
CYP3A5*3 and SLCO1B1 c.521T>C Polymorphisms Influence the Pharmacokinetics of Atorvastatin and 2-Hydroxy Atorvastatin

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Study Design
2.3. SLCO1B1 and CYP3A5 Genotyping
2.4. Bioanalysis
2.5. Pharmacokinetic Analysis
2.6. Statistical Analysis
3. Results
3.1. Demographics and Genotyping
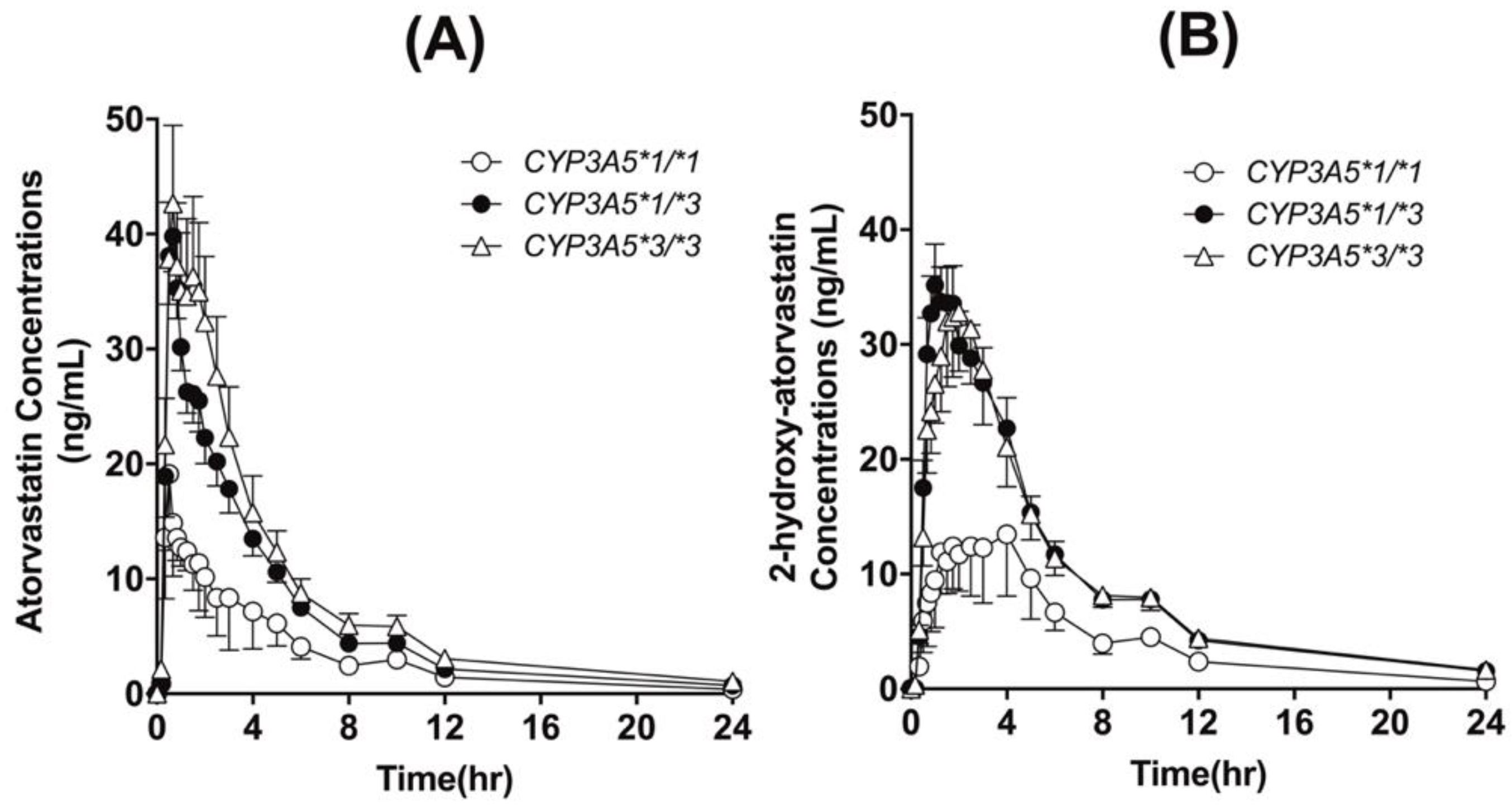
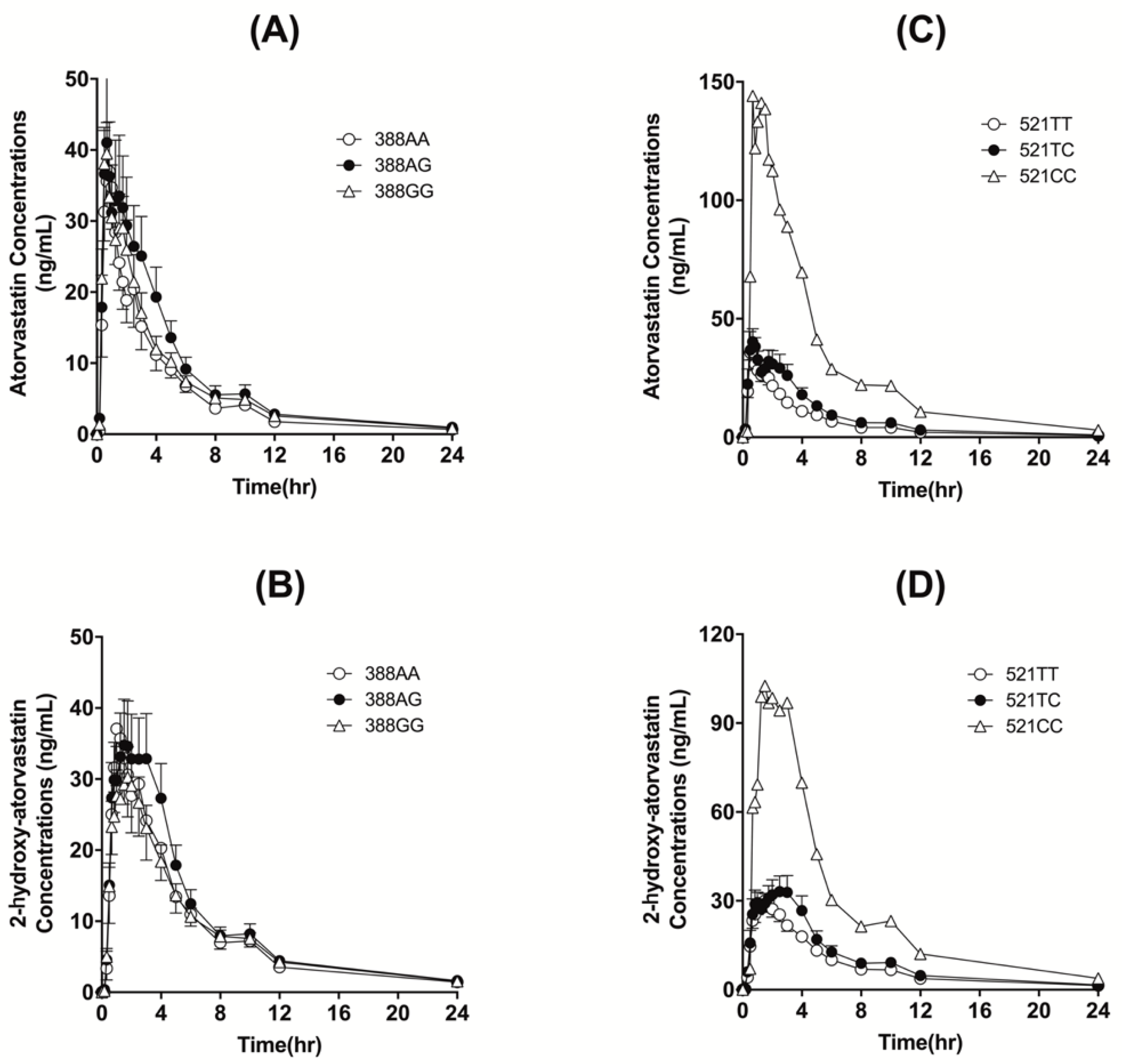
3.2. Effects of Polymorphic SLCO1B1 and CYP3A5 Genotypes
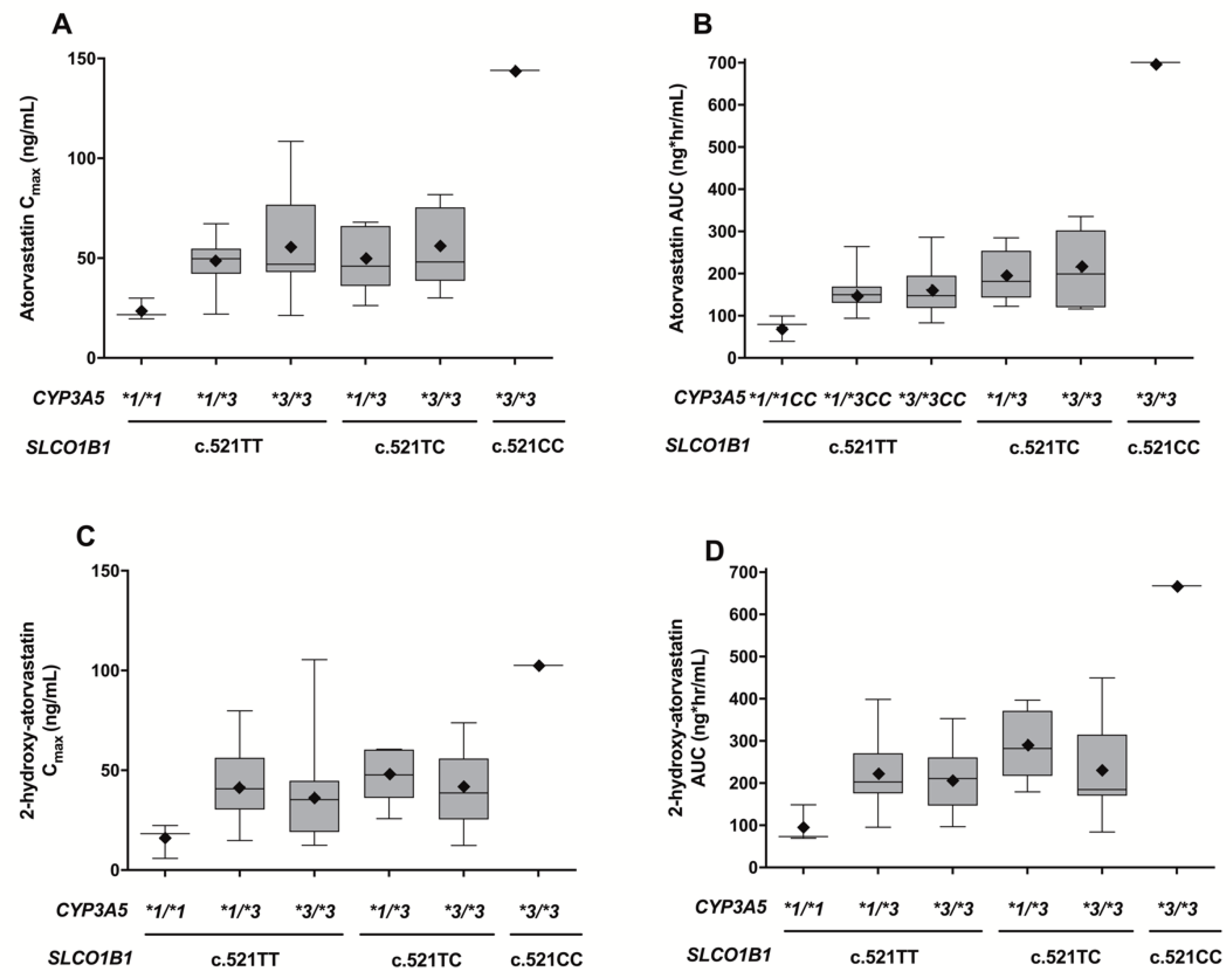
3.3. Simultaneous Effects of SLCO1B1 c.521T>C and CYP3A5*3 Polymorphisms
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Athyros, V.G.; Tziomalos, K.; Karagiannis, A.; Mikhailidis, D.P. Atorvastatin: Safety and tolerability. Expert Opin. Drug Saf. 2010, 9, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Poli, A. Atorvastatin: Pharmacological characteristics and lipid-lowering effects. Drugs 2007, 67 (Suppl. 1), 3–15. [Google Scholar] [CrossRef] [PubMed]
- Hermann, M.; Bogsrud, M.P.; Molden, E.; Asberg, A.; Mohebi, B.U.; Ose, L.; Retterstøl, K. Exposure of atorvastatin is unchanged but lactone and acid metabolites are increased several-fold in patients with atorvastatin-induced myopathy. Clin. Pharmacol. Ther. 2006, 79, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Wilke, R.A.; Moore, J.H.; Burmester, J.K. Relative impact of CYP3A genotype and concomitant medication on the severity of atorvastatin-induced muscle damage. Pharm. Genom. 2005, 15, 415–421. [Google Scholar] [CrossRef]
- Shin, J.; Pauly, D.F.; Pacanowski, M.A.; Langaee, T.; Frye, R.F.; Johnson, J.A. Effect of cytochrome P450 3A5 genotype on atorvastatin pharmacokinetics and its interaction with clarithromycin. Pharmacotherapy 2011, 31, 942–950. [Google Scholar] [CrossRef] [Green Version]
