
Comparison of Nozzle-Based and Nozzle-Free Electrospinning for Preparation of Fast-Dissolving Nanofibers Loaded with Ciprofloxacin

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Electrospinning Solutions
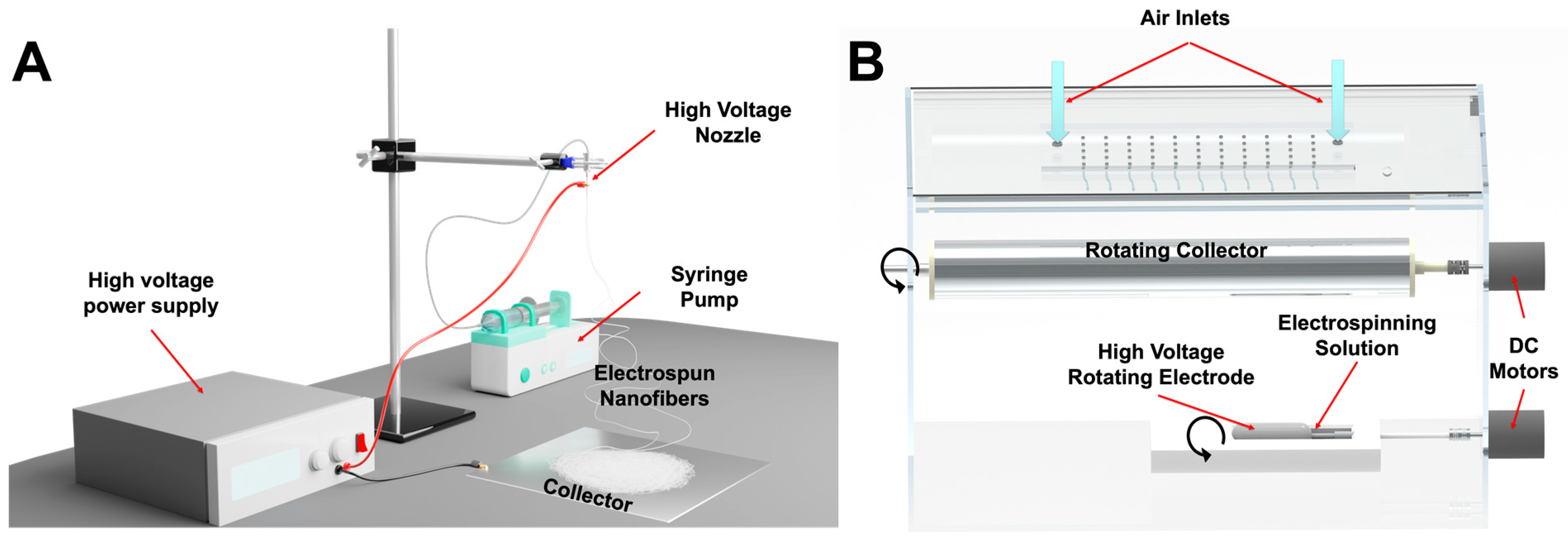
2.3. Electrospinning Using a Nozzle-Based Setup
2.4. Electrospinning Using a Nozzle-Free Setup
2.5. Preparation of the Physical Mixture
