
Transport Mechanisms at the Blood–Brain Barrier and in Cellular Compartments of the Neurovascular Unit: Focus on CNS Delivery of Small Molecule Drugs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of ATP-Binding Cassette (ABC) and Solute Carrier (SLC) Transporters at the Blood–Brain Barrier (BBB)
2.1. ABC Transporters
2.1.1. P-glycoprotein (P-gp)
2.1.2. Breast Cancer Resistance Protein (BCRP/Bcrp)
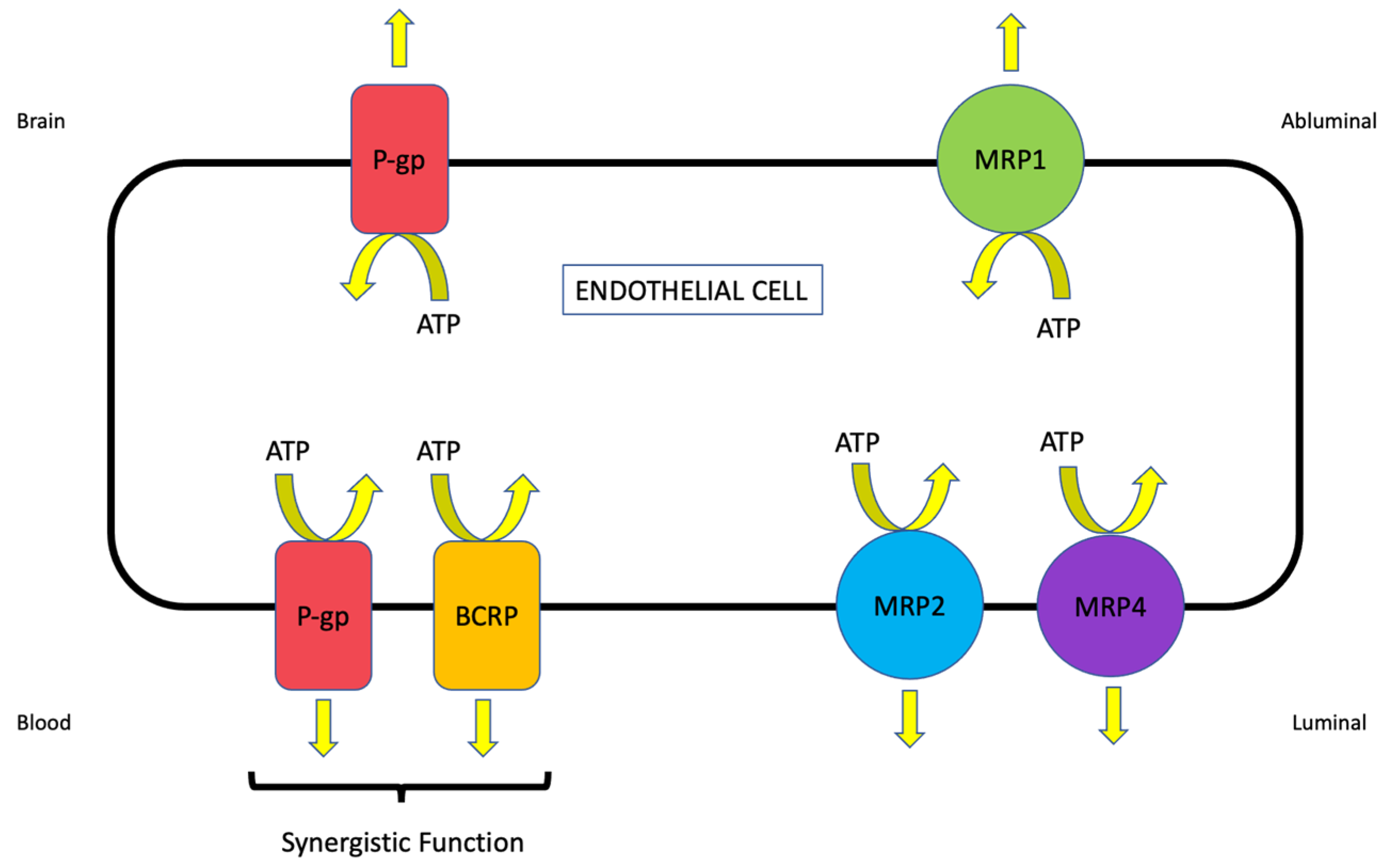
2.1.3. Multidrug Resistance Proteins (MRPs/Mrps)
2.2. Solute Carrier (SLC) Transporters
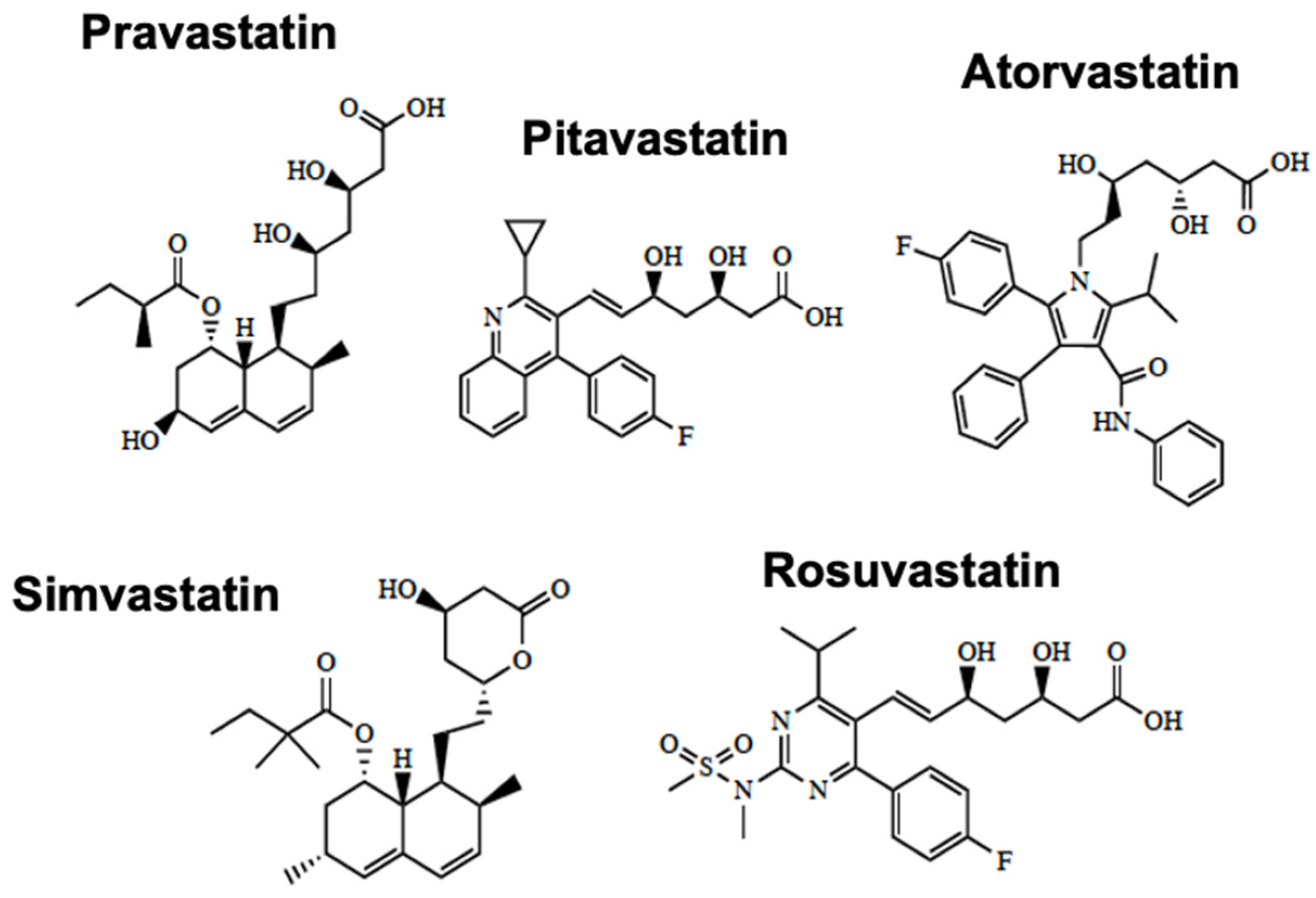
2.2.1. Organic Anion-Transporting Polypeptides (Oatps)
2.2.2. Organic Cation Transporters (Octs)
3. Case Study on Transport at the Blood–Brain Barrier (BBB)—Ischemic Stroke
4. Transport Mechanisms in Other Cell Types of the Neurovascular Unit
4.1. Astrocytes
4.2. Microglia
4.3. Pericytes
4.4. Neurons
5. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boado, R.J.; Ka-Wai Hui, E.; Zhiqiang Lu, J.; Pardridge, W.M. Insulin receptor antibody-iduronate 2-sulfatase fusion protein: Pharmacokinetics, anti-drug antibody, and safety pharmacology in Rhesus monkeys. Biotechnol. Bioeng. 2014, 111, 2317–2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardridge, W.M. Delivery of Biologics Across the Blood-Brain Barrier with Molecular Trojan Horse Technology. BioDrugs 2017, 31, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Al Maghribi, A.; Vanderpoel, V.; Vasilevko, V.; Cribbs, D.H.; Boado, R.; Pardridge, W.M.; Sumbria, R.K. Brain Penetrating Bifunctional Erythropoietin-Transferrin Receptor Antibody Fusion Protein for Alzheimer’s Disease. Mol. Pharm. 2018, 15, 4963–4973. [Google Scholar] [CrossRef] [PubMed]
- Haqqani, A.S.; Delaney, C.E.; Brunette, E.; Baumann, E.; Farrington, G.K.; Sisk, W.; Eldredge, J.; Ding, W.; Tremblay, T.L.; Stanimirovic, D.B. Endosomal trafficking regulates receptor-mediated transcytosis of antibodies across the blood brain barrier. J. Cereb Blood Flow Metab. 2018, 38, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Kariolis, M.S.; Wells, R.C.; Getz, J.A.; Kwan, W.; Mahon, C.S.; Tong, R.; Kim, D.J.; Srivastava, A.; Bedard, C.; Henne, K.R.; et al. Brain delivery of therapeutic proteins using an Fc fragment blood-brain barrier transport vehicle in mice and monkeys. Sci. Transl. Med. 2020, 12, eaay1359. [Google Scholar] [CrossRef] [PubMed]
- Mikitsh, J.L.; Chacko, A.M. Pathways for small molecule delivery to the central nervous system across the blood-brain barrier. Perspect Medicin. Chem. 2014, 6, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Mahar Doan, K.M.; Humphreys, J.E.; Webster, L.O.; Wring, S.A.; Shampine, L.J.; Serabjit-Singh, C.J.; Adkison, K.K.; Polli, J.W. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J. Pharmacol. Exp. Ther. 2002, 303, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol 2009, 9 (Suppl. S1), S3. [Google Scholar] [CrossRef] [Green Version]
- Abdullahi, W.; Davis, T.P.; Ronaldson, P.T. Functional Expression of P-glycoprotein and Organic Anion Transporting Polypeptides at the Blood-Brain Barrier: Understanding Transport Mechanisms for Improved CNS Drug Delivery? AAPS J. 2017, 19, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Reilly, B.G.; Davis, T.P.; Ronaldson, P.T. Modulation of Opioid Transport at the Blood-Brain Barrier by Altered ATP-Binding Cassette (ABC) Transporter Expression and Activity. Pharmaceutics 2018, 10, 192. [Google Scholar] [CrossRef] [Green Version]
- Potschka, H. Modulating P-glycoprotein regulation: Future perspectives for pharmacoresistant epilepsies? Epilepsia 2010, 51, 1333–1347. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef] [PubMed]
- Binkhathlan, Z.; Lavasanifar, A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Aller, S.G.; Beis, K.; Carpenter, E.P.; Chang, G.; Chen, L.; Dassa, E.; Dean, M.; Duong Van Hoa, F.; Ekiert, D.; et al. Structural and functional diversity calls for a new classification of ABC transporters. FEBS Lett. 2020, 594, 3767–3775. [Google Scholar] [CrossRef]
- Sanchez-Covarrubias, L.; Slosky, L.M.; Thompson, B.J.; Davis, T.P.; Ronaldson, P.T. Transporters at CNS barrier sites: Obstacles or opportunities for drug delivery? Curr. Pharm. Des. 2014, 20, 1422–1449. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M.; Hrycyna, C.A.; Schoenlein, P.V.; Germann, U.A.; Pastan, I. Genetic analysis of the multidrug transporter. Annu. Rev. Genet. 1995, 29, 607–649. [Google Scholar] [CrossRef]
- Nilles, K.L.; Williams, E.I.; Betterton, R.D.; Davis, T.P.; Ronaldson, P.T. Blood-Brain Barrier Transporters: Opportunities for Therapeutic Development in Ischemic Stroke. Int. J. Mol. Sci. 2022, 23, 1898. [Google Scholar] [CrossRef]
- Ling, V.; Thompson, L.H. Reduced permeability in CHO cells as a mechanism of resistance to colchicine. J. Cell Physiol. 1974, 83, 103–116. [Google Scholar] [CrossRef]
- Beaulieu, E.; Demeule, M.; Ghitescu, L.; Beliveau, R. P-glycoprotein is strongly expressed in the luminal membranes of the endothelium of blood vessels in the brain. Biochem. J. 1997, 326, 539–544. [Google Scholar] [CrossRef] [Green Version]
- Bendayan, R.; Ronaldson, P.T.; Gingras, D.; Bendayan, M. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J. Histochem. Cytochem. 2006, 54, 1159–1167. [Google Scholar] [CrossRef] [Green Version]
- Schinkel, A.H.; Smit, J.J.; van Tellingen, O.; Beijnen, J.H.; Wagenaar, E.; van Deemter, L.; Mol, C.A.; van der Valk, M.A.; Robanus-Maandag, E.C.; te Riele, H.P.; et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 1994, 77, 491–502. [Google Scholar] [CrossRef]
- Breuil, L.; Marie, S.; Goutal, S.; Auvity, S.; Truillet, C.; Saba, W.; Langer, O.; Caille, F.; Tournier, N. Comparative vulnerability of PET radioligands to partial inhibition of P-glycoprotein at the blood-brain barrier: A criterion of choice? J. Cereb. Blood Flow Metab. 2022, 42, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Dauchy, S.; Miller, F.; Couraud, P.O.; Weaver, R.J.; Weksler, B.; Romero, I.A.; Scherrmann, J.M.; De Waziers, I.; Decleves, X. Expression and transcriptional regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral microvascular endothelial cells. Biochem. Pharmacol. 2009, 77, 897–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInerney, M.P.; Volitakis, I.; Bush, A.I.; Banks, W.A.; Short, J.L.; Nicolazzo, J.A. Ionophore and Biometal Modulation of P-glycoprotein Expression and Function in Human Brain Microvascular Endothelial Cells. Pharm. Res. 2018, 35, 83. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson, P.T.; Brzica, H.; Abdullahi, W.; Reilly, B.G.; Davis, T.P. Transport Properties of Statins by Organic Anion Transporting Polypeptide 1A2 and Regulation by Transforming Growth Factor-beta Signaling in Human Endothelial Cells. J. Pharmacol. Exp. Ther. 2021, 376, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Virgintino, D.; Robertson, D.; Errede, M.; Benagiano, V.; Girolamo, F.; Maiorano, E.; Roncali, L.; Bertossi, M. Expression of P-glycoprotein in human cerebral cortex microvessels. J. Histochem. Cytochem. 2002, 50, 1671–1676. [Google Scholar] [CrossRef] [Green Version]
