
Enhanced Oral Bioavailability of MT-102, a New Anti-inflammatory Agent, via a Ternary Solid Dispersion Formulation



Abstract
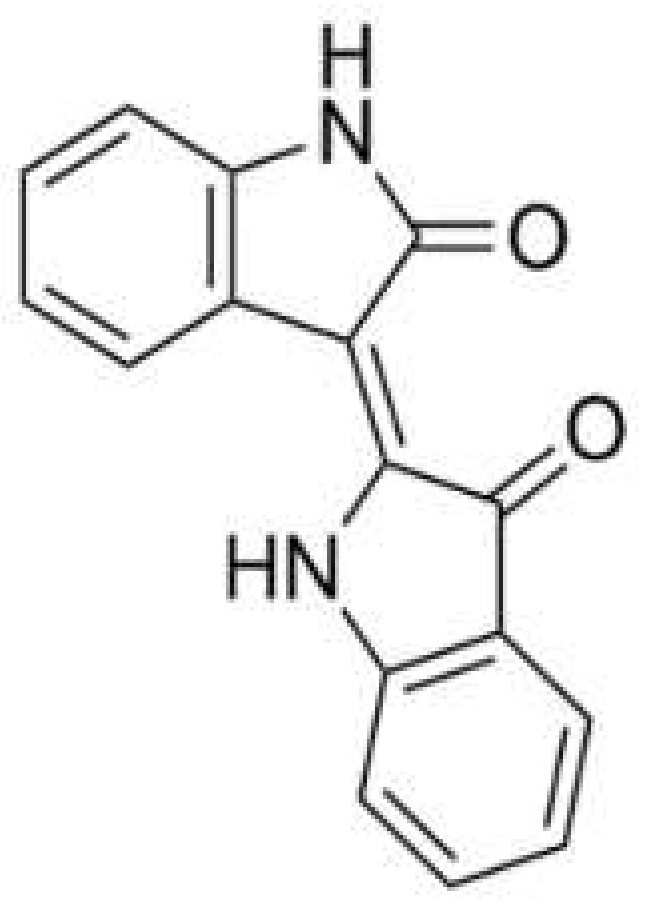
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Screening of Carriers and Preparation of SDs
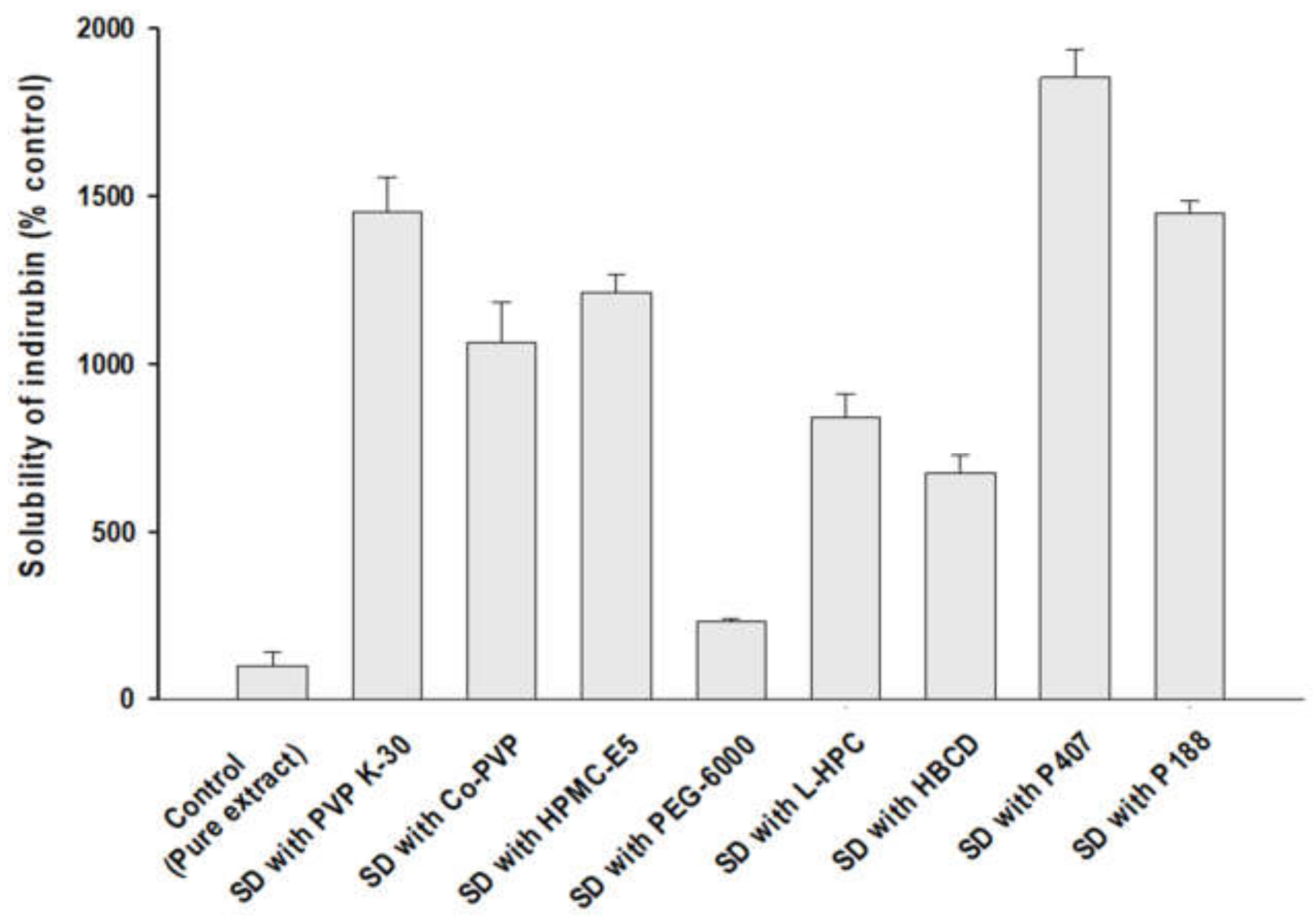
2.3. Solubility Studies for Carrier Selection
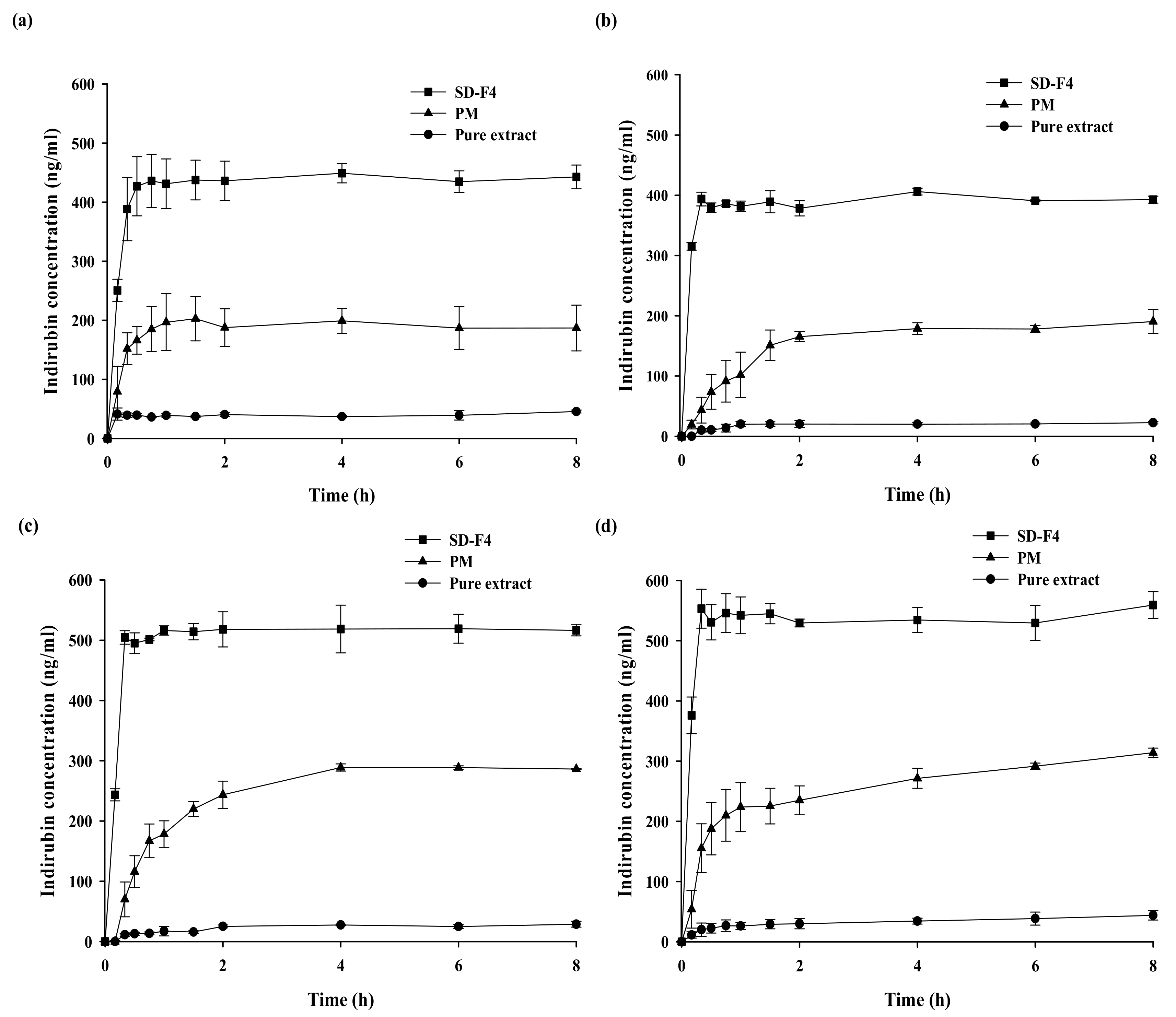
2.4. In Vitro Drug Release Studies
2.5. Morphology
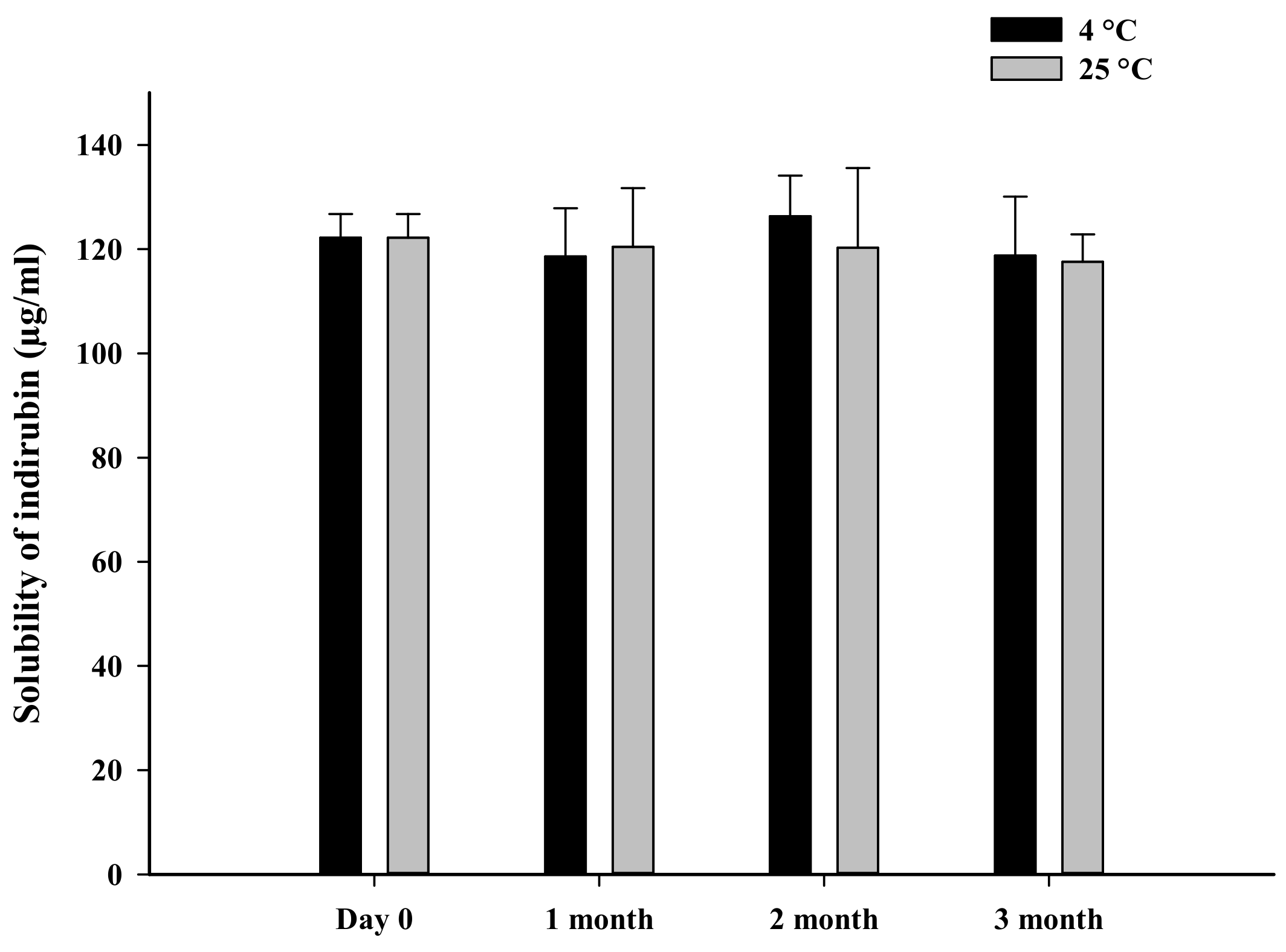
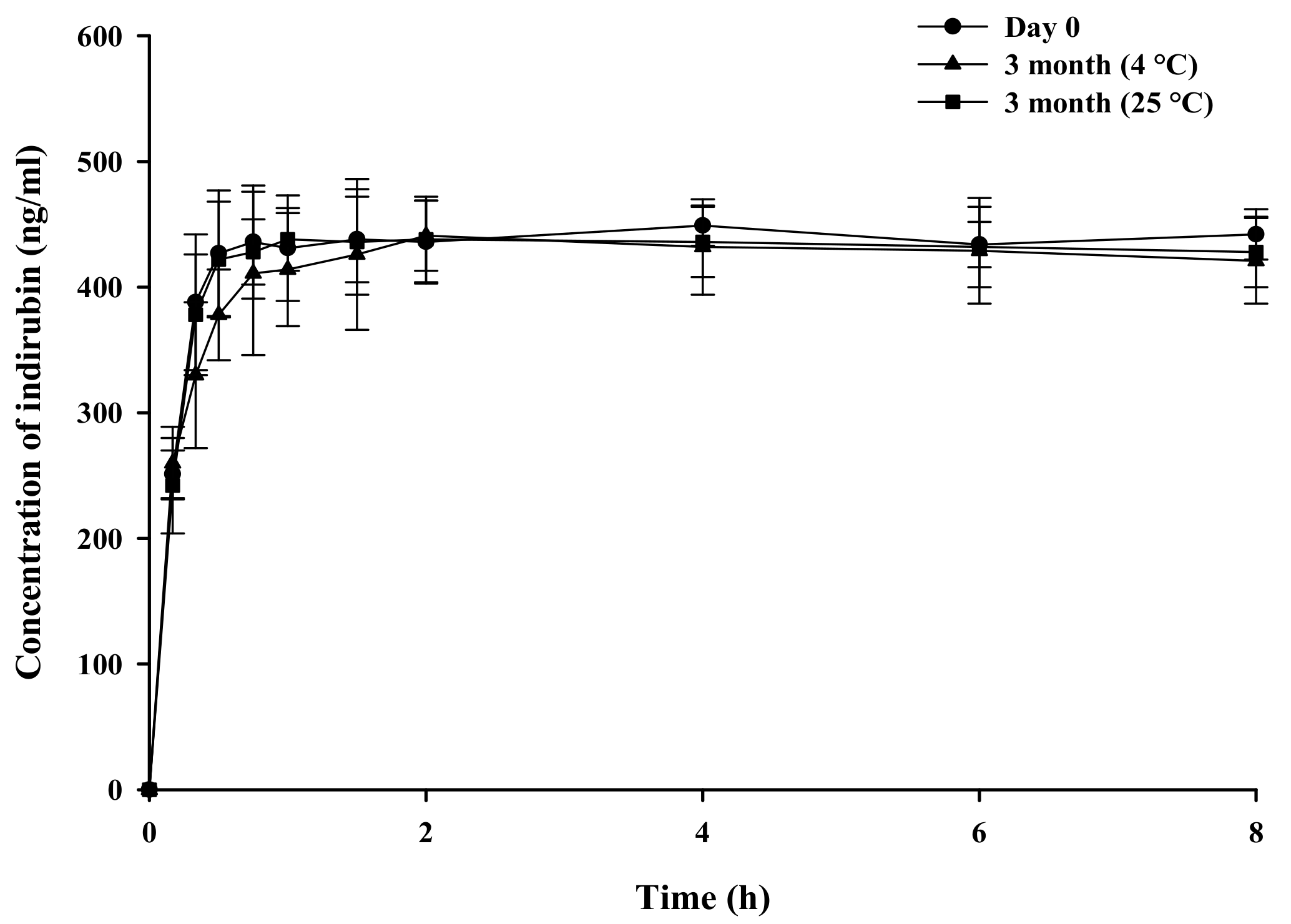
2.6. Stability Studies
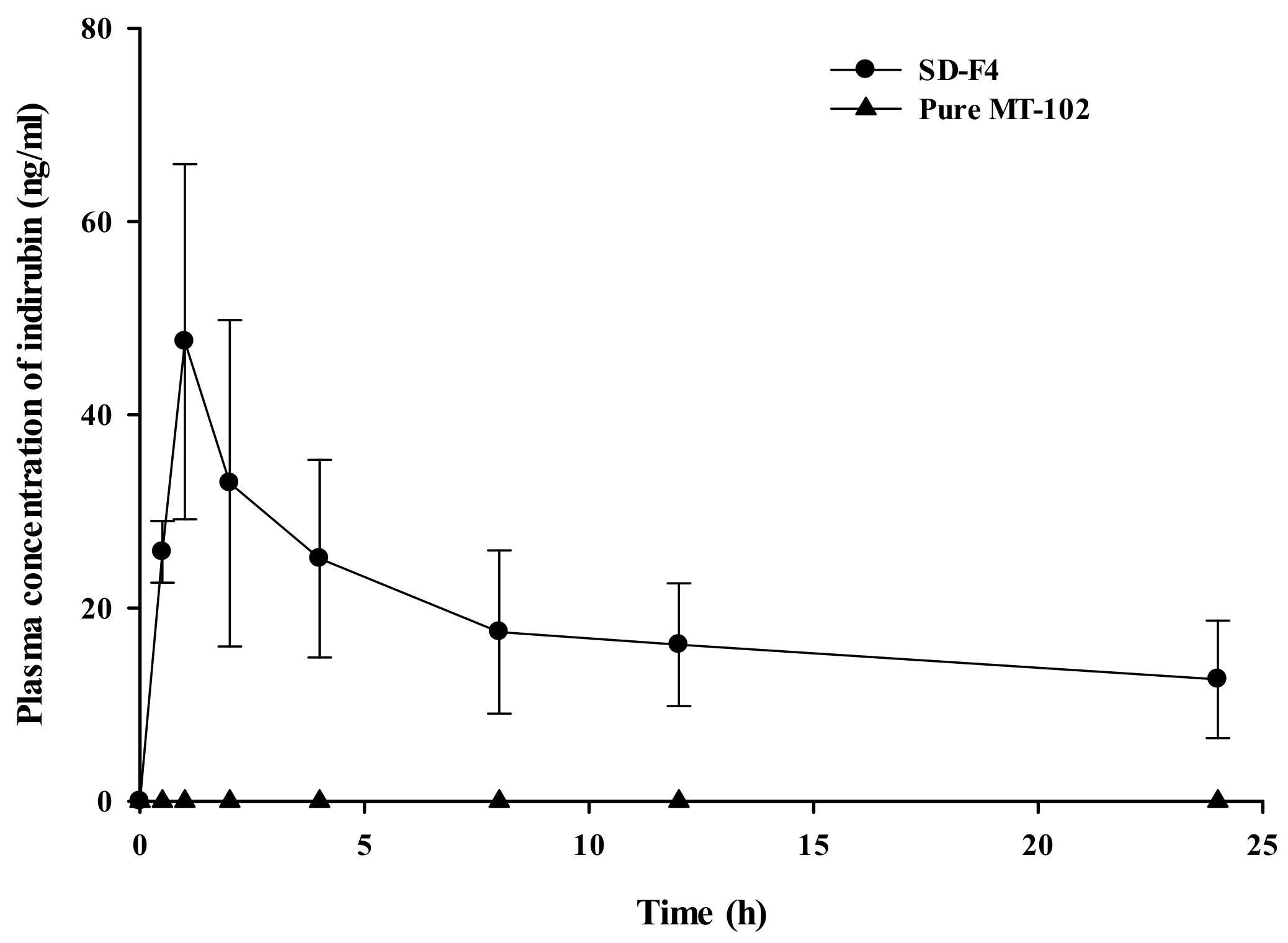
2.7. Pharmacokinetic Studies in Rats
2.8. Analytical Methods
2.9. Pharmacokinetic and Statistical Analysis
3. Results and Discussions
3.1. Selection of Excipients
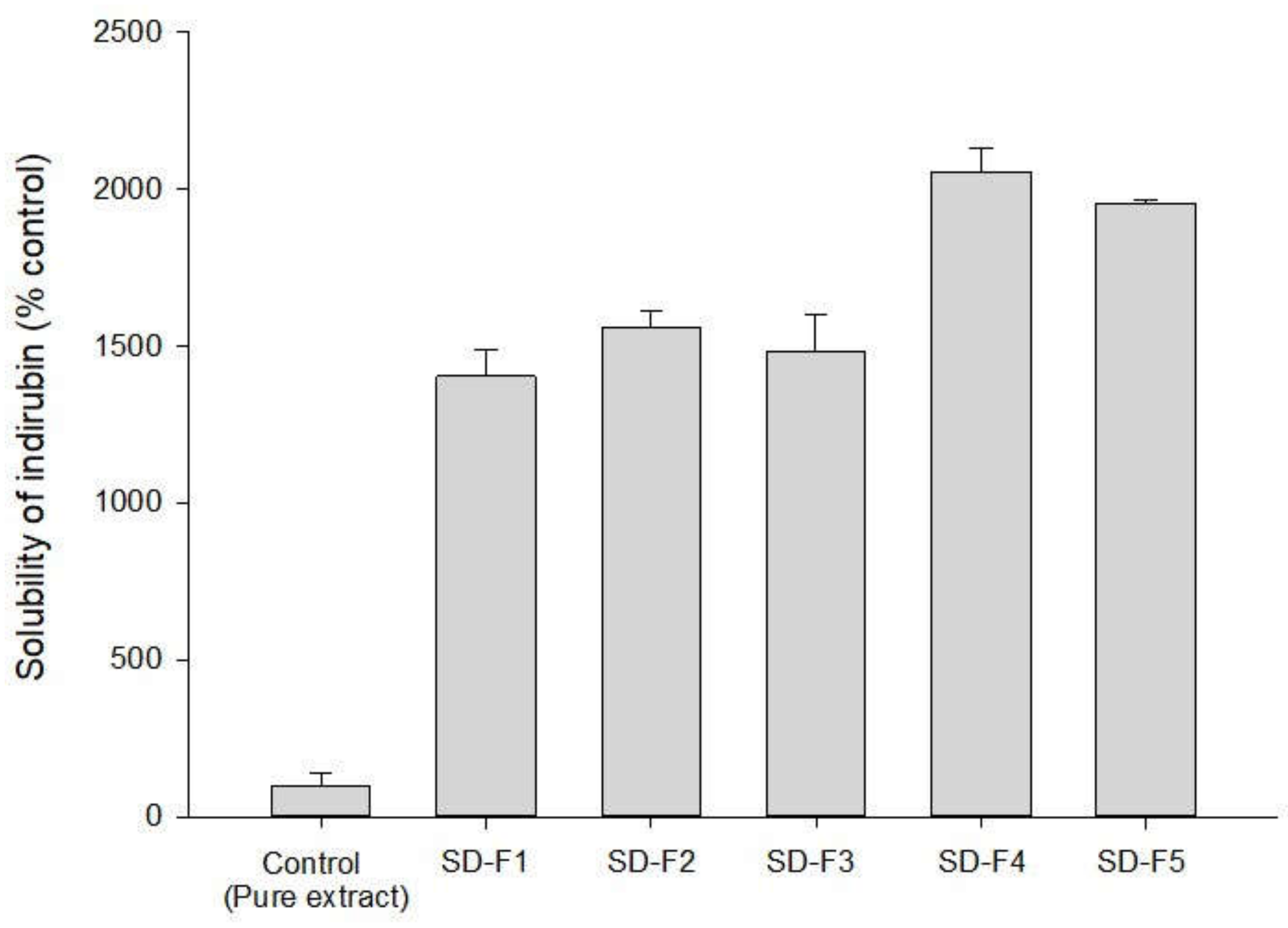
3.2. Optimization of SD Formulations
3.3. Storage Stability
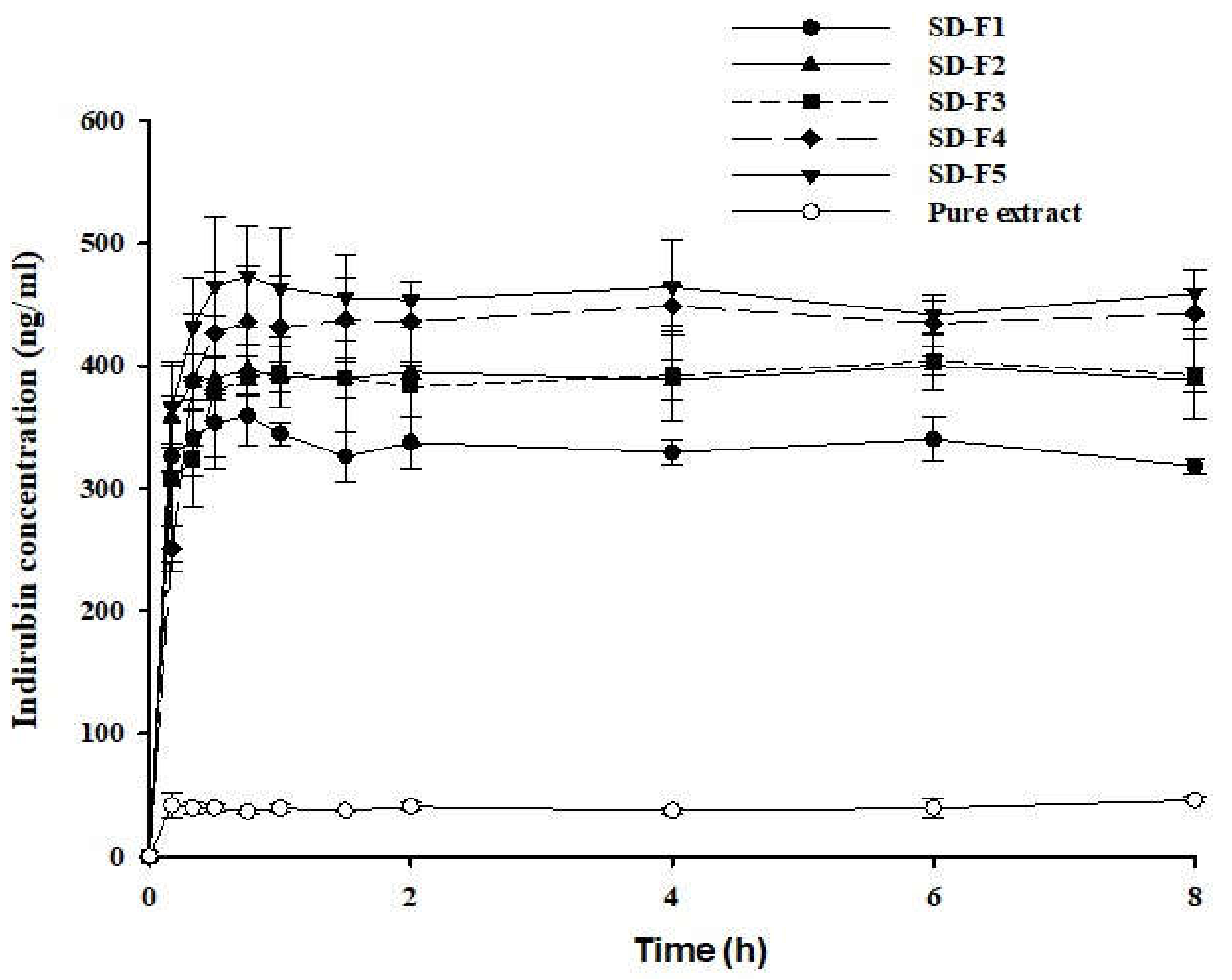
3.4. Pharmacokinetic Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Riansuwan, W.; Limsrivilai, J. Current status of IBD and surgery of Crohn’s disease in Thailand. Ann. Gastroenterol. Surg. 2021, 5, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Eliakim, R. Non steroidal anti-inflammatory drugs and inflammatory bowel disease. Pharmaceuticals 2010, 3, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Ju, S.; Park, S.; Jung, Y. 