.A.; Mahmoud, E.A.; Al-Karmalawy, A.A.; Santali, E.Y.; Alshehri, S.; Elsadek, M.E.M.; et al. Tailoring of Rosuvastatin Calcium and Atenolol Bilayer Tablets for the Management of Hyperlipidemia Associated with Hypertension: A Preclinical Study. Pharmaceutics 2022, 14, 1629. https://doi.org/10.3390/pharmaceutics14081629
Elsayed MMA, Aboelez MO, Mohamed MS, Mahmoud RA, El-Shenawy AA, Mahmoud EA, Al-Karmalawy AA, Santali EY, Alshehri S, Elsadek MEM, et al. Tailoring of Rosuvastatin Calcium and Atenolol Bilayer Tablets for the Management of Hyperlipidemia Associated with Hypertension: A Preclinical Study. Pharmaceutics. 2022; 14(8):1629. https://doi.org/10.3390/pharmaceutics14081629
Chicago/Turabian StyleElsayed, Mahmoud M. A., Moustafa O. Aboelez, Mohamed S. Mohamed, Reda A. Mahmoud, Ahmed A. El-Shenawy, Essam A. Mahmoud, Ahmed A. Al-Karmalawy, Eman Y. Santali, Sameer Alshehri, Mahmoud Elkot Mostafa Elsadek, and et al. 2022. "Tailoring of Rosuvastatin Calcium and Atenolol Bilayer Tablets for the Management of Hyperlipidemia Associated with Hypertension: A Preclinical Study" Pharmaceutics 14, no. 8: 1629. https://doi.org/10.3390/pharmaceutics14081629