2.6. Characterization of the Nanofibers
2.6.1. Scanning Electron Microscopy (SEM)
2.6.2. Differential Scanning Calorimetry (DSC)
2.6.3. Fourier-Transform Infrared Spectroscopy (FTIR)
2.6.4. X-ray Powder Diffraction (XRPD)
2.6.5. Homogeneity
2.6.6. In Vitro Drug Release Studies
2.6.7. Study of Drug-Release Kinetics and Mechanism
2.6.8. Long-Term Storage Stability Tests
2.6.9. In Vitro Cytotoxicity
2.6.10. Statistical Analysis
3. Results and Discussion
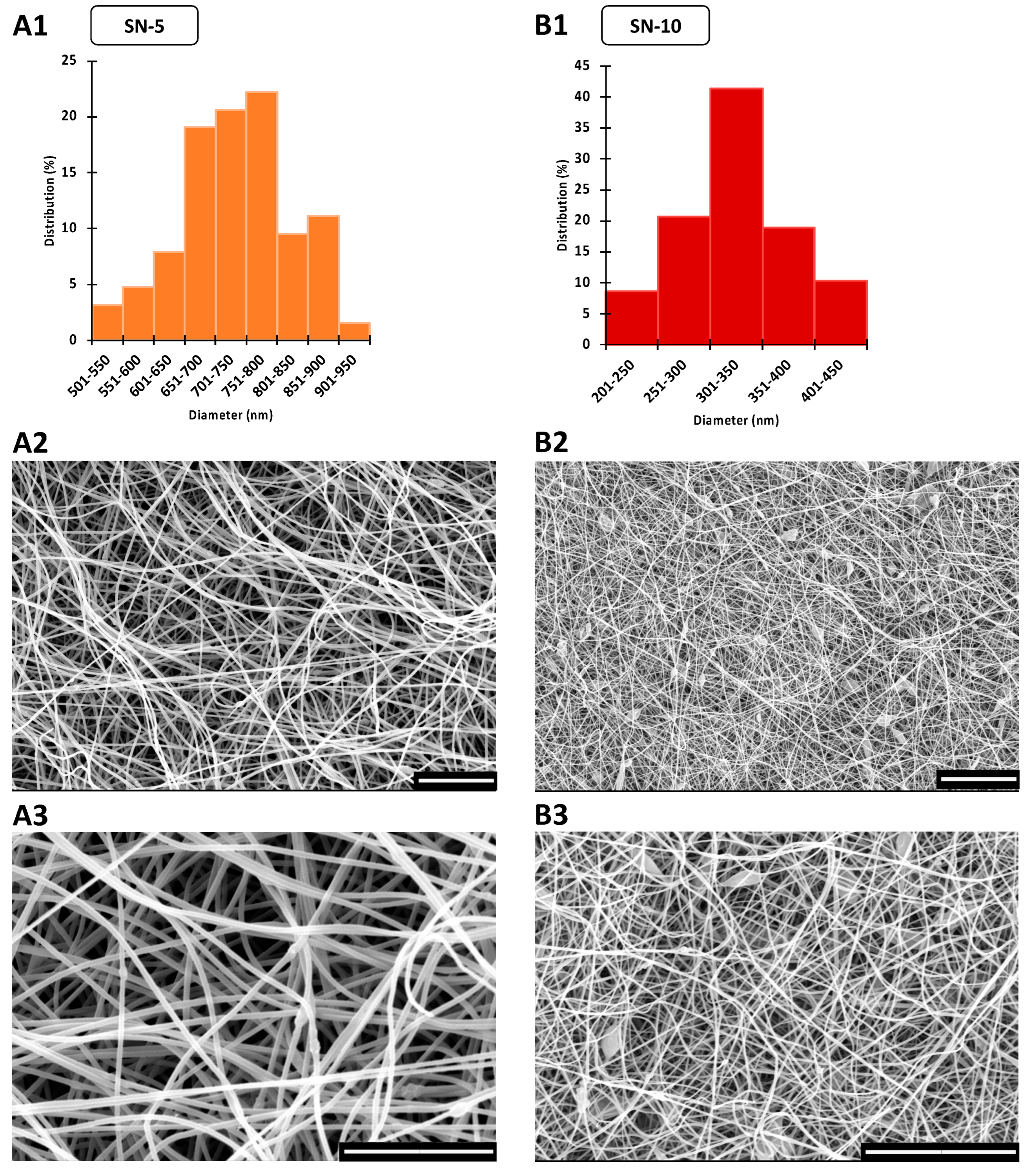
3.1. Morphology of CIP-Loaded Nanofibers Made Using a Nozzle-Based ES Apparatus
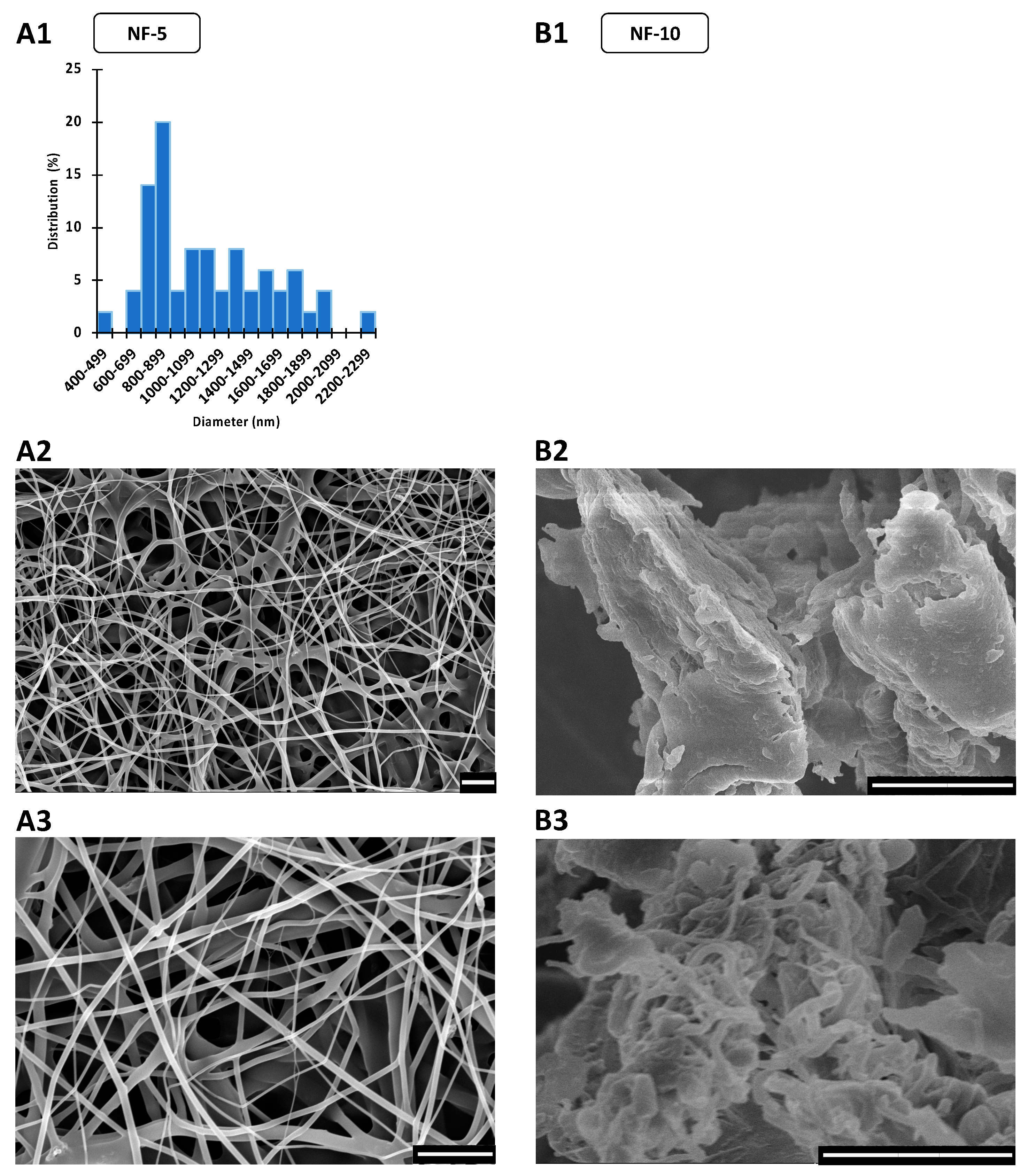
3.2. Morphology of CIP-Loaded Nanofibers Made Using a Nozzle-Free ES Apparatus
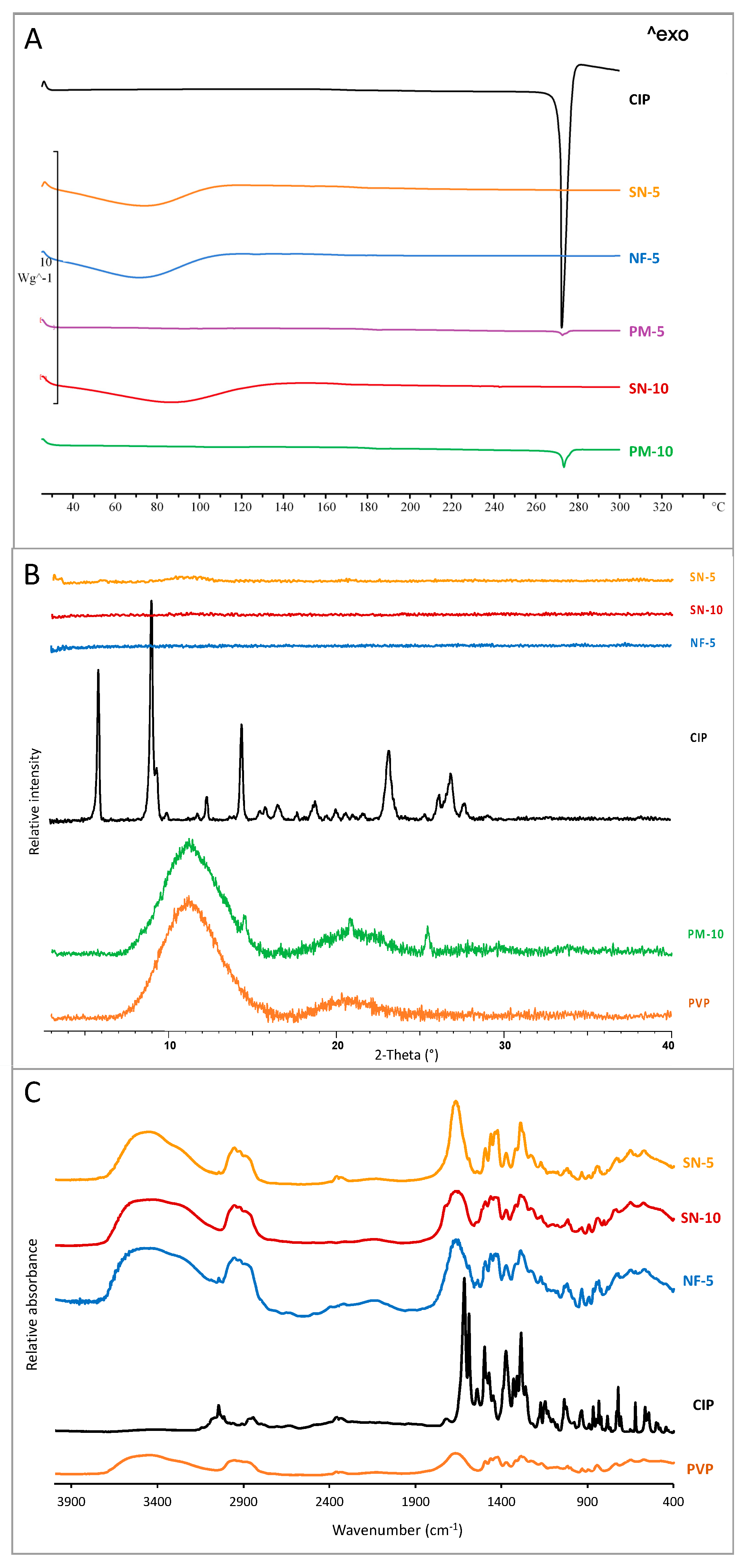
3.3. Structural Characterization
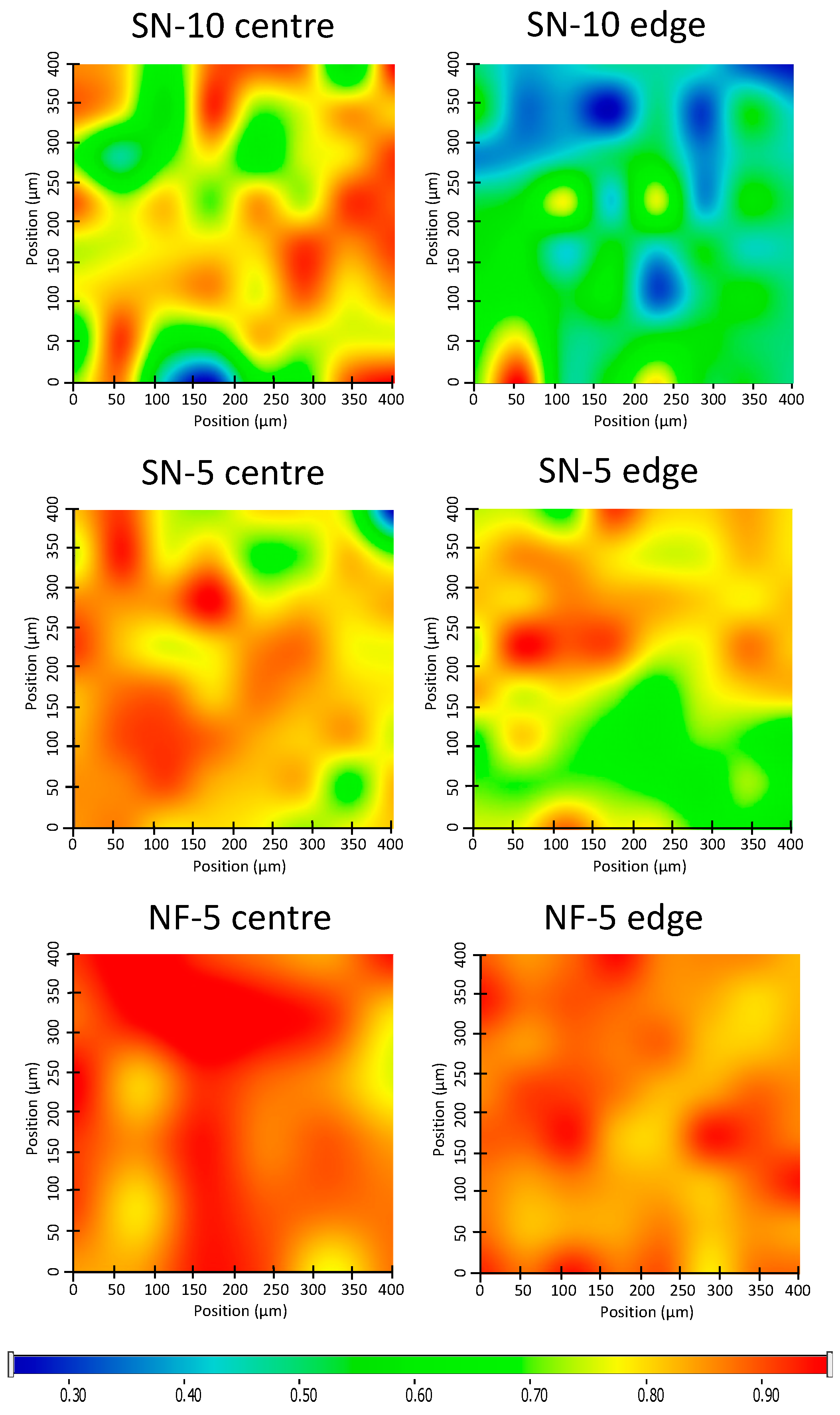
3.4. Homogeneity