- Zubiaur, P.; Benedicto, M.D.; Villapalos-García, G.; Navares-Gómez, M.; Mejía-Abril, G.; Román, M.; Martín-Vílchez, S.; Ochoa, D.; Abad-Santos, F. SLCO1B1 Phenotype and CYP3A5 Polymorphism Significantly Affect Atorvastatin Bioavailability. J. Pers. Med. 2021, 11, 204. [Google Scholar] [CrossRef]
- Lennernäs, H. Clinical pharmacokinetics of atorvastatin. Clin. Pharmacokinet. 2003, 42, 1141–1160. [Google Scholar] [CrossRef]
- Wang, Y.; Tian, Y.; Lv, P.; Chen, L.; Luo, W.; Jing, X.; Li, H.; Tan, Z.; Wang, Y.; Zhou, H.; et al. The effect of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and 2-hydroxyatorvastatin in healthy Chinese people. Pharmazie 2017, 72, 365–368. [Google Scholar]
- Rajput, T.A.; Naveed, A.K.; Farooqi, Z.R.; Khan, S. Effects of two functionally important SLCO1B1 gene polymorphisms on pharmacokinetics of atorvastatin. Pak. J. Pharm. Sci. 2017, 30, 1363–1370. [Google Scholar]
- Daka, A.; Dimovski, A.; Kapedanovska, A.; Vavlukis, M.; Eftimov, A.; Labachevski, N.; Jakjovski, K.; Geshkovska, M.N.; Nebija, D.; Mladenovska, K. Effects of single nucleotide polymorphisms and haplotypes of the SLCO1B1 gene on the pharmacokinetic profile of atorvastatin in healthy Macedonian volunteers. Pharmazie 2015, 70, 480–488. [Google Scholar]
- Prado, Y.; Saavedra, N.; Zambrano, T.; Lagos, J.; Rosales, A.; Salazar, L.A. SLCO1B1 c.388A>G Polymorphism Is Associated with HDL-C Levels in Response to Atorvastatin in Chilean Individuals. Int. J. Mol. Sci. 2015, 16, 20609–20619. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, A.C.; Perin, P.M.; Purim, S.G.; Silbiger, V.N.; Genvigir, F.D.; Willrich, M.A.; Arazi, S.S.; Luchessi, A.D.; Hirata, M.H.; Bernik, M.M.; et al. Pharmacogenetics of OATP transporters reveals that SLCO1B1 c.388A>G variant is determinant of increased atorvastatin response. Int. J. Mol. Sci. 2011, 12, 5815–5827. [Google Scholar] [PubMed] [Green Version]