- Al-Majdoub, Z.M.; Al Feteisi, H.; Achour, B.; Warwood, S.; Neuhoff, S.; Rostami-Hodjegan, A.; Barber, J. Proteomic Quantification of Human Blood-Brain Barrier SLC and ABC Transporters in Healthy Individuals and Dementia Patients. Mol. Pharm. 2019, 16, 1220–1233. [Google Scholar] [CrossRef] [Green Version]
- Billington, S.; Salphati, L.; Hop, C.; Chu, X.; Evers, R.; Burdette, D.; Rowbottom, C.; Lai, Y.; Xiao, G.; Humphreys, W.G.; et al. Interindividual and Regional Variability in Drug Transporter Abundance at the Human Blood-Brain Barrier Measured by Quantitative Targeted Proteomics. Clin. Pharmacol. Ther. 2019, 106, 228–237. [Google Scholar] [CrossRef]
- Storelli, F.; Billington, S.; Kumar, A.R.; Unadkat, J.D. Abundance of P-Glycoprotein and Other Drug Transporters at the Human Blood-Brain Barrier in Alzheimer’s Disease: A Quantitative Targeted Proteomic Study. Clin. Pharmacol. Ther. 2021, 109, 667–675. [Google Scholar] [CrossRef]
- Wagner, C.C.; Bauer, M.; Karch, R.; Feurstein, T.; Kopp, S.; Chiba, P.; Kletter, K.; Loscher, W.; Muller, M.; Zeitlinger, M.; et al. A pilot study to assess the efficacy of tariquidar to inhibit P-glycoprotein at the human blood-brain barrier with (R)-11C-verapamil and PET. J. Nucl. Med. 2009, 50, 1954–1961. [Google Scholar] [CrossRef] [Green Version]
- Muzi, M.; Mankoff, D.A.; Link, J.M.; Shoner, S.; Collier, A.C.; Sasongko, L.; Unadkat, J.D. Imaging of cyclosporine inhibition of P-glycoprotein activity using 11C-verapamil in the brain: Studies of healthy humans. J. Nucl. Med. 2009, 50, 1267–1275. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Collier, A.C.; Link, J.M.; Domino, K.B.; Mankoff, D.A.; Eary, J.F.; Spiekerman, C.F.; Hsiao, P.; Deo, A.K.; Unadkat, J.D. Modulation of P-glycoprotein at the Human Blood-Brain Barrier by Quinidine or Rifampin Treatment: A Positron Emission Tomography Imaging Study. Drug Metab. Dispos. 2015, 43, 1795–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, M.L. P-glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights 2013, 7, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Seelbach, M.J.; Brooks, T.A.; Egleton, R.D.; Davis, T.P. Peripheral inflammatory hyperalgesia modulates morphine delivery to the brain: A role for P-glycoprotein. J. Neurochem. 2007, 102, 1677–1690. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson, P.T.; Finch, J.D.; Demarco, K.M.; Quigley, C.E.; Davis, T.P. Inflammatory pain signals an increase in functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier. J. Pharmacol. Exp. Ther. 2011, 336, 827–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bicker, J.; Fortuna, A.; Alves, G.; Soares-da-Silva, P.; Falcao, A. Elucidation of the Impact of P-glycoprotein and Breast Cancer Resistance Protein on the Brain Distribution of Catechol-O-Methyltransferase Inhibitors. Drug Metab. Dispos. 2017, 45, 1282–1291. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.N.; Mickley, L.A.; Schwartz, A.M.; Acton, E.M.; Hwang, J.L.; Fojo, A.T. Characterization of adriamycin-resistant human breast cancer cells which display overexpression of a novel resistance-related membrane protein. J. Biol. Chem. 1990, 265, 10073–10080. [Google Scholar] [CrossRef]
- Nakagawa, M.; Schneider, E.; Dixon, K.H.; Horton, J.; Kelley, K.; Morrow, C.; Cowan, K.H. Reduced intracellular drug accumulation in the absence of P-glycoprotein (mdr1) overexpression in mitoxantrone-resistant human MCF-7 breast cancer cells. Cancer Res. 1992, 52, 6175–6181. [Google Scholar]
- Lee, J.S.; Scala, S.; Matsumoto, Y.; Dickstein, B.; Robey, R.; Zhan, Z.; Altenberg, G.; Bates, S.E. Reduced drug accumulation and multidrug resistance in human breast cancer cells without associated P-glycoprotein or MRP overexpression. J. Cell Biochem. 1997, 65, 513–526. [Google Scholar] [CrossRef]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef] [Green Version]
- Graf, G.A.; Yu, L.; Li, W.P.; Gerard, R.; Tuma, P.L.; Cohen, J.C.; Hobbs, H.H. ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J. Biol. Chem. 2003, 278, 48275–48282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graf, G.A.; Li, W.P.; Gerard, R.D.; Gelissen, I.; White, A.; Cohen, J.C.; Hobbs, H.H. Coexpression of ATP-binding cassette proteins ABCG5 and ABCG8 permits their transport to the apical surface. J. Clin. Investig. 2002, 110, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Eisenblatter, T.; Huwel, S.; Galla, H.J. Characterisation of the brain multidrug resistance protein (BMDP/ABCG2/BCRP) expressed at the blood-brain barrier. Brain Res. 2003, 971, 221–231. [Google Scholar] [CrossRef]
- Zhang, W.; Mojsilovic-Petrovic, J.; Andrade, M.F.; Zhang, H.; Ball, M.; Stanimirovic, D.B. The expression and functional characterization of ABCG2 in brain endothelial cells and vessels. FASEB J. 2003, 17, 2085–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronica, E.; Gorter, J.A.; Redeker, S.; van Vliet, E.A.; Ramkema, M.; Scheffer, G.L.; Scheper, R.J.; van der Valk, P.; Leenstra, S.; Baayen, J.C.; et al. Localization of breast cancer resistance protein (BCRP) in microvessel endothelium of human control and epileptic brain. Epilepsia 2005, 46, 849–857. [Google Scholar] [CrossRef]
- Ohtsuki, S.; Ikeda, C.; Uchida, Y.; Sakamoto, Y.; Miller, F.; Glacial, F.; Decleves, X.; Scherrmann, J.M.; Couraud, P.O.; Kubo, Y.; et al. Quantitative targeted absolute proteomic analysis of transporters, receptors and junction proteins for validation of human cerebral microvascular endothelial cell line hCMEC/D3 as a human blood-brain barrier model. Mol. Pharm. 2013, 10, 289–296. [Google Scholar] [CrossRef]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J. Neurochem. 2011, 117, 333–345. [Google Scholar] [CrossRef]
- Polli, J.W.; Olson, K.L.; Chism, J.P.; John-Williams, L.S.; Yeager, R.L.; Woodard, S.M.; Otto, V.; Castellino, S.; Demby, V.E. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino }methyl)-2-furyl]-4-quinazolinamine; GW572016). Drug Metab. Dispos. 2009, 37, 439–442. [Google Scholar] [CrossRef]
- Betterton, R.D.; Abdullahi, W.; Williams, E.I.; Lochhead, J.L.; Brzica, H.; Stanton, J.A.; Reddell, E.; Ogbonnaya, C.; Davis, T.P.; Ronaldson, P.T. Regulation of Blood-Brain Barrier Transporters by Transforming Growth Factor-beta /Activin Receptor-Like Kinase 1 (TGF-beta /ALK1) Signaling: Relevance to the Brain Disposition of 3-Hydroxy-3-Methylglutaryl Coenzyme A (HMG-CoA) Reductase Inhibitors (i.e., Statins). Drug Metab. Dispos. 2022, 50, 942–956. [Google Scholar] [CrossRef]