5-[(3-Carboxy-4-hydroxyphenyl) diazenyl] nicotinic acid, an azo-linked mesalazine-nicotinic acid conjugate, is a colon-targeted mutual prodrug against dextran sulfate sodium-induced colitis in mice. J. Pharm. Investig. 2021, 51, 317–325. [Google Scholar] [CrossRef]
- Guo, B.-J.; Bian, Z.-X.; Qiu, H.-C.; Wang, Y.-T.; Wang, Y. Biological and clinical implications of herbal medicine and natural products for the treatment of inflammatory bowel disease. Ann. N. Y. Acad. Sci. 2017, 1401, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-J.; Chang, Y.-C.; Lu, K.-Z.; Tsou, Y.-Y.; Lin, C.-W. Antiviral activity of Isatis indigotica extract and its derived indirubin against Japanese encephalitis virus. Evid. Based Complement. Alternat. Med. 2012, 2012, 925830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, F.; Wang, Z.; Yang, Y.; Ji, Y.; Lv, H.; Han, K.; Liu, D.; Shang, X.; He, X.; Zeng, N. Juglans mandshurica maxim.: A review of its traditional usages, phytochemical constituents, and pharmacological properties. Front. Pharmacol. 2020, 11, 569800. [Google Scholar]
- Sweed, N.M.; Fayez, A.M.; El-Emam, S.Z.; Dawoud, M.H.S. Response surface optimization of self nano-emulsifying drug delivery system of rosuvastatin calcium for hepatocellular carcinoma. J. Pharm. Investig. 2021, 51, 85–101. [Google Scholar] [CrossRef]