- Waring, R.H. Cytochrome P450: Genotype to phenotype. Xenobiotica 2020, 50, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Saiz-Rodríguez, M.; Almenara, S.; Navares-Gómez, M.; Ochoa, D.; Román, M.; Zubiaur, P.; Koller, D.; Santos, M.; Mejía, G.; Borobia, A.M.; et al. Effect of the Most Relevant CYP3A4 and CYP3A5 Polymorphisms on the Pharmacokinetic Parameters of 10 CYP3A Substrates. Biomedicines 2020, 8, 94. [Google Scholar] [CrossRef] [PubMed]
- Lolodi, O.; Wang, Y.M.; Wright, W.C.; Chen, T. Differential Regulation of CYP3A4 and CYP3A5 and its Implication in Drug Discovery. Curr. Drug Metab. 2017, 18, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- Yee, J.; Kim, H.; Heo, Y.; Yoon, H.Y.; Song, G.; Gwak, H.S. Association between CYP3A5 Polymorphism and Statin-Induced Adverse Events: A Systemic Review and Meta-Analysis. J. Pers. Med. 2021, 11, 677. [Google Scholar] [CrossRef]
- Xie, H.G.; Wood, A.J.; Kim, R.B.; Stein, C.M.; Wilkinson, G.R. Genetic variability in CYP3A5 and its possible consequences. Pharmacogenomics 2004, 5, 243–272. [Google Scholar] [CrossRef]
- De Jonge, H.; de Loor, H.; Verbeke, K.; Vanrenterghem, Y.; Kuypers, D.R. In vivo CYP3A4 activity, CYP3A5 genotype, and hematocrit predict tacrolimus dose requirements and clearance in renal transplant patients. Clin. Pharmacol. Ther. 2012, 92, 366–375. [Google Scholar] [CrossRef]
- Thummel, K.E. Does the CYP3A5*3 polymorphism affect in vivo drug elimination? Pharmacogenetics 2003, 13, 585–587. [Google Scholar] [CrossRef]
- Park, J.E.; Kim, K.B.; Bae, S.K.; Moon, B.S.; Liu, K.H.; Shin, J.G. Contribution of cytochrome P450 3A4 and 3A5 to the metabolism of atorvastatin. Xenobiotica 2008, 38, 1240–1251. [Google Scholar] [CrossRef]