- Agarwal, S.; Hartz, A.M.; Elmquist, W.F.; Bauer, B. Breast cancer resistance protein and P-glycoprotein in brain cancer: Two gatekeepers team up. Curr. Pharm. Des. 2011, 17, 2793–2802. [Google Scholar] [CrossRef] [Green Version]
- Laramy, J.K.; Kim, M.; Parrish, K.E.; Sarkaria, J.N.; Elmquist, W.F. Pharmacokinetic Assessment of Cooperative Efflux of the Multitargeted Kinase Inhibitor Ponatinib Across the Blood-Brain Barrier. J. Pharmacol. Exp. Ther. 2018, 365, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Mittapalli, R.K.; Vaidhyanathan, S.; Sane, R.; Elmquist, W.F. Impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) on the brain distribution of a novel BRAF inhibitor: Vemurafenib (PLX4032). J. Pharmacol. Exp. Ther. 2012, 342, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Elmquist, W.F. Insight into the cooperation of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) at the blood-brain barrier: A case study examining sorafenib efflux clearance. Mol. Pharm. 2012, 9, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.T.; Kis, O.; De Rosa, M.F.; Bendayan, R. Raltegravir permeability across blood-tissue barriers and the potential role of drug efflux transporters. Antimicrob. Agents Chemother. 2015, 59, 2572–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helms, H.C.; Hersom, M.; Kuhlmann, L.B.; Badolo, L.; Nielsen, C.U.; Brodin, B. An electrically tight in vitro blood-brain barrier model displays net brain-to-blood efflux of substrates for the ABC transporters, P-gp, Bcrp and Mrp-1. AAPS J. 2014, 16, 1046–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brechbuhl, H.M.; Gould, N.; Kachadourian, R.; Riekhof, W.R.; Voelker, D.R.; Day, B.J. Glutathione transport is a unique function of the ATP-binding cassette protein ABCG2. J. Biol. Chem. 2010, 285, 16582–16587. [Google Scholar] [CrossRef] [Green Version]
- Ifergan, I.; Shafran, A.; Jansen, G.; Hooijberg, J.H.; Scheffer, G.L.; Assaraf, Y.G. Folate deprivation results in the loss of breast cancer resistance protein (BCRP/ABCG2) expression. A role for BCRP in cellular folate homeostasis. J. Biol. Chem. 2004, 279, 25527–25534. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Morris, M.E. Membrane transport of dietary phenethyl isothiocyanate by ABCG2 (breast cancer resistance protein). Mol. Pharm. 2005, 2, 414–419. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Su, W.; Pasternak, G.W. The role of multidrug resistance-associated protein in the blood-brain barrier and opioid analgesia. Synapse 2013, 67, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Kanamitsu, K.; Kusuhara, H.; Schuetz, J.D.; Takeuchi, K.; Sugiyama, Y. Investigation of the Importance of Multidrug Resistance-Associated Protein 4 (Mrp4/Abcc4) in the Active Efflux of Anionic Drugs Across the Blood-Brain Barrier. J. Pharm. Sci. 2017, 106, 2566–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronaldson, P.T.; Bendayan, R. HIV-1 viral envelope glycoprotein gp120 produces oxidative stress and regulates the functional expression of multidrug resistance protein-1 (Mrp1) in glial cells. J. Neurochem. 2008, 106, 1298–1313. [Google Scholar] [CrossRef] [PubMed]
- Akanuma, S.I.; Hashimoto, K.; Yoshida, Y.; Kubo, Y.; Hosoya, K.I. Inflammation-Induced Attenuation of Prostaglandin D2 Elimination across Rat Blood-Brain Barrier: Involvement of the Downregulation of Organic Anion Transporter 3 and Multidrug Resistance-Associated Protein 4. Biol. Pharm. Bull 2020, 43, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Kalvass, J.C.; Polli, J.W.; Bourdet, D.L.; Feng, B.; Huang, S.M.; Liu, X.; Smith, Q.R.; Zhang, L.K.; Zamek-Gliszczynski, M.J.; International Transporter, C. Why clinical modulation of efflux transport at the human blood-brain barrier is unlikely: The ITC evidence-based position. Clin. Pharmacol. Ther. 2013, 94, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Colas, C.; Ung, P.M.; Schlessinger, A. SLC Transporters: Structure, Function, and Drug Discovery. Medchemcomm 2016, 7, 1069–1081. [Google Scholar] [CrossRef] [Green Version]
- Schumann, T.; Konig, J.; Henke, C.; Willmes, D.M.; Bornstein, S.R.; Jordan, J.; Fromm, M.F.; Birkenfeld, A.L. Solute Carrier Transporters as Potential Targets for the Treatment of Metabolic Disease. Pharmacol. Rev. 2020, 72, 343–379. [Google Scholar] [CrossRef]
- Brzica, H.; Abdullahi, W.; Ibbotson, K.; Ronaldson, P.T. Role of Transporters in Central Nervous System Drug Delivery and Blood-Brain Barrier Protection: Relevance to Treatment of Stroke. J. Cent. Nerv. Syst. Dis. 2017, 9, 1179573517693802. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Davis, T.P. Targeted drug delivery to treat pain and cerebral hypoxia. Pharmacol. Rev. 2013, 65, 291–314. [Google Scholar] [CrossRef] [Green Version]
- Betterton, R.D.; Davis, T.P.; Ronaldson, P.T. Organic Cation Transporter (OCT/OCTN) Expression at Brain Barrier Sites: Focus on CNS Drug Delivery. Handb. Exp. Pharmacol. 2021, 266, 301–328. [Google Scholar] [CrossRef]
- Thompson, B.J.; Sanchez-Covarrubias, L.; Slosky, L.M.; Zhang, Y.; Laracuente, M.L.; Ronaldson, P.T. Hypoxia/reoxygenation stress signals an increase in organic anion transporting polypeptide 1a4 (Oatp1a4) at the blood-brain barrier: Relevance to CNS drug delivery. J. Cereb. Blood Flow Metab. 2014, 34, 699–707. [Google Scholar] [CrossRef]
- Abdullahi, W.; Brzica, H.; Hirsch, N.A.; Reilly, B.G.; Ronaldson, P.T. Functional Expression of Organic Anion Transporting Polypeptide 1a4 Is Regulated by Transforming Growth Factor-beta/Activin Receptor-like Kinase 1 Signaling at the Blood-Brain Barrier. Mol. Pharmacol. 2018, 94, 1321–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullahi, W.; Brzica, H.; Ibbotson, K.; Davis, T.P.; Ronaldson, P.T. Bone morphogenetic protein-9 increases the functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier via the activin receptor-like kinase-1 receptor. J. Cereb. Blood Flow Metab. 2017, 37, 2340–2345. [Google Scholar] [CrossRef] [PubMed]
- Brzica, H.; Abdullahi, W.; Reilly, B.G.; Ronaldson, P.T. Sex-specific differences in organic anion transporting polypeptide 1a4 (Oatp1a4) functional expression at the blood-brain barrier in Sprague-Dawley rats. Fluids Barriers CNS 2018, 15, 25. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Glaeser, H.; Smith, L.H.; Roberts, R.L.; Moeckel, G.W.; Gervasini, G.; Leake, B.F.; Kim, R.B. Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): Implications for altered drug disposition and central nervous system drug entry. J. Biol. Chem. 2005, 280, 9610–9617. [Google Scholar] [CrossRef] [Green Version]