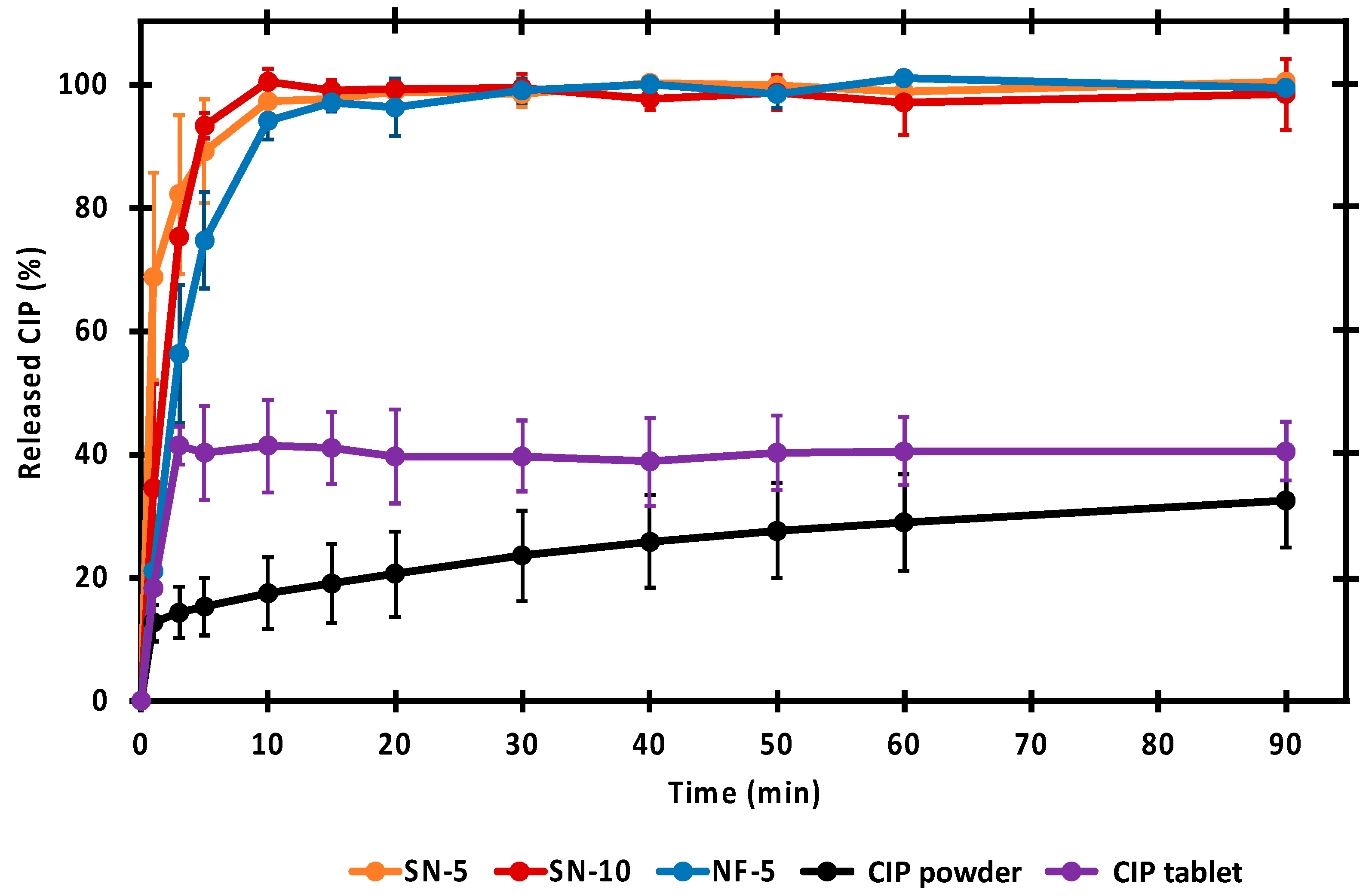
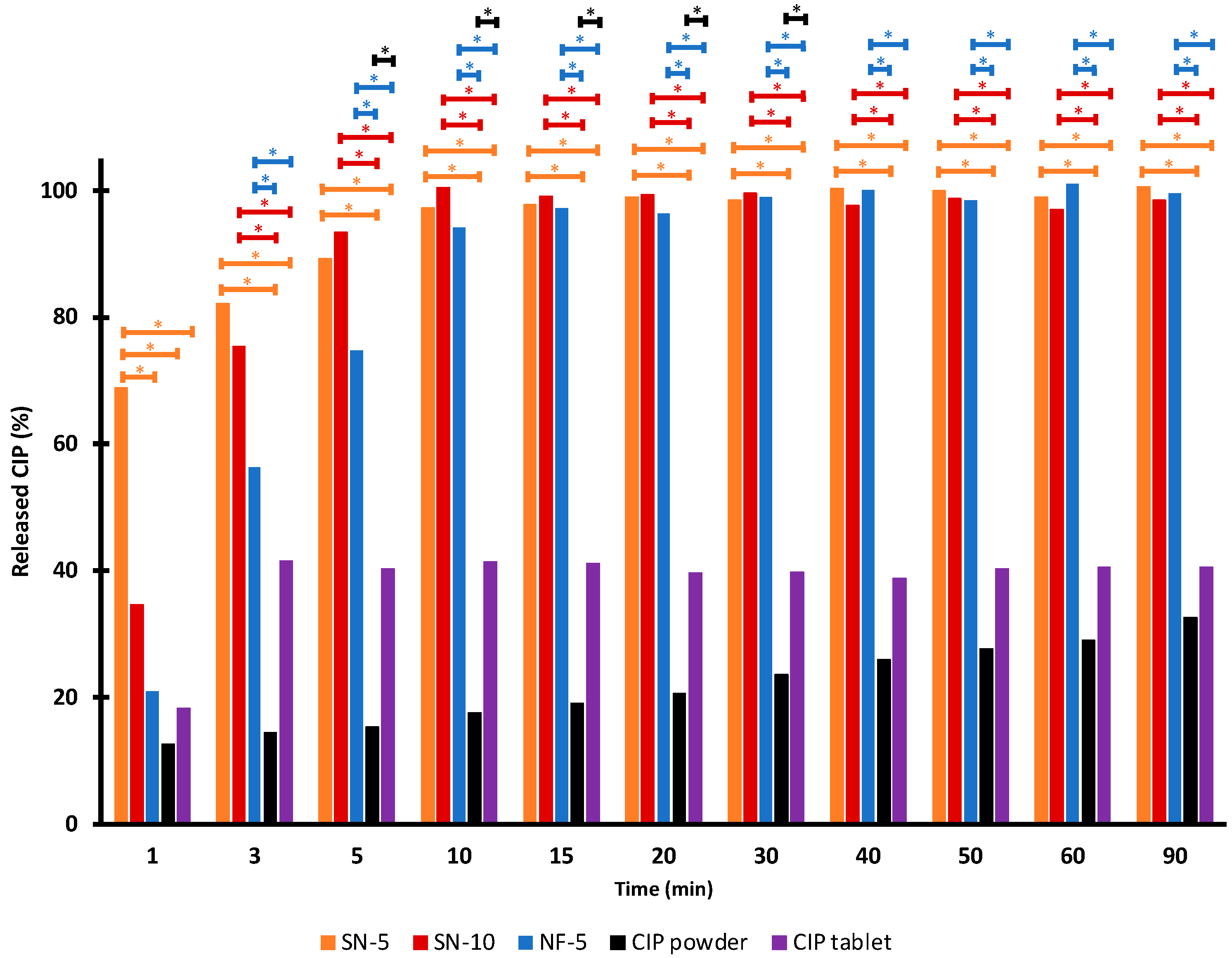
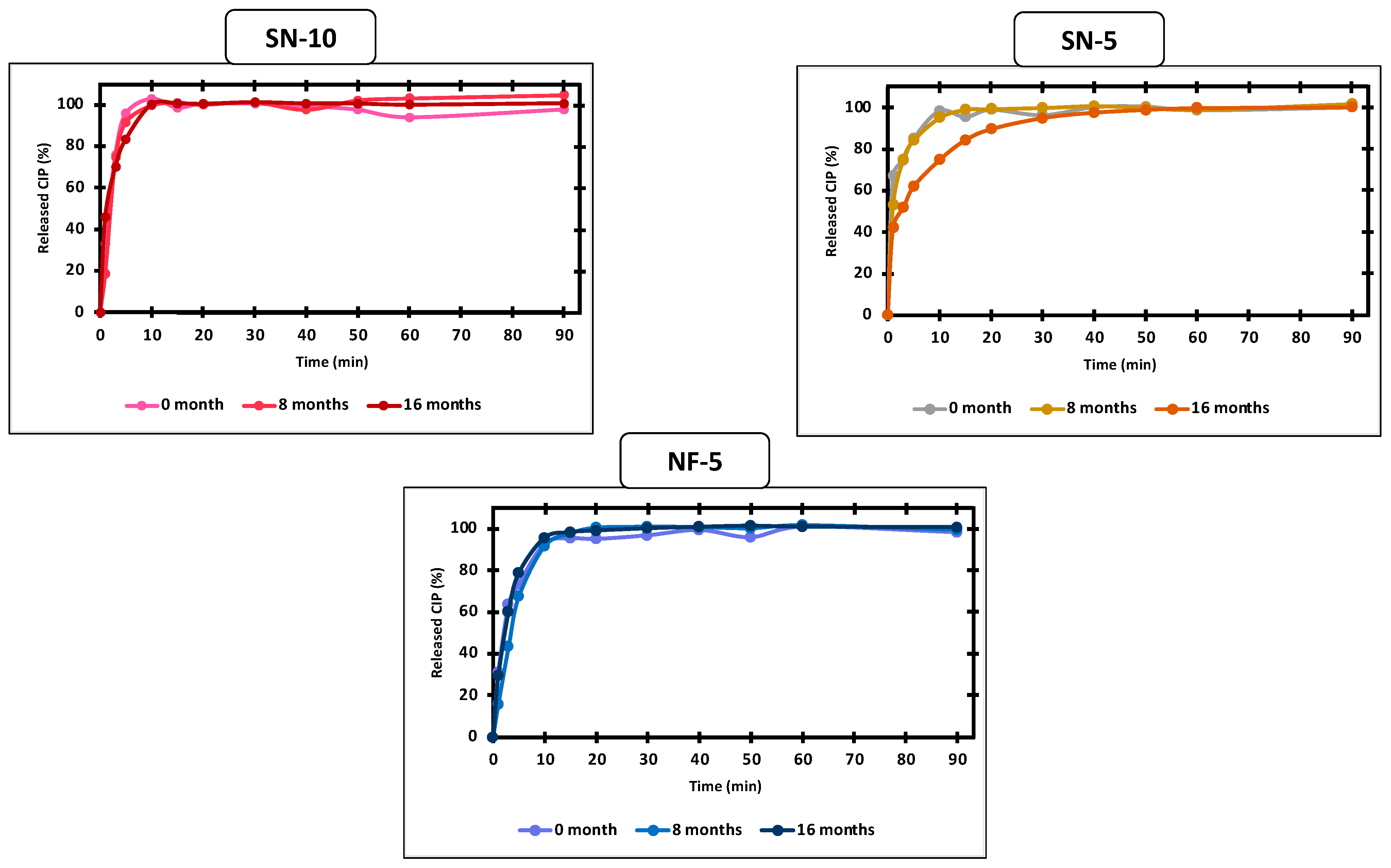
3.5. In Vitro Drug-Release Studies
3.6. Study of Drug-Release Kinetics
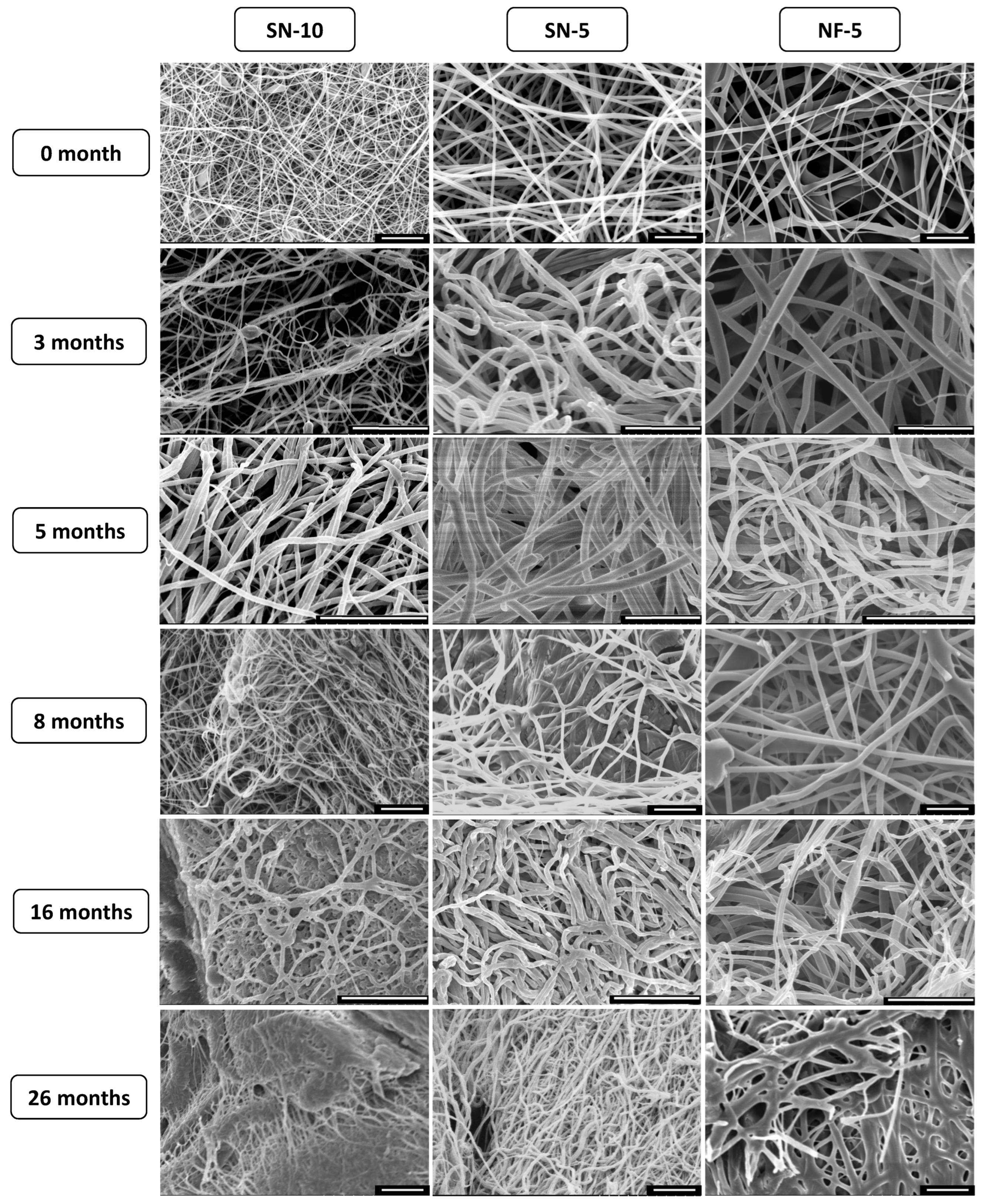
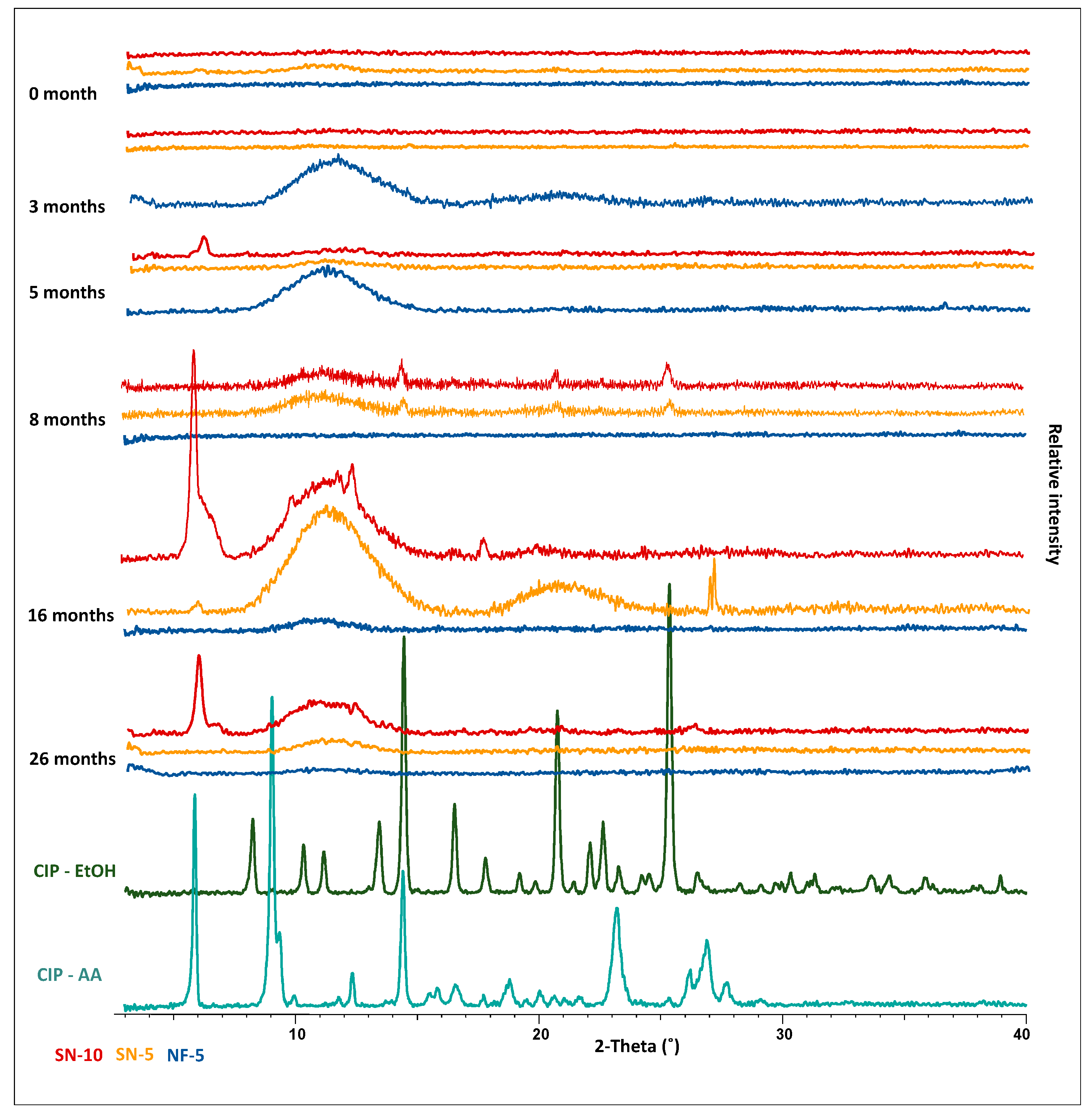
3.7. Long-Term Storage Stability
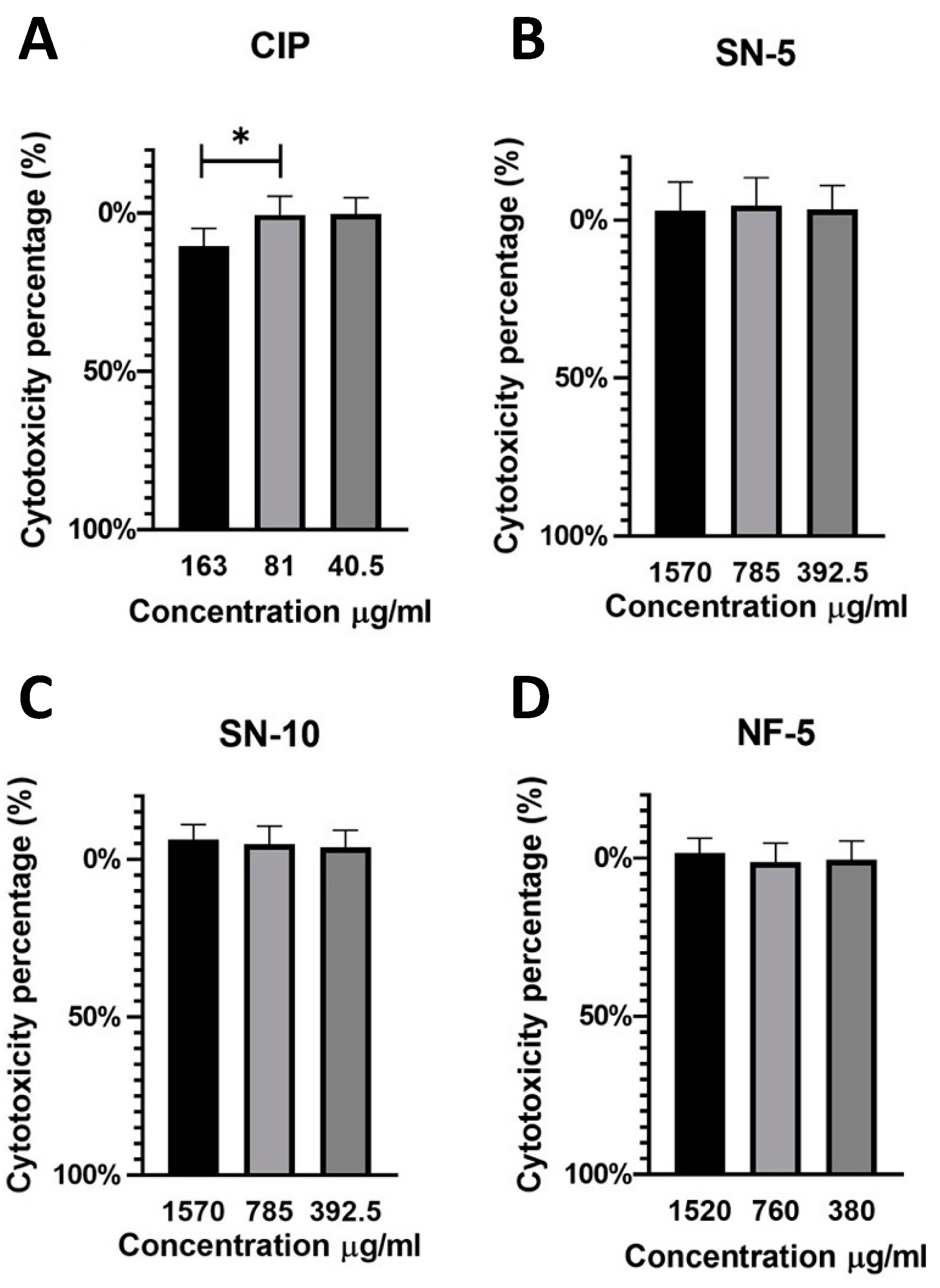
3.8. In Vitro Cytotoxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lipinski, C. Poor Aqueous Solubility—An Industry Wide Problem in Drug Discovery. Am. Pharm. Rev. 2002, 5, 82–85. [Google Scholar]
- Sharma, M.; Sharma, R.; Jain, D.K. Nanotechnology Based Approaches for Enhancing Oral Bioavailability of Poorly Water Soluble Antihypertensive Drugs. Available online: https://new.hindawi.com/journals/scientifica/2016/8525679/ (accessed on 9 January 2020).