- Kivistö, K.T.; Niemi, M.; Schaeffeler, E.; Pitkälä, K.; Tilvis, R.; Fromm, M.F.; Schwab, M.; Eichelbaum, M.; Strandberg, T. Lipid-lowering response to statins is affected by CYP3A5 polymorphism. Pharmacogenetics 2004, 14, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Park, I.H.; Kim, J.M.; Noh, J.H.; Kim, K.A.; Park, J.Y. Rapid detection of FMO3 single nucleotide polymorphisms using a pyrosequencing method. Mol. Med. Rep. 2022, 25, 1–7. [Google Scholar] [CrossRef]
- Kwak, H.D.; Kim, S.H.; Seo, Y.S.; Song, K.J. Detecting hepatitis B virus in surgical smoke emitted during laparoscopic surgery. Occup. Environ. Med. 2016, 73, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Cho, D.Y.; Shin, H.J.; Lee, S.S.; Shon, J.H.; Shin, J.G.; Shin, S.G. Duplex pyrosequencing assay of the 388A>G and 521T>C SLCO1B1 polymorphisms in three Asian populations. Clin. Chim. Acta 2008, 388, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Joo, H.J.; Lee, H.M.; Park, J.Y. Influence of ABCB1 and CYP3A5 genetic polymorphisms on the pharmacokinetics of quetiapine in healthy volunteers. Pharm. Genom. 2014, 24, 35–42. [Google Scholar] [CrossRef]
- Borek-Dohalský, V.; Huclová, J.; Barrett, B.; Nemec, B.; Ulc, I.; Jelínek, I. Validated HPLC-MS-MS method for simultaneous determination of atorvastatin and 2-hydroxyatorvastatin in human plasma-pharmacokinetic study. Anal. Bioanal. Chem. 2006, 386, 275–285. [Google Scholar] [CrossRef]
- Heatherington, A.C.; Vicini, P.; Golde, H. A pharmacokinetic/pharmacodynamic comparison of SAAM II and PC/WinNonlin modeling software. J. Pharm. Sci. 1998, 87, 1255–1263. [Google Scholar] [CrossRef]