- Bronger, H.; Konig, J.; Kopplow, K.; Steiner, H.H.; Ahmadi, R.; Herold-Mende, C.; Keppler, D.; Nies, A.T. ABCC drug efflux pumps and organic anion uptake transporters in human gliomas and the blood-tumor barrier. Cancer Res. 2005, 65, 11419–11428. [Google Scholar] [CrossRef] [Green Version]
- Roberts, L.M.; Black, D.S.; Raman, C.; Woodford, K.; Zhou, M.; Haggerty, J.E.; Yan, A.T.; Cwirla, S.E.; Grindstaff, K.K. Subcellular localization of transporters along the rat blood-brain barrier and blood-cerebral-spinal fluid barrier by in vivo biotinylation. Neuroscience 2008, 155, 423–438. [Google Scholar] [CrossRef]
- Gao, B.; Stieger, B.; Noe, B.; Fritschy, J.M.; Meier, P.J. Localization of the organic anion transporting polypeptide 2 (Oatp2) in capillary endothelium and choroid plexus epithelium of rat brain. J. Histochem. Cytochem. 1999, 47, 1255–1264. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Hagenbuch, B.; Kullak-Ublick, G.A.; Benke, D.; Aguzzi, A.; Meier, P.J. Organic anion-transporting polypeptides mediate transport of opioid peptides across blood-brain barrier. J. Pharmacol. Exp. Ther. 2000, 294, 73–79. [Google Scholar]
- Gao, B.; Vavricka, S.R.; Meier, P.J.; Stieger, B. Differential cellular expression of organic anion transporting peptides OATP1A2 and OATP2B1 in the human retina and brain: Implications for carrier-mediated transport of neuropeptides and neurosteriods in the CNS. Pflugers Arch. 2015, 467, 1481–1493. [Google Scholar] [CrossRef]
- Schafer, A.M.; Meyer Zu Schwabedissen, H.E.; Bien-Moller, S.; Hubeny, A.; Vogelgesang, S.; Oswald, S.; Grube, M. OATP1A2 and OATP2B1 Are Interacting with Dopamine-Receptor Agonists and Antagonists. Mol. Pharm. 2020, 17, 1987–1995. [Google Scholar] [CrossRef]
- Schafer, A.M.; Meyer Zu Schwabedissen, H.E.; Grube, M. Expression and Function of Organic Anion Transporting Polypeptides in the Human Brain: Physiological and Pharmacological Implications. Pharmaceutics 2021, 13, 834. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, C.D.; Aleksunes, L.M. Xenobiotic, bile acid, and cholesterol transporters: Function and regulation. Pharmacol. Rev. 2010, 62, 1–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grube, M.; Hagen, P.; Jedlitschky, G. Neurosteroid Transport in the Brain: Role of ABC and SLC Transporters. Front. Pharmacol. 2018, 9, 354. [Google Scholar] [CrossRef]
- Sung, J.H.; Yu, K.H.; Park, J.S.; Tsuruo, T.; Kim, D.D.; Shim, C.K.; Chung, S.J. Saturable distribution of tacrine into the striatal extracellular fluid of the rat: Evidence of involvement of multiple organic cation transporters in the transport. Drug Metab. Dispos. 2005, 33, 440–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.C.; Lu, Y.H.; Peng, Y.H.; Tsai, T.F.; Kao, Y.H.; Yang, H.T.; Lin, C.J. Decreased expression of organic cation transporters, Oct1 and Oct2, in brain microvessels and its implication to MPTP-induced dopaminergic toxicity in aged mice. J. Cereb. Blood Flow Metab. 2015, 35, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Sekhar, G.N.; Georgian, A.R.; Sanderson, L.; Vizcay-Barrena, G.; Brown, R.C.; Muresan, P.; Fleck, R.A.; Thomas, S.A. Organic cation transporter 1 (OCT1) is involved in pentamidine transport at the human and mouse blood-brain barrier (BBB). PLoS ONE 2017, 12, e0173474. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.J.; Tai, Y.; Huang, M.T.; Tsai, Y.F.; Hsu, H.J.; Tzen, K.Y.; Liou, H.H. Cellular localization of the organic cation transporters, OCT1 and OCT2, in brain microvessel endothelial cells and its implication for MPTP transport across the blood-brain barrier and MPTP-induced dopaminergic toxicity in rodents. J. Neurochem. 2010, 114, 717–727. [Google Scholar] [CrossRef]
- Sandoval, P.J.; Zorn, K.M.; Clark, A.M.; Ekins, S.; Wright, S.H. Assessment of Substrate-Dependent Ligand Interactions at the Organic Cation Transporter OCT2 Using Six Model Substrates. Mol. Pharmacol. 2018, 94, 1057–1068. [Google Scholar] [CrossRef] [Green Version]
- Hiasa, M.; Matsumoto, T.; Komatsu, T.; Moriyama, Y. Wide variety of locations for rodent MATE1, a transporter protein that mediates the final excretion step for toxic organic cations. Am. J. Physiol. Cell Physiol. 2006, 291, C678–C686. [Google Scholar] [CrossRef]
- Chaves, C.; Campanelli, F.; Chapy, H.; Gomez-Zepeda, D.; Glacial, F.; Smirnova, M.; Taghi, M.; Pallud, J.; Perriere, N.; Decleves, X.; et al. An Interspecies Molecular and Functional Study of Organic Cation Transporters at the Blood-Brain Barrier: From Rodents to Humans. Pharmaceutics 2020, 12, 308. [Google Scholar] [CrossRef] [Green Version]
- Goralski, K.B.; Lou, G.; Prowse, M.T.; Gorboulev, V.; Volk, C.; Koepsell, H.; Sitar, D.S. The cation transporters rOCT1 and rOCT2 interact with bicarbonate but play only a minor role for amantadine uptake into rat renal proximal tubules. J. Pharmacol. Exp. Ther. 2002, 303, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.C.; Short, J.L.; Nicolazzo, J.A. Memantine Transport across the Mouse Blood–Brain Barrier Is Mediated by a Cationic Influx H+ Antiporter. Mol. Pharm. 2013, 10, 4491–4498. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Stroke Collaborators. Global, regional, and national burden of stroke and its risk factors, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef]
- Liu, S.; Levine, S.R.; Winn, H.R. Targeting ischemic penumbra: Part I—from pathophysiology to therapeutic strategy. J. Exp. Stroke Transl. Med. 2010, 3, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol. Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef]
- Manning, N.W.; Campbell, B.C.; Oxley, T.J.; Chapot, R. Acute ischemic stroke: Time, penumbra, and reperfusion. Stroke 2014, 45, 640–644. [Google Scholar] [CrossRef] [Green Version]
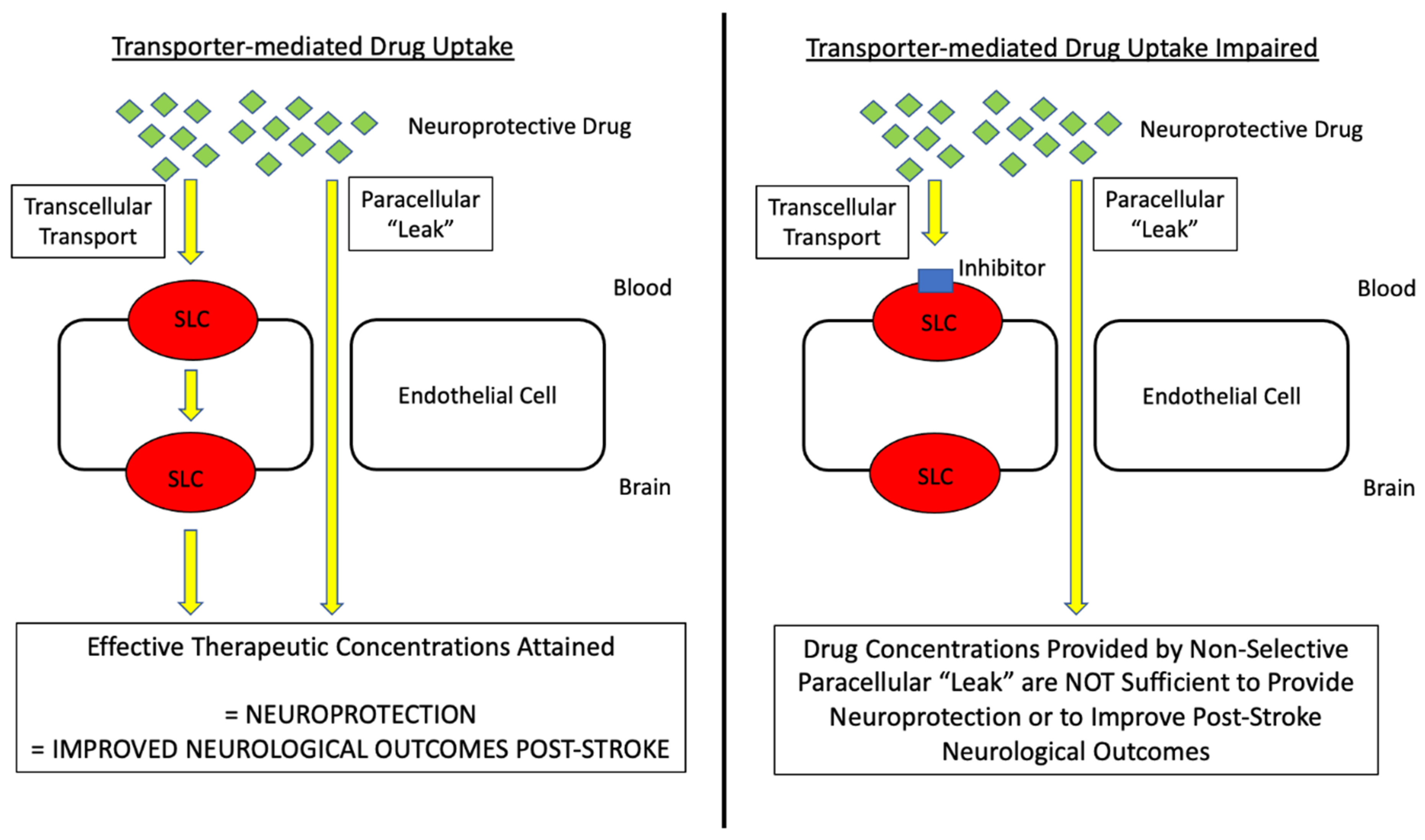
- Williams, E.I.; Betterton, R.D.; Davis, T.P.; Ronaldson, P.T. Transporter-Mediated Delivery of Small Molecule Drugs to the Brain: A Critical Mechanism That Can Advance Therapeutic Development for Ischemic Stroke. Pharmaceutics 2020, 12, 154. [Google Scholar] [CrossRef] [Green Version]
- National Institute of Neurological, D.; Stroke rt, P.A.S.S.G. Tissue plasminogen activator for acute ischemic stroke. N. Engl. J. Med. 1995, 333, 1581–1587. [Google Scholar] [CrossRef]