- Yu, D.-G.; Li, J.-J.; Williams, G.R.; Zhao, M. Electrospun Amorphous Solid Dispersions of Poorly Water-Soluble Drugs: A Review. J. Control. Release 2018, 292, 91–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrus, R.; Alshweiat, A.; Csóka, I.; Ovari, G.; Esmail, A.; Radacsi, N. 3D-Printed Electrospinning Setup for the Preparation of Loratadine Nanofibers with Enhanced Physicochemical Properties. Int. J. Pharm. 2019, 567, 118455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radacsi, N.; Giapis, K.P.; Ovari, G.; Szabó-Révész, P.; Ambrus, R. Electrospun Nanofiber-Based Niflumic Acid Capsules with Superior Physicochemical Properties. J. Pharm. Biomed. Anal. 2019, 166, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Greiner, A.; Wendorff, J.H. Electrospinning: A Fascinating Method for the Preparation of Ultrathin Fibers. Angew. Chem. Int. Ed. Engl. 2007, 46, 5670–5703. [Google Scholar] [CrossRef]
- Verreck, G.; Chun, I.; Peeters, J.; Rosenblatt, J.; Brewster, M.E. Preparation and Characterization of Nanofibers Containing Amorphous Drug Dispersions Generated by Electrostatic Spinning. Pharm. Res. 2003, 20, 810–817. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, Y.; Wu, J.; Shi, Z.; Zhao, P.; Su, H.; Wang, Q.; Jin, L. Nanofiber Orodispersible Films Based on Carboxymethyl Curdlan and PEO: New Delivery System for Amlodipine Besylate. Colloids Surf. A Physicochem. Eng. Asp. 2022, 635, 128096. [Google Scholar] [CrossRef]
- Chachlioutaki, K.; Tzimtzimis, E.K.; Tzetzis, D.; Chang, M.-W.; Ahmad, Z.; Karavasili, C.; Fatouros, D.G. Electrospun Orodispersible Films of Isoniazid for Pediatric Tuberculosis Treatment. Pharmaceutics 2020, 12, 470. [Google Scholar] [CrossRef]
- Park, S.; Park, K.; Yoon, H.; Son, J.; Min, T.; Kim, G. Apparatus for Preparing Electrospun Nanofibers: Designing an Electrospinning Process for Nanofiber Fabrication. Polym. Int. 2007, 56, 1361–1366. [Google Scholar] [CrossRef]
- Cleeton, C.; Keirouz, A.; Chen, X.; Radacsi, N. Electrospun Nanofibers for Drug Delivery and Biosensing. ACS Biomater. Sci. Eng. 2019, 5, 4183–4205. [Google Scholar] [CrossRef] [PubMed]
- Paaver, U.; Heinämäki, J.; Laidmäe, I.; Lust, A.; Kozlova, J.; Sillaste, E.; Kirsimäe, K.; Veski, P.; Kogermann, K. Electrospun Nanofibers as a Potential Controlled-Release Solid Dispersion System for Poorly Water-Soluble Drugs. Int. J. Pharm. 2015, 479, 252–260. [Google Scholar] [CrossRef]
- Abdel-Hady, F.; Alzahrany, A.; Hamed, M. Experimental Validation of Upward Electrospinning Process. ISRN Nanotechnol. 2011, 2011, 851317. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Wang, D.; Zhang, Z.; Pan, J.; Cui, Z.; Yu, D.-G.; Annie Bligh, S.-W. Testing of Fast Dissolution of Ibuprofen from Its Electrospun Hydrophilic Polymer Nanocomposites. Polym. Test. 2021, 93, 106872. [Google Scholar] [CrossRef]
- Begum, H.A.; Khan, K.R. Study on the Various Types of Needle Based and Needleless Electrospinning System for Nanofiber Production. Int. J. Text. Sci. 2017, 6, 8. [Google Scholar]
- SalehHudin, H.S.; Mohamad, E.N.; Mahadi, W.N.L.; Muhammad Afifi, A. Multiple-Jet Electrospinning Methods for Nanofiber Processing: A Review. Mater. Manuf. Processes 2018, 33, 479–498. [Google Scholar] [CrossRef]
- Nagy, Z.K.; Balogh, A.; Démuth, B.; Pataki, H.; Vigh, T.; Szabó, B.; Molnár, K.; Schmidt, B.T.; Horák, P.; Marosi, G.; et al. High Speed Electrospinning for Scaled-up Production of Amorphous Solid Dispersion of Itraconazole. Int. J. Pharm. 2015, 480, 137–142. [Google Scholar] [CrossRef]
- Radacsi, N.; Campos, F.D.; Chisholm, C.R.I.; Giapis, K.P. Spontaneous Formation of Nanoparticles on Electrospun Nanofibres. Nat. Commun. 2018, 9, 4740. [Google Scholar] [CrossRef] [Green Version]
- Erben, J.; Kalous, T.; Chvojka, J. Ac Bubble Electrospinning Technology for Preparation of Nanofibrous Mats. ACS Omega 2020, 5, 8268–8271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakkarainen, E.; Kõrkjas, T.; Laidmäe, I.; Lust, A.; Semjonov, K.; Kogermann, K.; Nieminen, H.J.; Salmi, A.; Korhonen, O.; Haeggström, E.; et al. Comparison of Traditional and Ultrasound-Enhanced Electrospinning in Fabricating Nanofibrous Drug Delivery Systems. Pharmaceutics 2019, 11, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarin, A.L.; Zussman, E. Upward Needleless Electrospinning of Multiple Nanofibers. Polymer 2004, 45, 2977–2980. [Google Scholar] [CrossRef]
- Molnar, K.; Nagy, Z.K. Corona-Electrospinning: Needleless Method for High-Throughput Continuous Nanofiber Production. Eur. Polym. J. 2016, 74, 279–286. [Google Scholar] [CrossRef]
- Storck, J.L.; Grothe, T.; Mamun, A.; Sabantina, L.; Klöcker, M.; Blachowicz, T.; Ehrmann, A. Orientation of Electrospun Magnetic Nanofibers Near Conductive Areas. Materials 2019, 13, 47. [Google Scholar] [CrossRef] [Green Version]
- Jirsak, O.; Sysel, P.; Sanetrnik, F.; Hruza, J.; Chaloupek, J. Polyamic Acid Nanofibers Produced by Needleless Electrospinning. J. Nanomater. 2010, 2010, 842831. [Google Scholar] [CrossRef] [PubMed]
- Waqas, M.; Keirouz, A.; Sanira Putri, M.K.; Fazal, F.; Diaz Sanchez, F.J.; Ray, D.; Koutsos, V.; Radacsi, N. Design and Development of a Nozzle-Free Electrospinning Device for the High-Throughput Production of Biomaterial Nanofibers. Med. Eng. Phys. 2021, 92, 80–87. [Google Scholar] [CrossRef]
- Keirouz, A.; Fortunato, G.; Zhang, M.; Callanan, A.; Radacsi, N. Nozzle-Free Electrospinning of Polyvinylpyrrolidone/Poly(Glycerol Sebacate) Fibrous Scaffolds for Skin Tissue Engineering Applications. Med. Eng. Phys. 2019, 71, 56–67. [Google Scholar] [CrossRef]
- Keirouz, A.; Zakharova, M.; Kwon, J.; Robert, C.; Koutsos, V.; Callanan, A.; Chen, X.; Fortunato, G.; Radacsi, N. High-Throughput Production of Silk Fibroin-Based Electrospun Fibers as Biomaterial for Skin Tissue Engineering Applications. Mater. Sci. Eng. C 2020, 112, 110939. [Google Scholar] [CrossRef]
- Olivera, M.; Manzo, R.H.; Junginger, H.; Midha, K.K.; Shah, V.P.; Stavchansky, S.A.; Dressman, J.; Barends, D.M. Biowaiver Monographs for Immediate Release Solid Oral Dosage Forms: Ciprofloxacin Hydrochloride. J. Pharm. Sci. 2011, 100, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Uhljar, L.É.; Kan, S.Y.; Radacsi, N.; Koutsos, V.; Szabó-Révész, P.; Ambrus, R. In Vitro Drug Release, Permeability, and Structural Test of Ciprofloxacin-Loaded Nanofibers. Pharmaceutics 2021, 13, 556. [Google Scholar] [CrossRef]