- Singh, G.; Singh, D.; Choudhari, M.; Kaur, S.D.; Dubey, S.K.; Arora, S.; Bedi, N. Exemestane encapsulated copolymers L121/F127/GL44 based mixed micelles: Solubility enhancement and in vitro cytotoxicity evaluation using MCF-7 breast cancer cells. J. Pharm. Investig. 2021, 51, 701–714. [Google Scholar] [CrossRef]
- Baral, K.C.; Song, J.-G.; Lee, S.H.; Bajracharya, R.; Sreenivasulu, G.; Kim, M.; Lee, K.; Han, H.-K. Enhanced bioavailability of AC1497, a novel anticancer drug candidate, via a self-nanoemulsifying drug delivery system. Pharmaceutics 2021, 13, 1142. [Google Scholar] [CrossRef]
- Wani, S.U.D.; Kakkar, V.; Gautam, S.P.; Hv, G.; Ali, M.; Masoodi, M.H.; Moin, A. Enhancing therapeutic potential of poor aqueous soluble herbal drugs through solid dispersion-An overview. Phytomed. Plus 2021, 1, 100069. [Google Scholar] [CrossRef]
- Han, H.K.; Lee, B.J.; Lee, H.K. Enhanced dissolution and bioavailability of biochanin A via the preparation of solid dispersion: In vitro and in vivo evaluation. Int. J. Pharm. 2011, 415, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Patlan, D.; Solis-Cruz, B.; Pontin, K.P.; Latorre, J.D.; Baxter, M.F.A.; Hernandez-Velasco, X.; Merino-Guzman, R.; Méndez-Albores, A.; Hargis, B.M.; Lopez-Arellano, R.; et al. Evaluation of a solid dispersion of curcumin with polyvinylpyrrolidone and boric acid against Salmonella enteritidis infection and intestinal permeability in broiler chickens: A pilot study. Front. Microbiol. 