- Kalliokoski, A.; Niemi, M. Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 2009, 158, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Mladenovska, K.; Grapci, A.D.; Vavlukis, M.; Kapedanovska, A.; Eftimov, A.; Geshkovska, N.M.; Nebija, D.; Dimovski, A.J. Influence of SLCO1B1 polymorphisms on atorvastatin efficacy and safety in Macedonian subjects. Pharmazie 2017, 72, 288–295. [Google Scholar]
- Lau, Y.Y.; Okochi, H.; Huang, Y.; Benet, L.Z. Multiple transporters affect the disposition of atorvastatin and its two active hydroxy metabolites: Application of in vitro and ex situ systems. J. Pharmacol. Exp. Ther. 2006, 316, 762–771. [Google Scholar] [CrossRef]
- Tirona, R.G.; Leake, B.F.; Merino, G.; Kim, R.B. Polymorphisms in OATP-C: Identification of multiple allelic variants associated with altered transport activity among European- and African-Americans. J. Biol. Chem. 2001, 276, 35669–35675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romaine, S.P.; Bailey, K.M.; Hall, A.S.; Balmforth, A.J. The influence of SLCO1B1 (OATP1B1) gene polymorphisms on response to statin therapy. Pharmacogenom. J. 2010, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, A.E.; Hayes, R.N.; Roth, B.D.; Woo, P.; Woolf, T.F. Metabolism and excretion of atorvastatin in rats and dogs. Drug Metab. Dispos. 1999, 27, 916–923. [Google Scholar] [PubMed]
- Backman, J.T.; Luurila, H.; Neuvonen, M.; Neuvonen, P.J. Rifampin markedly decreases and gemfibrozil increases the plasma concentrations of atorvastatin and its metabolites. Clin. Pharmacol. Ther. 2005, 78, 154–167. [Google Scholar] [CrossRef]
- Backman, J.T.; Kyrklund, C.; Kivistö, K.T.; Wang, J.S.; Neuvonen, P.J. Plasma concentrations of active simvastatin acid are increased by gemfibrozil. Clin. Pharmacol. Ther. 2000, 68, 122–129. [Google Scholar] [CrossRef]
