
Extracellular Alterations in pH and K+ Modify the Murine Brain Endothelial Cell Total and Phospho-Proteome

 , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Treatments
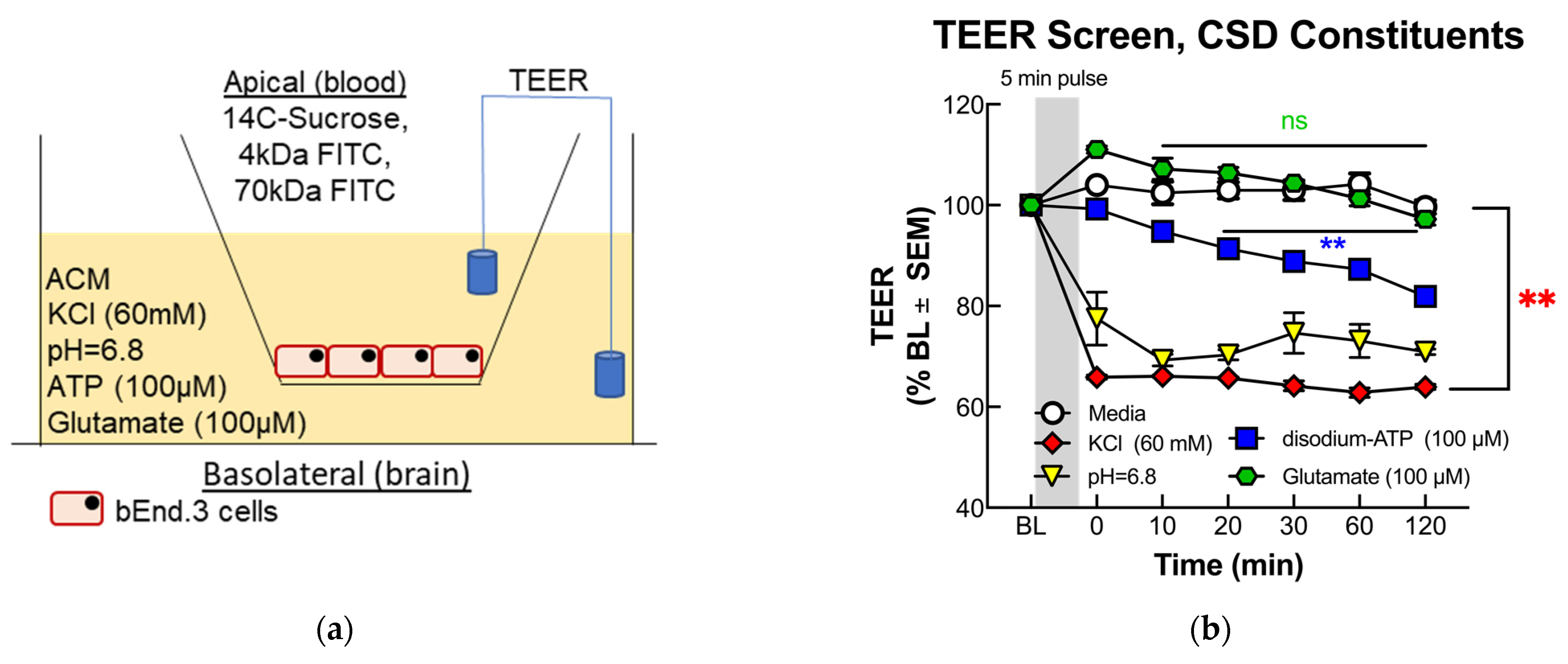
2.3. Transwell Cell Co-Cultures
2.4. Trans-Endothelial Electrical Resistance (TEER)
2.5. Immunocytochemistry