2018, 9, 1289. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, X.; Zhang, Y.; Huang, K.; Liu, H.; Xu, D.; Li, S.; Liu, Q.; Huang, J.; Yao, H.; et al. Improved solubility, dissolution rate, and oral bioavailability of main biflavonoids from Selaginella doederleinii extract by amorphous solid dispersion. Drug Deliv. 2020, 27, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Duan, J.; Xie, Y.; Lin, G.; Luo, H.; Guowen, L.; Yuan, X. Effects of solid dispersion and self-emulsifying formulations on the solubility, dissolution, permeability and pharmacokinetics of isorhamnetin, quercetin and kaempferol in total flavones of Hippophae rhamnoides L. Drug Dev. Ind. Pharm. 2013, 39, 1037–1045. [Google Scholar] [CrossRef]
- Nair, A.R.; Lakshman, Y.D.; Anand, V.S.K.; Sree, K.S.N.; Bhat, K.; Dengale, S.J. Overview of extensively employed polymeric carriers in solid dispersion technology. AAPS PharmSciTech 2020, 21, 309. [Google Scholar] [CrossRef]
- Dengale, S.J.; Grohganz, H.; Rades, T.; Löbmann, K. Recent advances in co-amorphous drug formulations. Adv. Drug Deliv. Rev. 2016, 100, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization and stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef]