- Caco, A.I.; Varanda, F.; de Melo, M.J.P.; Dias, A.M.A.; Dohrn, R.; Marrucho, I.M. Solubility of Antibiotics in Different Solvents. Part II. Non-Hydrochloride Forms of Tetracycline and Ciprofloxacin. Ind. Eng. Chem. Res. 2008, 47, 8083–8089. [Google Scholar] [CrossRef]
- Choi, J.M.; Park, K.; Lee, B.; Jeong, D.; Dindulkar, S.D.; Choi, Y.; Cho, E.; Park, S.; Yu, J.-H.; Jung, S. Solubility and Bioavailability Enhancement of Ciprofloxacin by Induced Oval-Shaped Mono-6-Deoxy-6-Aminoethylamino-β-Cyclodextrin. Carbohydr. Polym. 2017, 163, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Bonthagarala, B.; Pola, L.M.; Nama, S. Enhancement of Dissolution Rate of Ciprofloxacin by Using Various Solid Dispersion Techniques. Int. J. Pharm. Sci. Res. 2013, 4, 4376–4383. [Google Scholar]
- Francés, C.; Veiga, M.D.; Español, O.M.; Cadórniga, R. Preparation, Characterization and Dissolution of Ciprofloxacin/PEG 6000 Binary Systems. Int. J. Pharm. 1991, 77, 193–198. [Google Scholar] [CrossRef]
- Mesallati, H.; Umerska, A.; Paluch, K.J.; Tajber, L. Amorphous Polymeric Drug Salts as Ionic Solid Dispersion Forms of Ciprofloxacin. Mol. Pharm. 2017, 14, 2209–2223. [Google Scholar] [CrossRef] [Green Version]
- Shazly, G.A. Ciprofloxacin Controlled-Solid Lipid Nanoparticles: Characterization, In Vitro Release, and Antibacterial Activity Assessment. BioMed Res. Int. 2017, 2017, 2120734. [Google Scholar] [CrossRef] [Green Version]
- Sobhani, Z.; Mohammadi Samani, S.; Montaseri, H.; Khezri, E. Nanoparticles of Chitosan Loaded Ciprofloxacin: Fabrication and Antimicrobial Activity. Adv. Pharm. Bull. 2017, 7, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Masoumi, S.; Amiri, S.; Bahrami, S.H. PCL-Based Nanofibers Loaded with Ciprofloxacin/Cyclodextrin Container. J. Text. Inst. 2018, 109, 1044–1053. [Google Scholar] [CrossRef]
- Modgill, V.; Garg, T.; Goyal, A.K.; Rath, G. Permeability Study of Ciprofloxacin from Ultra-Thin Nanofibrous Film through Various Mucosal Membranes. Artif. Cells Nanomed. Biotechnol. 2014, 44, 122–127. [Google Scholar] [CrossRef]
- Kyzioł, A.; Michna, J.; Moreno, I.; Gamez, E.; Irusta, S. Preparation and Characterization of Electrospun Alginate Nanofibers Loaded with Ciprofloxacin Hydrochloride. Eur. Polym. J. 2017, 96, 350–360. [Google Scholar] [CrossRef] [Green Version]
- Unnithan, A.R.; Barakat, N.A.M.; Pichiah, P.B.T.; Gnanasekaran, G.; Nirmala, R.; Chad, Y.-S.; Jung, C.-H.; El-Newehy, M.; Kim, H.Y. Wound-Dressing Materials with Antibacterial Activity from Electrospun Polyurethane–Dextran Nanofiber Mats Containing Ciprofloxacin HCl. Carbohydr. Polym. 2012, 90, 1786–1793. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, L.; Wu, T.; Cai, T.; Gunn, E.M.; Yu, L. Stability of Amorphous Pharmaceutical Solids: Crystal Growth Mechanisms and Effect of Polymer Additives. AAPS J. 2012, 14, 380–388. [Google Scholar] [CrossRef]
- Brettmann, B.K.; Myerson, A.S.; Trout, B.L. Solid-State Nuclear Magnetic Resonance Study of the Physical Stability of Electrospun Drug and Polymer Solid Solutions. J. Pharm. Sci. 2012, 101, 2185–2193. [Google Scholar] [CrossRef]
- Vigh, T.; Horváthová, T.; Balogh, A.; Sóti, P.L.; Drávavölgyi, G.; Nagy, Z.K.; Marosi, G. Polymer-Free and Polyvinylpirrolidone-Based Electrospun Solid Dosage Forms for Drug Dissolution Enhancement. Eur. J. Pharm. Sci. 2013, 49, 595–602. [Google Scholar] [CrossRef]
- Lopez, F.L.; Shearman, G.C.; Gaisford, S.; Williams, G.R. Amorphous Formulations of Indomethacin and Griseofulvin Prepared by Electrospinning. Mol. Pharm. 2014, 11, 4327–4338. [Google Scholar] [CrossRef]
- Poller, B.; Strachan, C.; Broadbent, R.; Walker, G.F. A Minitablet Formulation Made from Electrospun Nanofibers. Eur. J. Pharm. Biopharm. 2017, 114, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Bukhary, H.; Williams, G.R.; Orlu, M. Electrospun Fixed Dose Formulations of Amlodipine Besylate and Valsartan. Int. J. Pharm. 2018, 549, 446–455. [Google Scholar] [CrossRef]
- Li, J.; Pan, H.; Ye, Q.; Shi, C.; Zhang, X.; Pan, W. Carvedilol-Loaded Polyvinylpyrrolidone Electrospun Nanofiber Film for Sublingual Delivery. J. Drug Deliv. Sci. Technol. 2020, 58, 101726. [Google Scholar] [CrossRef]
- Geng, Y.; Zhou, F.; Williams, G.R. Developing and Scaling up Fast-Dissolving Electrospun Formulations Based on Poly(Vinylpyrrolidone) and Ketoprofen. J. Drug Deliv. Sci. Technol. 2021, 61, 102138. [Google Scholar] [CrossRef]
- Kazsoki, A.; Szabó, P.; Domján, A.; Balázs, A.; Bozó, T.; Kellermayer, M.; Farkas, A.; Balogh-Weiser, D.; Pinke, B.; Darcsi, A.; et al. Microstructural Distinction of Electrospun Nanofibrous Drug Delivery Systems Formulated with Different Excipients. Mol. Pharm. 2018, 15, 4214–4225. [Google Scholar] [CrossRef]
- Tort, S.; Acartürk, F. Preparation and Characterization of Electrospun Nanofibers Containing Glutamine. Carbohydr. Polym. 2016, 152, 802–814. [Google Scholar] [CrossRef]
- Khoshbakht, S.; Asghari-Sana, F.; Fathi-Azarbayjani, A.; Sharifi, Y. Fabrication and Characterization of Tretinoin-Loaded Nanofiber for Topical Skin Delivery. Biomater. Res. 2020, 24, 8. [Google Scholar] [CrossRef] [Green Version]
- Borbás, E.; Balogh, A.; Bocz, K.; Müller, J.; Kiserdei, É.; Vigh, T.; Sinkó, B.; Marosi, A.; Halász, A.; Dohányos, Z.; et al. In Vitro Dissolution–Permeation Evaluation of an Electrospun Cyclodextrin-Based Formulation of Aripiprazole Using ΜFluxTM. Int. J. Pharm. 2015, 491, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Celebioglu, A.; Uyar, T. Hydrocortisone/Cyclodextrin Complex Electrospun Nanofibers for a Fast-Dissolving Oral Drug Delivery System. RSC Med. Chem. 2020, 11, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.K.; Balogh, A.; Vajna, B.; Farkas, A.; Patyi, G.; Kramarics, Á.; Marosi, G. Comparison of Electrospun and Extruded Soluplus®-Based Solid Dosage Forms of Improved Dissolution. J. Pharm. Sci. 2012, 101, 322–332. [Google Scholar] [CrossRef]
- Sóti, P.L.; Bocz, K.; Pataki, H.; Eke, Z.; Farkas, A.; Verreck, G.; Kiss, É.; Fekete, P.; Vigh, T.; Wagner, I.; et al. Comparison of Spray Drying, Electroblowing and Electrospinning for Preparation of Eudragit E and Itraconazole Solid Dispersions. Int. J. Pharm. 2015, 494, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Balogh, A.; Horváthová, T.; Fülöp, Z.; Loftsson, T.; Harasztos, A.H.; Marosi, G.; Nagy, Z.K. Electroblowing and Electrospinning of Fibrous Diclofenac Sodium-Cyclodextrin Complex-Based Reconstitution Injection. J. Drug Deliv. Sci. Technol. 2015, 26, 28–34. [Google Scholar] [CrossRef]