- O’Carroll, C.B.; Rubin, M.N.; Chong, B.W. What is the Role for Intra-Arterial Therapy in Acute Stroke Intervention? Neurohospitalist 2015, 5, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Tymianski, M. Combining Neuroprotection With Endovascular Treatment of Acute Stroke: Is There Hope? Stroke 2017, 48, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Rocha, M.; Leak, R.K.; Zhao, J.; Bhatia, T.N.; Mu, H.; Wei, Z.; Yu, F.; Weiner, S.L.; Ma, F.; et al. A new era for stroke therapy: Integrating neurovascular protection with optimal reperfusion. J. Cereb. Blood Flow Metab. 2018, 38, 2073–2091. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.; Menon, B.K.; van Zwam, W.H.; Dippel, D.W.; Mitchell, P.J.; Demchuk, A.M.; Davalos, A.; Majoie, C.B.; van der Lugt, A.; de Miquel, M.A.; et al. Endovascular thrombectomy after large-vessel ischaemic stroke: A meta-analysis of individual patient data from five randomised trials. Lancet 2016, 387, 1723–1731. [Google Scholar] [CrossRef]
- Nogueira, R.G.; Jadhav, A.P.; Haussen, D.C.; Bonafe, A.; Budzik, R.F.; Bhuva, P.; Yavagal, D.R.; Ribo, M.; Cognard, C.; Hanel, R.A.; et al. Thrombectomy 6 to 24 Hours after Stroke with a Mismatch between Deficit and Infarct. N. Engl. J. Med. 2018, 378, 11–21. [Google Scholar] [CrossRef]
- Pan, J.; Konstas, A.A.; Bateman, B.; Ortolano, G.A.; Pile-Spellman, J. Reperfusion injury following cerebral ischemia: Pathophysiology, MR imaging, and potential therapies. Neuroradiology 2007, 49, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Candelario-Jalil, E. Injury and repair mechanisms in ischemic stroke: Considerations for the development of novel neurotherapeutics. Curr. Opin. Investig. Drugs 2009, 10, 644–654. [Google Scholar]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion--from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Nour, M.; Scalzo, F.; Liebeskind, D.S. Ischemia-reperfusion injury in stroke. Interv. Neurol. 2013, 1, 185–199. [Google Scholar] [CrossRef] [Green Version]
- Lyden, P.; Pryor, K.E.; Coffey, C.S.; Cudkowicz, M.; Conwit, R.; Jadhav, A.; Sawyer, R.N., Jr.; Claassen, J.; Adeoye, O.; Song, S.; et al. Final Results of the RHAPSODY Trial: A Multi-Center, Phase 2 Trial Using a Continual Reassessment Method to Determine the Safety and Tolerability of 3K3A-APC, A Recombinant Variant of Human Activated Protein C, in Combination with Tissue Plasminogen Activator, Mechanical Thrombectomy or both in Moderate to Severe Acute Ischemic Stroke. Ann. Neurol. 2019, 85, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Turner, R.C.; Lucke-Wold, B.; Lucke-Wold, N.; Elliott, A.S.; Logsdon, A.F.; Rosen, C.L.; Huber, J.D. Neuroprotection for ischemic stroke: Moving past shortcomings and identifying promising directions. Int. J. Mol. Sci. 2013, 14, 1890–1917. [Google Scholar] [CrossRef]
- Spudich, A.; Kilic, E.; Xing, H.; Kilic, U.; Rentsch, K.M.; Wunderli-Allenspach, H.; Bassetti, C.L.; Hermann, D.M. Inhibition of multidrug resistance transporter-1 facilitates neuroprotective therapies after focal cerebral ischemia. Nat. Neurosci. 2006, 9, 487–488. [Google Scholar] [CrossRef] [PubMed]
- DeMars, K.M.; Yang, C.; Hawkins, K.E.; McCrea, A.O.; Siwarski, D.M.; Candelario-Jalil, E. Spatiotemporal Changes in P-glycoprotein Levels in Brain and Peripheral Tissues Following Ischemic Stroke in Rats. J. Exp. Neurosci. 2017, 11, 1179069517701741. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, Z.W.; Xue, C.C.; Li, X.X.; Zhou, S.F. Role of P-glycoprotein in restricting the brain penetration of tanshinone IIA, a major active constituent from the root of Salvia miltiorrhiza Bunge, across the blood-brain barrier. Xenobiotica 2007, 37, 635–678. [Google Scholar] [CrossRef] [PubMed]
- Tornabene, E.; Helms, H.C.C.; Pedersen, S.F.; Brodin, B. Effects of oxygen-glucose deprivation (OGD) on barrier properties and mRNA transcript levels of selected marker proteins in brain endothelial cells/astrocyte co-cultures. PLoS ONE 2019, 14, e0221103. [Google Scholar] [CrossRef]
- Ose, A.; Kusuhara, H.; Endo, C.; Tohyama, K.; Miyajima, M.; Kitamura, S.; Sugiyama, Y. Functional characterization of mouse organic anion transporting peptide 1a4 in the uptake and efflux of drugs across the blood-brain barrier. Drug Metab. Dispos. 2010, 38, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yu, N.; Lu, S.; Ito, S.; Zhang, X.; Prasad, B.; He, E.; Lu, X.; Li, Y.; Wang, F.; et al. Solute Carrier Family of the Organic Anion-Transporting Polypeptides 1A2- Madin-Darby Canine Kidney II: A Promising In Vitro System to Understand the Role of Organic Anion-Transporting Polypeptide 1A2 in Blood-Brain Barrier Drug Penetration. Drug Metab. Dispos. 2015, 43, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Sano, Y.; Mizuno, T.; Mochizuki, T.; Uchida, Y.; Umetsu, M.; Terasaki, T.; Kusuhara, H. Evaluation of Organic Anion Transporter 1A2-knock-in Mice as a Model of Human Blood-brain Barrier. Drug Metab. Dispos. 2018, 46, 1767–1775. [Google Scholar] [CrossRef]
- Albekairi, T.H.; Vaidya, B.; Patel, R.; Nozohouri, S.; Villalba, H.; Zhang, Y.; Lee, Y.S.; Al-Ahmad, A.; Abbruscato, T.J. Brain Delivery of a Potent Opioid Receptor Agonist, Biphalin during Ischemic Stroke: Role of Organic Anion Transporting Polypeptide (OATP). Pharmaceutics 2019, 11, 467. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Islam, M.R.; Karamyan, V.T.; Abbruscato, T.J. In vitro and in vivo efficacy of a potent opioid receptor agonist, biphalin, compared to subtype-selective opioid receptor agonists for stroke treatment. Brain Res. 2015, 1609, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Shah, K.; Wang, H.; Karamyan, V.T.; Abbruscato, T.J. Characterization of neuroprotective effects of biphalin, an opioid receptor agonist, in a model of focal brain ischemia. J. Pharmacol. Exp. Ther. 2011, 339, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wang, H.; Shah, K.; Karamyan, V.T.; Abbruscato, T.J. Opioid receptor agonists reduce brain edema in stroke. Brain Res. 2011, 1383, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Yang, L.; Lee, Y.S.; Hruby, V.J.; Karamyan, V.T.; Abbruscato, T.J. Enkephalin-Fentanyl Multifunctional Opioids as Potential Neuroprotectants for Ischemic Stroke Treatment. Curr. Pharm. Des. 2016, 22, 6459–6468. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Xu, H.; Lu, J.; Zhu, Y.; Jiang, H. Neuroprotection by the kappa-opioid receptor agonist, BRL52537, is mediated via up-regulating phosphorylated signal transducer and activator of transcription-3 in cerebral ischemia/reperfusion injury in rats. Neurochem. Res. 2013, 38, 2305–2312. [Google Scholar] [CrossRef]
- Eftekhar-Vaghefi, S.; Esmaeili-Mahani, S.; Elyasi, L.; Abbasnejad, M. Involvement of Mu Opioid Receptor Signaling in the Protective Effect of Opioid against 6-Hydroxydopamine-Induced SH-SY5Y Human Neuroblastoma Cells Apoptosis. Basic Clin. Neurosci. 2015, 6, 171–178. [Google Scholar] [PubMed]