- Shamblin, S.L.; Tang, X.; Chang, L.; Hancock, B.C.; Pikal, M.J. Characterization of the time scales of molecular motion in pharmaceutically important glasses. J. Phys. Chem. B 1999, 103, 4113–4121. [Google Scholar] [CrossRef]
- Craig, D.Q.; Royall, P.G.; Kett, V.L.; Hopton, M.L. The relevance of the amorphous state to pharmaceutical dosage forms: Glassy drugs and freeze dried systems. Int. J. Pharm. 1999, 179, 179–207. [Google Scholar] [CrossRef] [Green Version]
- Bhasin, N. Current trends in solid dispersion: A review. J. Drug Deliv. Ther. 2014, 4, 80–86. [Google Scholar] [CrossRef]
- Linn, M.; Collnot, E.M.; Djuric, D.; Hempel, K.; Fabian, E.; Kolter, K.; Lehr, C.M. Soluplus® as an effective absorption enhancer of poorly soluble drugs in vitro and in vivo. Eur. J. Pharm. Sci. 2012, 45, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Mande, P.P.; Bachhav, S.S.; Devarajan, P.V. Bioenhanced advanced third generation solid dispersion of tadalafil: Repurposing with improved therapy in pyelonephritis. Asian J. Pharm. Sci. 2017, 12, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Prasad, D.; Chauhan, H.; Atef, E. Role of molecular interactions for synergistic precipitation inhibition of poorly soluble drug in supersaturated drug-polymer-polymer ternary solution. Mol. Pharm. 