- Celebioglu, A. Electrospun Formulation of Acyclovir/Cyclodextrin Nanofibers for Fast-Dissolving Antiviral Drug Delivery. Mater. Sci. 2021, 118, 111514. [Google Scholar] [CrossRef]
- Kajdič, S.; Vrečer, F.; Kocbek, P. Preparation of Poloxamer-Based Nanofibers for Enhanced Dissolution of Carvedilol. Eur. J. Pharm. Sci. 2018, 117, 331–340. [Google Scholar] [CrossRef]
- Hirsch, E.; Pantea, E.; Vass, P.; Domján, J.; Molnár, M.; Suhajda, Á.; Andersen, S.K.; Vigh, T.; Verreck, G.; Marosi, G.J.; et al. Probiotic Bacteria Stabilized in Orally Dissolving Nanofibers Prepared by High-Speed Electrospinning. Food Bioprod. Processing 2021, 128, 84–94. [Google Scholar] [CrossRef]
- Démuth, B.; Farkas, A.; Pataki, H.; Balogh, A.; Szabó, B.; Borbás, E.; Sóti, P.L.; Vigh, T.; Kiserdei, É.; Farkas, B.; et al. Detailed Stability Investigation of Amorphous Solid Dispersions Prepared by Single-Needle and High Speed Electrospinning. Int. J. Pharm. 2016, 498, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Paarakh, M.P.; Jose, P.A.; Setty, C.; Peter, G.V. Release Kinetics—Concepts and Applications. Int. J. Pharm. Res. Technol. 2018, 8, 12. [Google Scholar]
- Ajmal, G.; Bonde, G.V.; Thokala, S.; Mittal, P.; Khan, G.; Singh, J.; Pandey, V.K.; Mishra, B. Ciprofloxacin HCl and Quercetin Functionalized Electrospun Nanofiber Membrane: Fabrication and Its Evaluation in Full Thickness Wound Healing. Artif. Cells Nanomed. Biotechnol. 2019, 47, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Contardi, M.; Heredia-Guerrero, J.A.; Perotto, G.; Valentini, P.; Pompa, P.P.; Spanò, R.; Goldoni, L.; Bertorelli, R.; Athanassiou, A.; Bayer, I.S. Transparent Ciprofloxacin-Povidone Antibiotic Films and Nanofiber Mats as Potential Skin and Wound Care Dressings. Eur. J. Pharm. Sci. 2017, 104, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Panda, B.P.; Wei, M.X.; Shivashekaregowda, N.K.H.; Patnaik, S. Design, Fabrication and Characterization of PVA/PLGA Electrospun Nanofibers Carriers for Improvement of Drug Delivery of Gliclazide in Type-2 Diabetes. In Proceedings of the 1st International Electronic Conference on Pharmaceutics, Online, 1–15 December 2020; MDPI: Basel, Switzerland, 2020; p. 14. [Google Scholar]
- Partheniadis, I.; Athanasiou, K.; Laidmäe, I.; Heinämäki, J.; Nikolakakis, I. Physicomechanical Characterization and Tablet Compression of Theophylline Nanofibrous Mats Prepared by Conventional and Ultrasound Enhanced Electrospinning. Int. J. Pharm. 2022, 616, 121558. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time Interval of the Study | Tested Properties of the Nanofibers | ||||
|---|---|---|---|---|---|
| Morphology | Crystallinity | Drug Loading | In vitro Drug Release | Reference | |
| 1 month | × | × | [49] | ||
| 1 month | × | × | [50] | ||
| 45 days | × | [51] | |||
| 3 months | × | [52] | |||
| 3 months | × | × | [53] | ||
| 3 months | × | × | [54,55] | ||
| 4 months | × | [46] | |||
| 6 months | × | [42,56] | |||
| 8 months | × | [44,57] | |||
| 10 months | × | [45] | |||
| 12 months | × | [58] | |||
| 12 months | × | [59] | |||
| 12 months | × | × | [60] | ||
| Sample | Volume Ratio | ||||||
|---|---|---|---|---|---|---|---|
| CIP Solution in Acetic Acid (mg/mL) | PVP Solution in Ethanol (w/v%) | PVP | CIP | Viscosity of the ES Solution (mPa s) | ES Setup | CIP-Loading (wt%) | |
| SN-5 | 20 | 10 | 4 | 1 | 79 ± 2 | single-nozzle | 5 |
| SN-10 | 20 | 5 | 4 | 1 | 336 ± 19 | single-nozzle | 10 |
| NF-5 | 20 | 10 | 4 | 1 | 79 ± 2 | nozzle-free | 5 |
| NF-10 | 20 | 5 | 4 | 1 | 336 ± 19 | nozzle-free | 10 |
| Electrospinning Setup | Sample | Fiber Morphology | Av Diameter ± S.D. (nm) | |
|---|---|---|---|---|
| single-nozzle | SN-5 | continuous, smooth-surfaced | mainly uniform, some beads | 323 ± 51 |
| SN-10 | continuous, smooth-surfaced | large number of beads and bags | 735 ± 91 | |
| nozzle-free | NF-5 | continuous, smooth-surfaced | thicker and thinner fibers | 1167 ± 415 |
| NF-10 | merged, rough-surfaced | more film than nanofiber | unmeasurable | |
| ES Setup | Sample | Theoretical CIP Content (%) | DL (%) ± S.D. | EE (%) ± S.D. |
|---|---|---|---|---|
| single-nozzle | SN-10 center | 10 | 8.72 ± 1.42 | 87.2 ± 14.2 |
| SN-10 edge | 10 | 7.01 ± 1.64 | 70.1 ± 16.4 | |
| SN-5 center | 5 | 3.70 ± 1.62 | 73.9 ± 32.5 | |
| SN-5 edge | 5 | 3.36 ± 0.61 | 67.1 ± 12.1 | |
| nozzle-free | NF-5 center | 5 | 4.43 ± 0.47 | 88.7 ± 14.2 |
| NF-5 edge | 5 | 4.67 ± 0.75 | 93.3 ± 15.0 |
| Model | SN-5 | SN-10 | NF-5 | CIP Powder | CIP Tablet |
|---|---|---|---|---|---|
| Zero order | 0.5297 | 0.7317 | 0.7874 | 0.7510 | 0.5636 |
| First order | 0.9352 | 0.8809 | 0.9870 | 0.8062 | 0.5794 |
| Higuchi | 0.8103 | 0.9327 | 0.9491 | 0.9148 | 0.8243 |
| Hixson–Crowell | 0.8061 | 0.9876 | 0.9437 | 0.7880 | 0.5745 |
| Korsmeyer–Peppas | 0.9994 | 0.8657 | 0.9836 | 0.9033 | 0.7334 |
| Average Fiber Diameter ± Standard Deviation (nm) | |||
|---|---|---|---|
| Time | SN-10 | SN-5 | NF-5 |
| 0 month | 323 ± 51 | 735 ± 91 | 1167 ± 415 |
| 3 months | 328 ± 84 | 739 ± 105 | 1195 ± 464 |
| 5 months | 345 ± 108 | 866 ± 233 | 1159 ± 406 |
| 8 months | 304 ± 53 | 805 ± 133 | 1418 ± 343 |
| 16 months | 434 ± 69 | 783 ± 162 | 1444 ± 478 |
| 26 months | 442 ± 89 | 707 ± 185 | 2148 ± 853 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uhljar, L.É.; Alshweiat, A.; Katona, G.; Chung, M.; Radacsi, N.; Kókai, D.; Burián, K.; Ambrus, R. Comparison of Nozzle-Based and Nozzle-Free Electrospinning for Preparation of Fast-Dissolving Nanofibers Loaded with Ciprofloxacin. Pharmaceutics 2022, 14, 1559. https://doi.org/10.3390/pharmaceutics14081559
Uhljar LÉ, Alshweiat A, Katona G, Chung M, Radacsi N, Kókai D, Burián K, Ambrus R. Comparison of Nozzle-Based and Nozzle-Free Electrospinning for Preparation of Fast-Dissolving Nanofibers Loaded with Ciprofloxacin. Pharmaceutics. 2022; 14(8):1559. https://doi.org/10.3390/pharmaceutics14081559
Chicago/Turabian StyleUhljar, Luca Éva, Areen Alshweiat, Gábor Katona, Michael Chung, Norbert Radacsi, Dávid Kókai, Katalin Burián, and Rita Ambrus. 2022. "Comparison of Nozzle-Based and Nozzle-Free Electrospinning for Preparation of Fast-Dissolving Nanofibers Loaded with Ciprofloxacin" Pharmaceutics 14, no. 8: 1559. https://doi.org/10.3390/pharmaceutics14081559