- Prueksaritanont, T.; Zhao, J.J.; Ma, B.; Roadcap, B.A.; Tang, C.; Qiu, Y.; Liu, L.; Lin, J.H.; Pearson, P.G.; Baillie, T.A. Mechanistic studies on metabolic interactions between gemfibrozil and statins. J. Pharmacol. Exp. Ther. 2002, 301, 1042–1051. [Google Scholar] [CrossRef] [Green Version]
- Hermann, M.; Asberg, A.; Christensen, H.; Holdaas, H.; Hartmann, A.; Reubsaet, J.L. Substantially elevated levels of atorvastatin and metabolites in cyclosporine-treated renal transplant recipients. Clin. Pharmacol. Ther. 2004, 76, 388–391. [Google Scholar] [CrossRef]
- Asberg, A.; Hartmann, A.; Fjeldså, E.; Bergan, S.; Holdaas, H. Bilateral pharmacokinetic interaction between cyclosporine A and atorvastatin in renal transplant recipients. Am. J. Transplant. 2001, 1, 382–386. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Genotype | Frequency (%) | Allele | Frequency (%) | |
|---|---|---|---|---|
| CYP3A5*3 | *1/*1 | 3 (6.5%) | *1 | 30.4% |
| (rs776746) | *1/*3 | 22 (47.8%) | *3 | 69.6% |
| *3/*3 | 21 (45.7%) | |||
| SLCO1B1 c.521T>C | TT | 33 (71.7%) | T | 84.8% |
| (rs4149056) | TC | 12 (26.1%) | C | 15.2% |
| CC | 1 (2.2%) | |||
| SLCO1B1 c.388A>G | AA | 6 (13.0%) | A | 28.3% |
| (rs2306283) | AG | 15 (30.4%) | G | 71.7% |
| GG | 27 (56.5%) |
| CYP3A5 Genotype | CYP3A5*1/*1 | CYP3A5*1/*3 | CYP3A5*3/*3 | p-Value |
|---|---|---|---|---|
| Individuals (n) | 3 | 22 | 21 | |
| Atorvastatin | ||||
| Half-life (h) | 5.18 ± 2.42 | 7.87 ± 2.17 | 8.28 ± 1.74 | 0.052 |
| Tmax (h) | 0.86 ± 0.77 | 0.94 ± 0.88 | 1.00 ± 0.61 | 0.483 |
| Cmax (ng/mL) | 23.77 ± 5.50 | 49.04 ± 11.90 | 61.15 ± 27.99 | 0.012 a,* |
| AUCall (ng·h/mL) | 72.78 ± 30.57 | 161.03 ± 47.85 | 209.74 ± 131.03 | 0.010 a,* |
| AUCinf (ng·h/mL) | 75.86 ± 26.98 | 162.79 ± 49.62 | 211.56 ± 131.45 | 0.010 a,* |
| CL/F (L/h) | 1164.3 ± 470.4 | 532.2 ± 151.4 | 463.0 ± 173.6 | <0.001 a,b,* |
| 2-OH atorvastatin | ||||