2016, 13, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Tokuyasu, N.; Shomori, K.; Amano, K.; Honjo, S.; Sakamoto, T.; Watanabe, J.; Amisaki, M.; Morimoto, M.; Uchinaka, E.; Yagyu, T.; et al. Indirubin, a constituent of the Chinese herbal medicine Qing-Dai, attenuates dextran sulfate sodium-induced murine colitis. Yonago Acta Med. 2018, 61, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.-Q.; Liu, Y.; Zhao, J.-H.; Wang, L.; Feng, N.-P. Improved oral bioavailability of poorly water-soluble indirubin by a supersaturatable self-microemulsifying drug delivery system. Int. J. Nanomed. 2012, 7, 1115–1125. [Google Scholar]
- Vasconcelos, T.; Prezotti, F.; Araújo, F.; Lopes, C.; Loureiro, A.; Marques, S.; Sarmento, B. Third-generation solid dispersion combining soluplus and poloxamer 407 enhances the oral bioavailability of resveratrol. Int. J. Pharm. 2021, 595, 120245. [Google Scholar] [CrossRef]
- Bajracharya, R.; Lee, S.H.; Song, J.G.; Kim, M.; Lee, K.; Han, H.-K. Development of a ternary solid dispersion formulation of LW6 to improve the in vivo activity as a BCRP Inhibitor: Preparation and in vitro/in vivo characterization. Pharmaceutics 2019, 11, 206. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.A.; Oh, H.K.; Lee, J.C.; Choi, Y.H.; Jeong, S.H. Comparison of solubility enhancement by solid dispersion and micronized butein and its correlation with in vivo study. J. Pharm. Investig. 2021, 51, 53–60. [Google Scholar] [CrossRef]