- Nozohouri, S.; Zhang, Y.; Albekairi, T.H.; Vaidya, B.; Abbruscato, T.J. Glutamate Buffering Capacity and Blood-Brain Barrier Protection of Opioid Receptor Agonists Biphalin and Nociceptin. J. Pharmacol. Exp. Ther. 2021, 379, 260–269. [Google Scholar] [CrossRef]
- Busch, A.E.; Karbach, U.; Miska, D.; Gorboulev, V.; Akhoundova, A.; Volk, C.; Arndt, P.; Ulzheimer, J.C.; Sonders, M.S.; Baumann, C.; et al. Human neurons express the polyspecific cation transporter hOCT2, which translocates monoamine neurotransmitters, amantadine, and memantine. Mol. Pharmacol. 1998, 54, 342–352. [Google Scholar] [CrossRef] [Green Version]
- Stanton, J.A.; Williams, E.I.; Betterton, R.D.; Davis, T.P.; Ronaldson, P.T. Targeting Organic Cation Transporters at the Blood-Brain Barrier To Treat Ischemic Stroke in Rats. Exp. Neurol. 2022; in press. [Google Scholar]
- McCaffrey, G.; Willis, C.L.; Staatz, W.D.; Nametz, N.; Quigley, C.A.; Hom, S.; Lochhead, J.J.; Davis, T.P. Occludin oligomeric assemblies at tight junctions of the blood-brain barrier are altered by hypoxia and reoxygenation stress. J. Neurochem. 2009, 110, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, J.J.; McCaffrey, G.; Quigley, C.E.; Finch, J.; DeMarco, K.M.; Nametz, N.; Davis, T.P. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J. Cereb. Blood Flow Metab. 2010, 30, 1625–1636. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Campbell, M.; Tachibana, K.; Okada, Y.; Kondoh, M. Claudin-5: A Pharmacological Target to Modify the Permeability of the Blood-Brain Barrier. Biol. Pharm. Bull 2021, 44, 1380–1390. [Google Scholar] [CrossRef]
- Winkler, L.; Blasig, R.; Breitkreuz-Korff, O.; Berndt, P.; Dithmer, S.; Helms, H.C.; Puchkov, D.; Devraj, K.; Kaya, M.; Qin, Z.; et al. Tight junctions in the blood-brain barrier promote edema formation and infarct size in stroke—Ambivalent effects of sealing proteins. J. Cereb. Blood Flow Metab. 2021, 41, 132–145. [Google Scholar] [CrossRef]
- Seo, J.H.; Maki, T.; Miyamoto, N.; Choi, Y.K.; Chung, K.K.; Hamanaka, G.; Park, J.H.; Mandeville, E.T.; Takase, H.; Hayakawa, K.; et al. AKAP12 Supports Blood-Brain Barrier Integrity against Ischemic Stroke. Int. J. Mol. Sci. 2020, 21, 9078. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yan, F.; Han, X.; Huang, X.; Cheng, X.; Geng, Y.; Jiang, X.; Han, Y.; Zhao, M.; Zhu, L. NB-3 expression in endothelial cells contributes to the maintenance of blood brain barrier integrity in a mouse high-altitude cerebral edema model. Exp. Neurol. 2022, 354, 114116. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Campbell, M. Tight junction modulation at the blood-brain barrier: Current and future perspectives. Biochim. et Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183298. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Hanley, N.; Campbell, M. Claudin-5: Gatekeeper of neurological function. Fluids Barriers CNS 2019, 16, 3. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, J.J.; Yang, J.; Ronaldson, P.T.; Davis, T.P. Structure, Function, and Regulation of the Blood-Brain Barrier Tight Junction in Central Nervous System Disorders. Front. Physiol. 2020, 11, 914. [Google Scholar] [CrossRef]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef]
- McCaffrey, G.; Seelbach, M.J.; Staatz, W.D.; Nametz, N.; Quigley, C.; Campos, C.R.; Brooks, T.A.; Davis, T.P. Occludin oligomeric assembly at tight junctions of the blood-brain barrier is disrupted by peripheral inflammatory hyperalgesia. J. Neurochem. 2008, 106, 2395–2409. [Google Scholar] [CrossRef] [Green Version]
- Haseloff, R.F.; Dithmer, S.; Winkler, L.; Wolburg, H.; Blasig, I.E. Transmembrane proteins of the tight junctions at the blood-brain barrier: Structural and functional aspects. Semin. Cell Dev. Biol. 2015, 38, 16–25. [Google Scholar] [CrossRef]
- Zeniya, S.; Kuwahara, H.; Daizo, K.; Watari, A.; Kondoh, M.; Yoshida-Tanaka, K.; Kaburagi, H.; Asada, K.; Nagata, T.; Nagahama, M.; et al. Angubindin-1 opens the blood-brain barrier in vivo for delivery of antisense oligonucleotide to the central nervous system. J. Control Release 2018, 283, 126–134. [Google Scholar] [CrossRef]
- Kakogiannos, N.; Ferrari, L.; Giampietro, C.; Scalise, A.A.; Maderna, C.; Rava, M.; Taddei, A.; Lampugnani, M.G.; Pisati, F.; Malinverno, M.; et al. JAM-A Acts via C/EBP-alpha to Promote Claudin-5 Expression and Enhance Endothelial Barrier Function. Circ. Res. 2020, 127, 1056–1073. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Qian, F.; Guo, J.; Sha, Y.; Qian, Y. Selegiline Protects Against Lipopolysaccharide (LPS)-Induced Impairment of the Blood-Brain Barrier Through Regulating the NF-kappaB/MLCK/p-MLC Signaling Pathway. Neurotox. Res. 2022, 40, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Qu, M.; Li, Y.; Wang, L.; Zhang, L.; Wang, Y.; Tang, Y.; Tian, H.L.; Zhang, Z.; Yang, G.Y. MicroRNA-126-3p/-5p Overexpression Attenuates Blood-Brain Barrier Disruption in a Mouse Model of Middle Cerebral Artery Occlusion. Stroke 2020, 51, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, S.; D’Mello, V.; Caruso, D.; Wallerstein, A.; Abdul-Muneer, P.M. Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury. Exp. Neurol. 2019, 317, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.P.; He, Q.W.; Li, Y.N.; Chen, S.C.; Huang, M.; Wang, Y.; Gao, Y.; Huang, Y.; Wang, M.D.; Mao, L.; et al. Recombinant human sonic hedgehog protein regulates the expression of ZO-1 and occludin by activating angiopoietin-1 in stroke damage. PLoS ONE 2013, 8, e68891. [Google Scholar] [CrossRef] [Green Version]
- Huber, J.D.; Witt, K.A.; Hom, S.; Egleton, R.D.; Mark, K.S.; Davis, T.P. Inflammatory pain alters blood-brain barrier permeability and tight junctional protein expression. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1241–H1248. [Google Scholar] [CrossRef]
- Campos, C.R.; Ocheltree, S.M.; Hom, S.; Egleton, R.D.; Davis, T.P. Nociceptive inhibition prevents inflammatory pain induced changes in the blood-brain barrier. Brain Res. 2008, 1221, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Ronaldson, P.T.; Demarco, K.M.; Sanchez-Covarrubias, L.; Solinsky, C.M.; Davis, T.P. Transforming growth factor-beta signaling alters substrate permeability and tight junction protein expression at the blood-brain barrier during inflammatory pain. J. Cereb. Blood Flow Metab. 2009, 29, 1084–1098. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Ronaldson, P.T.; Davis, T. Blood-Brain Barrier Integrity and Glial Support: Mechanisms that can be Targeted for Novel Therapeutic Approaches in Stroke. Curr. Pharm. Des. 2012, 18, 3624–3644. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaeffer, S.; Iadecola, C. Revisiting the neurovascular unit. Nat. Neurosci. 2021, 24, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Willis, C.L.; Nolan, C.C.; Reith, S.N.; Lister, T.; Prior, M.J.; Guerin, C.J.; Mavroudis, G.; Ray, D.E. Focal astrocyte loss is followed by microvascular damage, with subsequent repair of the blood-brain barrier in the apparent absence of direct astrocytic contact. Glia 2004, 45, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Cantrill, C.A.; Skinner, R.A.; Rothwell, N.J.; Penny, J.I. An immortalised astrocyte cell line maintains the in vivo phenotype of a primary porcine in vitro blood-brain barrier model. Brain Res. 2012, 1479, 17–30. [Google Scholar] [CrossRef]