| Half-life (h) | 7.47 ± 3.41 | 8.98 ± 1.90 | 9.39 ± 2.27 | 0.644 |
| Tmax (h) | 2.02 ± 1.72 | 1.45 ± 0.89 | 1.50 ± 0.72 | 0.789 |
| Cmax (ng/mL) | 15.50 ± 8.58 | 43.34 ± 16.66 | 42.71 ± 25.05 | 0.097 |
| AUCall (ng·h/mL) | 96.911 ± 44.67 | 238.50 ± 85.82 | 244.80 ± 128.63 | 0.030 a,b,* |
| AUCinf (ng·h/mL) | 109.39 ± 38.47 | 242.49 ± 88.65 | 249.22 ± 129.18 | 0.040 a,b,* |
| AUC Ratio | 1.54 ± 0.61 | 1.49 ± 0.39 | 1.21 ± 0.33 | 0.049 a,* |
| (2-OH atorvastatin/atorvastatin) |
| c.388AA | c.388AG | c.388GG | p-Value | |
|---|---|---|---|---|
| Individuals (n) | 6 | 14 | 26 | |
| Atorvastatin | ||||
| Half-life (h) | 0.09 ± 0.02 | 0.08 ± 0.01 | 0.10 ± 0.04 | 0.619 |
| Tmax (h) | 7.74 ± 1.75 | 8.35 ± 1.73 | 7.67 ± 2.36 | 0.390 |
| Cmax (ng/mL) | 0.819 ± 0.309 | 1.166 ± 1.090 | 0.839 ± 0.564 | 0.529 |
| AUCall (ng·h/mL) | 42.751 ± 11.328 | 52.018 ± 31.393 | 54.716 ± 19.825 | 0.464 |
| AUCinf (ng·h/mL) | 146.14 ± 39.051 | 201.82 ± 152.98 | 168.23 ± 73.396 | 0.458 |
| CL/F (L/h) | 147.32 ± 39.909 | 203.95 ± 153.58 | 170.14 ± 73.819 | 0.747 |
| 2-OH atorvastatin | ||||
| Half-life (h) | 9.52 ± 1.49 | 9.65 ± 2.61 | 8.68 ± 2.03 | 0.358 |
| Tmax (h) | 1.02 ± 0.18 | 1.79 ± 1.24 | 1.42 ± 0.68 | 0.174 |
| Cmax (ng/mL) | 40.21 ± 18.94 | 44.07 ± 24.33 | 38.90 ± 21.67 | 0.782 |
| AUCall (ng·h/mL) | 223.33 ± 96.387 | 256.59 ± 146.55 | 214.05 ± 94.209 | 0.526 |
| AUCinf (ng·h/mL) | 227.24 ± 98.987 | 261.32 ± 146.88 | 218.89 ± 94.842 | 0.530 |
| AUC Ratio | 1.55 ± 0.71 | 1.34 ± 0.27 | 1.34 ± 0.36 | 0.482 |
| (2-OH atorvastatin/atorvastatin) |
| c.521TT | c.521TC | c.521CC | c.521TC or c.521CC | p-Value | |
|---|---|---|---|---|---|
| Individuals (n) | 33 | 12 | 1 | 13 | |
| Atorvastatin | |||||
| Half-life (h) | 7.84 ± 2.38 | 8.07 ± 1.20 | 7.31 | 8.01 ± 1.17 | 0.812 |
| Tmax (h) | 0.86 ± 0.68 | 1.15 ± 0.90 | 0.66 | 1.12 ± 0.88 | 0.295 |