- Ullah, K.H.; Raza, F.; Munawar, S.M.; Sohail, M.; Zafar, H.; Zafar, M.I.; Ur-Rehman, T. Poloxamer 407 based gel formulations for transungual delivery of hydrophobic drugs: Selection and optimization of potential additives. Polymers 2021, 13, 3376. [Google Scholar] [CrossRef]
- Gómez-Guillén, M.C.; Montero, M.P. Enhancement of oral bioavailability of natural compounds and probiotics by mucoadhesive tailored biopolymer-based nanoparticles: A review. Food Hydrocoll. 2021, 118, 106772. [Google Scholar] [CrossRef]
- Hua, S. Advances in oral drug delivery for regional targeting in the gastrointestinal tract—Influence of physiological, pathophysiological and pharmaceutical factors. Front. Pharmacol. 2020, 11, 524. [Google Scholar] [CrossRef] [PubMed]
- Pangeni, R.; Kang, S.; Jha, S.K.; Subedi, L.; Park, J.W. Intestinal membrane transporter-mediated approaches to improve oral drug delivery. J. Pharm. Investig. 2021, 51, 137–158. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Composition (w/w/w) |
|---|---|
| SD-F1 | Drug: P407: PVP K30 = 1:1:1 |
| SD-F2 | Drug: P407: PVP K30 = 1:1:2 |
| SD-F3 | Drug: P407: PVP K30 = 1:1:3 |