- Canfield, S.G.; Stebbins, M.J.; Morales, B.S.; Asai, S.W.; Vatine, G.D.; Svendsen, C.N.; Palecek, S.P.; Shusta, E.V. An isogenic blood-brain barrier model comprising brain endothelial cells, astrocytes, and neurons derived from human induced pluripotent stem cells. J. Neurochem. 2017, 140, 874–888. [Google Scholar] [CrossRef]
- Janzer, R.C.; Raff, M.C. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature 1987, 325, 253–257. [Google Scholar] [CrossRef]
- Willis, C.L.; Leach, L.; Clarke, G.J.; Nolan, C.C.; Ray, D.E. Reversible disruption of tight junction complexes in the rat blood-brain barrier, following transitory focal astrocyte loss. Glia 2004, 48, 1–13. [Google Scholar] [CrossRef]
- Kucharz, K.; Kutuzov, N.; Zhukov, O.; Mathiesen Janiurek, M.; Lauritzen, M. Shedding Light on the Blood-Brain Barrier Transport with Two-Photon Microscopy In Vivo. Pharm. Res. 2022, 39, 1457–1468. [Google Scholar] [CrossRef]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef]
- Ren, Z.; Iliff, J.J.; Yang, L.; Yang, J.; Chen, X.; Chen, M.J.; Giese, R.N.; Wang, B.; Shi, X.; Nedergaard, M. ‘Hit & Run’ model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J. Cereb. Blood Flow Metab. 2013, 33, 834–845. [Google Scholar] [CrossRef]
- Kimelberg, H.K.; Goderie, S.K.; Higman, S.; Pang, S.; Waniewski, R.A. Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J. Neurosci. 1990, 10, 1583–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.S.; Bach, M.D.; Ashkavand, Z.; Norman, K.R.; Martino, N.; Adam, A.P.; Mongin, A.A. Metabolic constraints of swelling-activated glutamate release in astrocytes and their implication for ischemic tissue damage. J. Neurochem. 2019, 151, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Schlachetzki, F.; Pardridge, W.M. P-glycoprotein and caveolin-1α in endothelium and astrocytes of primate brain. NeuroReport 2003, 14, 2041–2046. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson, P.T.; Bendayan, M.; Gingras, D.; Piquette-Miller, M.; Bendayan, R. Cellular localization and functional expression of P-glycoprotein in rat astrocyte cultures. J. Neurochem. 2004, 89, 788–800. [Google Scholar] [CrossRef]
- van Vliet, E.A.; Iyer, A.M.; Mesarosova, L.; Colakoglu, H.; Anink, J.J.; van Tellingen, O.; Maragakis, N.J.; Shefner, J.; Bunt, T.; Aronica, E. Expression and Cellular Distribution of P-Glycoprotein and Breast Cancer Resistance Protein in Amyotrophic Lateral Sclerosis Patients. J. Neuropathol. Exp. Neurol. 2020, 79, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Nies, A.; Jedlitschky, G.; König, J.; Herold-Mende, C.; Steiner, H.; Schmitt, H.-P.; Keppler, D. Expression and immunolocalization of the multidrug resistance proteins, MRP1–MRP6 (ABCC1–ABCC6), in human brain. Neuroscience 2004, 129, 349–360. [Google Scholar] [CrossRef]
- Jördens, M.S.; Keitel, V.; Karababa, A.; Zemtsova, I.; Bronger, H.; Häussinger, D.; Görg, B. Multidrug resistance-associated protein 4 expression in ammonia-treated cultured rat astrocytes and cerebral cortex of cirrhotic patients with hepatic encephalopathy. Glia 2015, 63, 2092–2105. [Google Scholar] [CrossRef]
- Huber, R.D.; Gao, B.; Sidler Pfandler, M.A.; Zhang-Fu, W.; Leuthold, S.; Hagenbuch, B.; Folkers, G.; Meier, P.J.; Stieger, B. Characterization of two splice variants of human organic anion transporting polypeptide 3A1 isolated from human brain. Am. J. Physiol. Cell Physiol. 2007, 292, C795–C806. [Google Scholar] [CrossRef]
- Sweet, D.H. Organic Cation Transporter Expression and Function in the CNS. Handb. Exp. Pharmacol. 2021, 266, 41–80. [Google Scholar] [CrossRef]
- Hladky, S.B.; Barrand, M.A. The glymphatic hypothesis: The theory and the evidence. Fluids Barriers CNS 2022, 19, 9. [Google Scholar] [CrossRef]
- Thomas, J.H. Theoretical analysis of wake/sleep changes in brain solute transport suggests a flow of interstitial fluid. Fluids Barriers CNS 2022, 19, 30. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Culebras, A.; Duran-Laforet, V.; Pena-Martinez, C.; Ballesteros, I.; Pradillo, J.M.; Diaz-Guzman, J.; Lizasoain, I.; Moro, M.A. Myeloid cells as therapeutic targets in neuroinflammation after stroke: Specific roles of neutrophils and neutrophil-platelet interactions. J. Cereb. Blood Flow Metab. 2018, 38, 2150–2164. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Federoff, H.J. Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 176. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Davis, T.P. Regulation of blood-brain barrier integrity by microglia in health and disease: A therapeutic opportunity. J. Cereb. Blood Flow Metab. 2020, 40, S6–S24. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Akhmetzyanova, E.; Kletenkov, K.; Mukhamedshina, Y.; Rizvanov, A. Different Approaches to Modulation of Microglia Phenotypes After Spinal Cord Injury. Front. Syst. Neurosci. 2019, 13, 37. [Google Scholar] [CrossRef] [Green Version]
- Bachiller, S.; Jimenez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Hamanaka, G.; Lo, E.H.; Arai, K. Heterogeneity of microglia and their differential roles in white matter pathology. CNS Neurosci. Ther. 2019, 25, 1290–1298. [Google Scholar] [CrossRef]
- Barisano, G.; Montagne, A.; Kisler, K.; Schneider, J.A.; Wardlaw, J.M.; Zlokovic, B.V. Blood-brain barrier link to human cognitive impairment and Alzheimer’s Disease. Nat. Cardiovasc. Res. 2022, 1, 108–115. [Google Scholar] [CrossRef]
- Bankstahl, M.; Breuer, H.; Leiter, I.; Markel, M.; Bascunana, P.; Michalski, D.; Bengel, F.M.; Loscher, W.; Meier, M.; Bankstahl, J.P.; et al. Blood-Brain Barrier Leakage during Early Epileptogenesis Is Associated with Rapid Remodeling of the Neurovascular Unit. eNeuro 2018, 5, ENEURO.0123-18.2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.; Schlichter, L.; Bendayan, M.; Bendayan, R. Functional expression of P-glycoprotein in rat brain microglia. J. Pharmacol. Exp. Ther. 2001, 299, 204–212. [Google Scholar] [PubMed]