| Cmax (ng/mL) | 49.16 ± 18.81 | 53.41 ± 18.78 | 144.0 | 60.39 ± 30.92 | 0.139 |
| AUCall (ng·h/mL) | 148.35 ± 51.206 | 206.67 ± 78.036 | 700.80 | 244.68 ± 156.09 | 0.003 * |
| AUCinf (ng·h/mL) | 150.27 ± 52.008 | 208.26 ± 78.133 | 704.74 | 246.46 ± 156.70 | 0.003 * |
| CL/F (L/h) | 607.22 ± 264.20 | 438.18 ± 163.14 | 113.51 | 413.21 ± 180.30 | 0.019 * |
| 2-OH atorvastatin | |||||
| Half-life (h) | 9.18 ± 2.34 | 8.92 ± 1.80 | 7.90 | 8.85 ± 1.75 | 0.641 |
| Tmax (h) | 1.41 ± 0.85 | 1.69 ± 0.97 | 1.5 | 1.68 ± 0.94 | 0.361 |
| Cmax (ng/mL) | 37.47 ± 20.79 | 44.20 ± 17.39 | 102.5 | 48.70 ± 23.22 | 0.118 |
| AUCall (ng·h/mL) | 204.57 ± 82.641 | 256.57 ± 106.30 | 667.80 | 288.21 ±152.86 | 0.02 * |
| AUCinf (ng·h/mL) | 209.70 ± 83.89 | 259.91 ± 106.47 | 673.98 | 291.77 ± 153.56 | 0.024 * |
| AUC Ratio | 1.42 ± 0.41 | 1.26 ± 0.32 | 0.96 | 1.24 ± 0.41 | 0.172 |
| (2-OH atorvastatin/atorvastatin) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.-W.; Kim, J.-M.; Lee, H.-Y.; Noh, J.; Kim, K.-A.; Park, J.-Y. CYP3A5*3 and SLCO1B1 c.521T>C Polymorphisms Influence the Pharmacokinetics of Atorvastatin and 2-Hydroxy Atorvastatin. Pharmaceutics 2022, 14, 1491. https://doi.org/10.3390/pharmaceutics14071491
Park J-W, Kim J-M, Lee H-Y, Noh J, Kim K-A, Park J-Y. CYP3A5*3 and SLCO1B1 c.521T>C Polymorphisms Influence the Pharmacokinetics of Atorvastatin and 2-Hydroxy Atorvastatin. Pharmaceutics. 2022; 14(7):1491. https://doi.org/10.3390/pharmaceutics14071491
Chicago/Turabian StylePark, Jin-Woo, Jong-Min Kim, Hwa-Young Lee, Jihyeon Noh, Kyoung-Ah Kim, and Ji-Young Park. 2022. "CYP3A5*3 and SLCO1B1 c.521T>C Polymorphisms Influence the Pharmacokinetics of Atorvastatin and 2-Hydroxy Atorvastatin" Pharmaceutics 14, no. 7: 1491. https://doi.org/10.3390/pharmaceutics14071491