| SD-F4 | Drug: P407: PVP K30 = 1:2:2 |
| SD-F5 | Drug: P407: PVP K30 = 1:3:2 |
| Temp. (°C) | Indirubin Concentration (ng/mL) | |||
|---|---|---|---|---|
| Day 0 | 1 Month | 2 Months | 3 Months | |
| 4 | 449 ± 16 | 436 ± 24 | 437 ± 22 | 441 ± 28 |
| 25 | 449 ± 16 | 438 ± 23 | 436 ± 26 | 438 ± 25 |
| Formulation | AUC (ng × h/mL) | Cmax (ng/mL) | Tmax (h) |
|---|---|---|---|
| SD-F4 | 448.5 ± 156.8 | 49.28 ± 15.43 | 0.9 ± 0.2 |
| Pure MT-102 | ND | ND | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajracharya, R.; Song, J.G.; Lee, S.H.; Jeong, S.H.; Han, H.-K. Enhanced Oral Bioavailability of MT-102, a New Anti-inflammatory Agent, via a Ternary Solid Dispersion Formulation. Pharmaceutics 2022, 14, 1510. https://doi.org/10.3390/pharmaceutics14071510
Bajracharya R, Song JG, Lee SH, Jeong SH, Han H-K. Enhanced Oral Bioavailability of MT-102, a New Anti-inflammatory Agent, via a Ternary Solid Dispersion Formulation. Pharmaceutics. 2022; 14(7):1510. https://doi.org/10.3390/pharmaceutics14071510
Chicago/Turabian StyleBajracharya, Rajiv, Jae Geun Song, Sang Hoon Lee, Seong Hoon Jeong, and Hyo-Kyung Han. 2022. "Enhanced Oral Bioavailability of MT-102, a New Anti-inflammatory Agent, via a Ternary Solid Dispersion Formulation" Pharmaceutics 14, no. 7: 1510. https://doi.org/10.3390/pharmaceutics14071510