- Dallas, S.; Zhu, X.; Baruchel, S.; Schlichter, L.; Bendayan, R. Functional expression of the multidrug resistance protein 1 in microglia. J. Pharmacol. Exp. Ther. 2003, 307, 282–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallas, S.; Schlichter, L.; Bendayan, R. Multidrug resistance protein (MRP) 4- and MRP 5-mediated efflux of 9-(2-phosphonylmethoxyethyl)adenine by microglia. J. Pharmacol. Exp. Ther. 2004, 309, 1221–1229. [Google Scholar] [CrossRef] [Green Version]
- Ishimoto, T.; Nakamichi, N.; Nishijima, H.; Masuo, Y.; Kato, Y. Carnitine/Organic Cation Transporter OCTN1 Negatively Regulates Activation in Murine Cultured Microglial Cells. Neurochem. Res. 2018, 43, 116–128. [Google Scholar] [CrossRef]
- Dore-Duffy, P.; Cleary, K. Morphology and properties of pericytes. Methods Mol. Biol. 2011, 686, 49–68. [Google Scholar] [CrossRef]
- Halliday, M.R.; Rege, S.V.; Ma, Q.; Zhao, Z.; Miller, C.A.; Winkler, E.A.; Zlokovic, B.V. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2016, 36, 216–227. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef]
- Grant, R.I.; Hartmann, D.A.; Underly, R.G.; Berthiaume, A.A.; Bhat, N.R.; Shih, A.Y. Organizational hierarchy and structural diversity of microvascular pericytes in adult mouse cortex. J. Cereb. Blood Flow Metab. 2019, 39, 411–425. [Google Scholar] [CrossRef] [Green Version]
- Berthiaume, A.A.; Grant, R.I.; McDowell, K.P.; Underly, R.G.; Hartmann, D.A.; Levy, M.; Bhat, N.R.; Shih, A.Y. Dynamic Remodeling of Pericytes In Vivo Maintains Capillary Coverage in the Adult Mouse Brain. Cell. Rep. 2018, 22, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dore-Duffy, P.; Katychev, A.; Wang, X.; Van Buren, E. CNS microvascular pericytes exhibit multipotential stem cell activity. J. Cereb. Blood Flow Metab. 2006, 26, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, S.; Ohtsuki, S.; Hosoya, K.; Nakashima, E.; Terasaki, T. A pericyte-derived angiopoietin-1 multimeric complex induces occludin gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. J. Neurochem. 2004, 89, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villasenor, R.; Kuennecke, B.; Ozmen, L.; Ammann, M.; Kugler, C.; Gruninger, F.; Loetscher, H.; Freskgard, P.O.; Collin, L. Region-specific permeability of the blood-brain barrier upon pericyte loss. J. Cereb. Blood Flow Metab. 2017, 37, 3683–3694. [Google Scholar] [CrossRef]
- Dohgu, S.; Takata, F.; Yamauchi, A.; Nakagawa, S.; Egawa, T.; Naito, M.; Tsuruo, T.; Sawada, Y.; Niwa, M.; Kataoka, Y. Brain pericytes contribute to the induction and up-regulation of blood-brain barrier functions through transforming growth factor-beta production. Brain Res. 2005, 1038, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Berezowski, V.; Landry, C.; Dehouck, M.P.; Cecchelli, R.; Fenart, L. Contribution of glial cells and pericytes to the mRNA profiles of P-glycoprotein and multidrug resistance-associated proteins in an in vitro model of the blood-brain barrier. Brain Res. 2004, 1018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.G.; Holzl, G.; Dobrowolny, H.; Hildebrandt, J.; Trubner, K.; Krohn, M.; Bogerts, B.; Pahnke, J. Vascular and extravascular distribution of the ATP-binding cassette transporters ABCB1 and ABCC1 in aged human brain and pituitary. Mech. Ageing Dev. 2014, 141–142, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, M.; Pittman, Q.J. Talking back: Dendritic neurotransmitter release. Trends Neurosci. 2003, 26, 255–261. [Google Scholar] [CrossRef]
- Nishio, T.; Adachi, H.; Nakagomi, R.; Tokui, T.; Sato, E.; Tanemoto, M.; Fujiwara, K.; Okabe, M.; Onogawa, T.; Suzuki, T.; et al. Molecular identification of a rat novel organic anion transporter moat1, which transports prostaglandin D(2), leukotriene C(4), and taurocholate. Biochem. Biophys. Res. Commun. 2000, 275, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Feurstein, D.; Kleinteich, J.; Heussner, A.H.; Stemmer, K.; Dietrich, D.R. Investigation of microcystin congener-dependent uptake into primary murine neurons. Environ. Health Perspect 2010, 118, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Scafidi, S.; Douglas, R.M.; Farahani, R.; Banasiak, K.J.; Haddad, G.G. Prostaglandin transporter expression in mouse brain during development and in response to hypoxia. Neuroscience 2007, 146, 1150–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courousse, T.; Bacq, A.; Belzung, C.; Guiard, B.; Balasse, L.; Louis, F.; Le Guisquet, A.M.; Gardier, A.M.; Schinkel, A.H.; Giros, B.; et al. Brain organic cation transporter 2 controls response and vulnerability to stress and GSK3beta signaling. Mol. Psychiatry 2015, 20, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Bacq, A.; Balasse, L.; Biala, G.; Guiard, B.; Gardier, A.M.; Schinkel, A.; Louis, F.; Vialou, V.; Martres, M.P.; Chevarin, C.; et al. Organic cation transporter 2 controls brain norepinephrine and serotonin clearance and antidepressant response. Mol. Psychiatry 2012, 17, 926–939. [Google Scholar] [CrossRef] [Green Version]
- Lazarowski, A.; Caltana, L.; Merelli, A.; Rubio, M.D.; Ramos, A.J.; Brusco, A. Neuronal mdr-1 gene expression after experimental focal hypoxia: A new obstacle for neuroprotection? J. Neurol. Sci. 2007, 258, 84–92. [Google Scholar] [CrossRef]
- Merelli, A.; Ramos, A.J.; Lazarowski, A.; Auzmendi, J. Convulsive Stress Mimics Brain Hypoxia and Promotes the P-Glycoprotein (P-gp) and Erythropoietin Receptor Overexpression. Recombinant Human Erythropoietin Effect on P-gp Activity. Front. Neurosci. 2019, 13, 750. [Google Scholar] [CrossRef] [Green Version]
- Fonseca-Barriendos, D.; Perez-Perez, D.; Fuentes-Mejia, M.; Orozco-Suarez, S.; Alonso-Vanegas, M.; Martinez-Juarez, I.E.; Guevara-Guzman, R.; Castaneda-Cabral, J.L.; Rocha, L. Protein expression of P-glycoprotein in neocortex from patients with frontal lobe epilepsy. Epilepsy Res. 2022, 181, 106892. [Google Scholar] [CrossRef]
- Chen, L.; Chen, H.; Xing, Y.; Li, J. ABCC1 regulates cocaine-associated memory, spine plasticity and GluA1 and GluA2 surface expression. Neuroreport 2021, 32, 833–839. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ronaldson, P.T.; Davis, T.P. Transport Mechanisms at the Blood–Brain Barrier and in Cellular Compartments of the Neurovascular Unit: Focus on CNS Delivery of Small Molecule Drugs. Pharmaceutics 2022, 14, 1501. https://doi.org/10.3390/pharmaceutics14071501
Ronaldson PT, Davis TP. Transport Mechanisms at the Blood–Brain Barrier and in Cellular Compartments of the Neurovascular Unit: Focus on CNS Delivery of Small Molecule Drugs. Pharmaceutics. 2022; 14(7):1501. https://doi.org/10.3390/pharmaceutics14071501
Chicago/Turabian StyleRonaldson, Patrick T., and Thomas P. Davis. 2022. "Transport Mechanisms at the Blood–Brain Barrier and in Cellular Compartments of the Neurovascular Unit: Focus on CNS Delivery of Small Molecule Drugs" Pharmaceutics 14, no. 7: 1501. https://doi.org/10.3390/pharmaceutics14071501