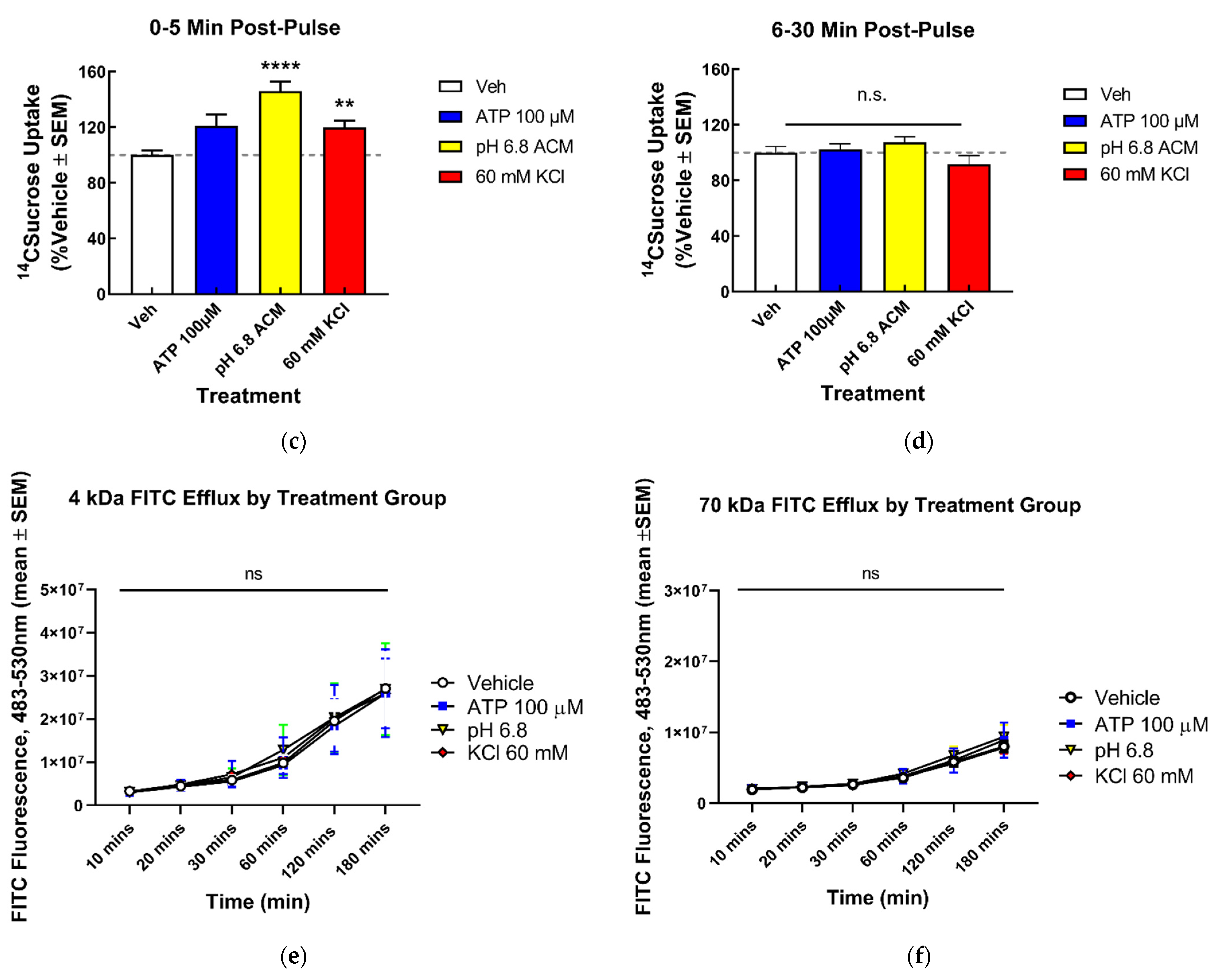
2.6. 14C-Sucrose Transport Assays
2.7. Fluorescein Isothiocyanate-Dextran Transport Assays
2.8. Western Immunoblotting
2.9. Biotynylation
2.10. Proteomics
2.11. Phospho-Proteomics
2.12. Statistics
3. Results
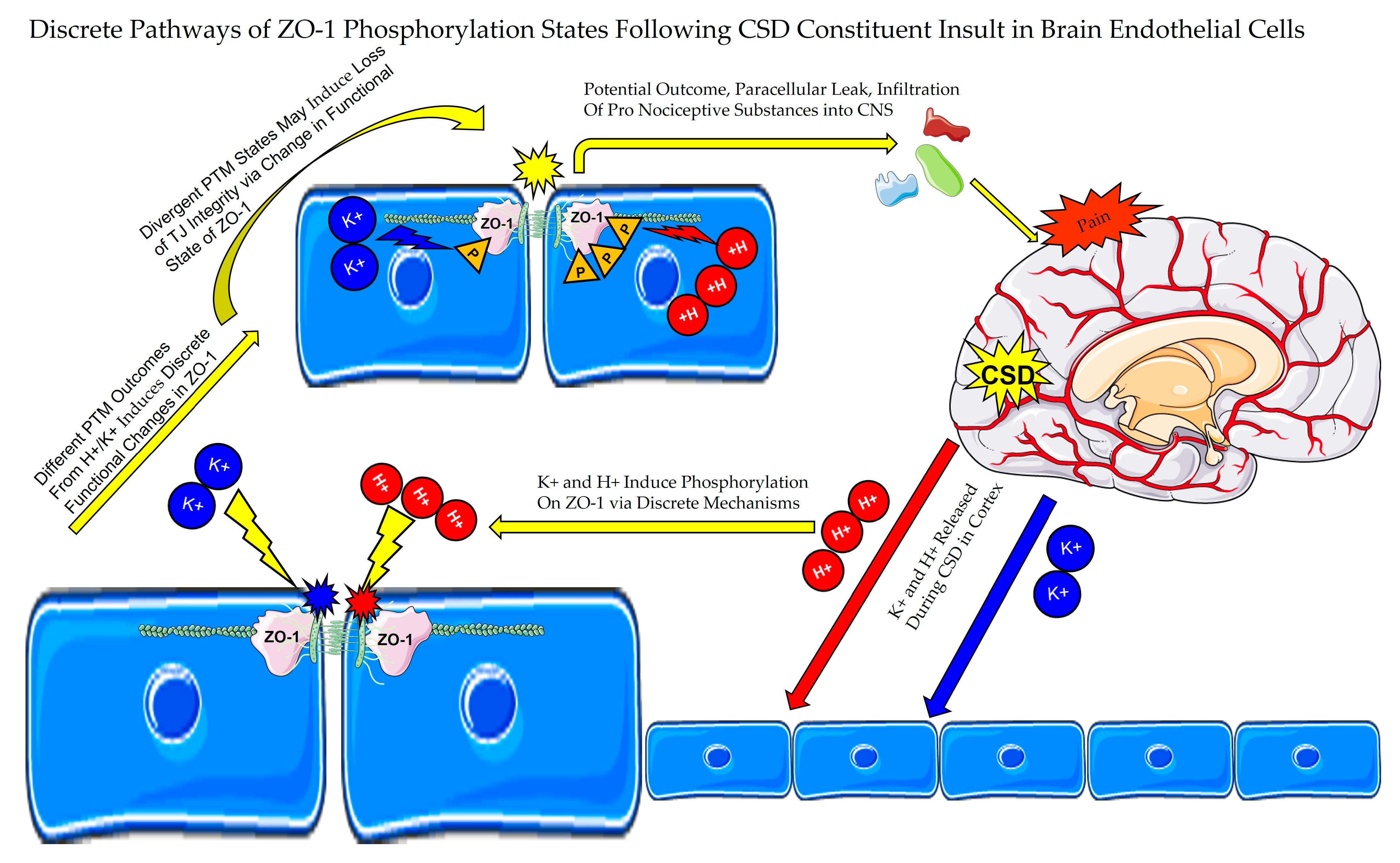
3.1. In Vitro Modeling and Functional Significance of CSD Induced Paracellular Leak in BEB
3.1.1. TEER Screening of BEB Demonstrates Rapid Induction of Paracellular Leak by Abluminal K+ and H+ Ion Treatment in a Transwell In Vitro Model of the BEB
3.1.2. Functional BEB Integrity Is Disrupted by KCl, Acidified pH, and ATP, but Not High Concentrations of Abluminal Glutamate
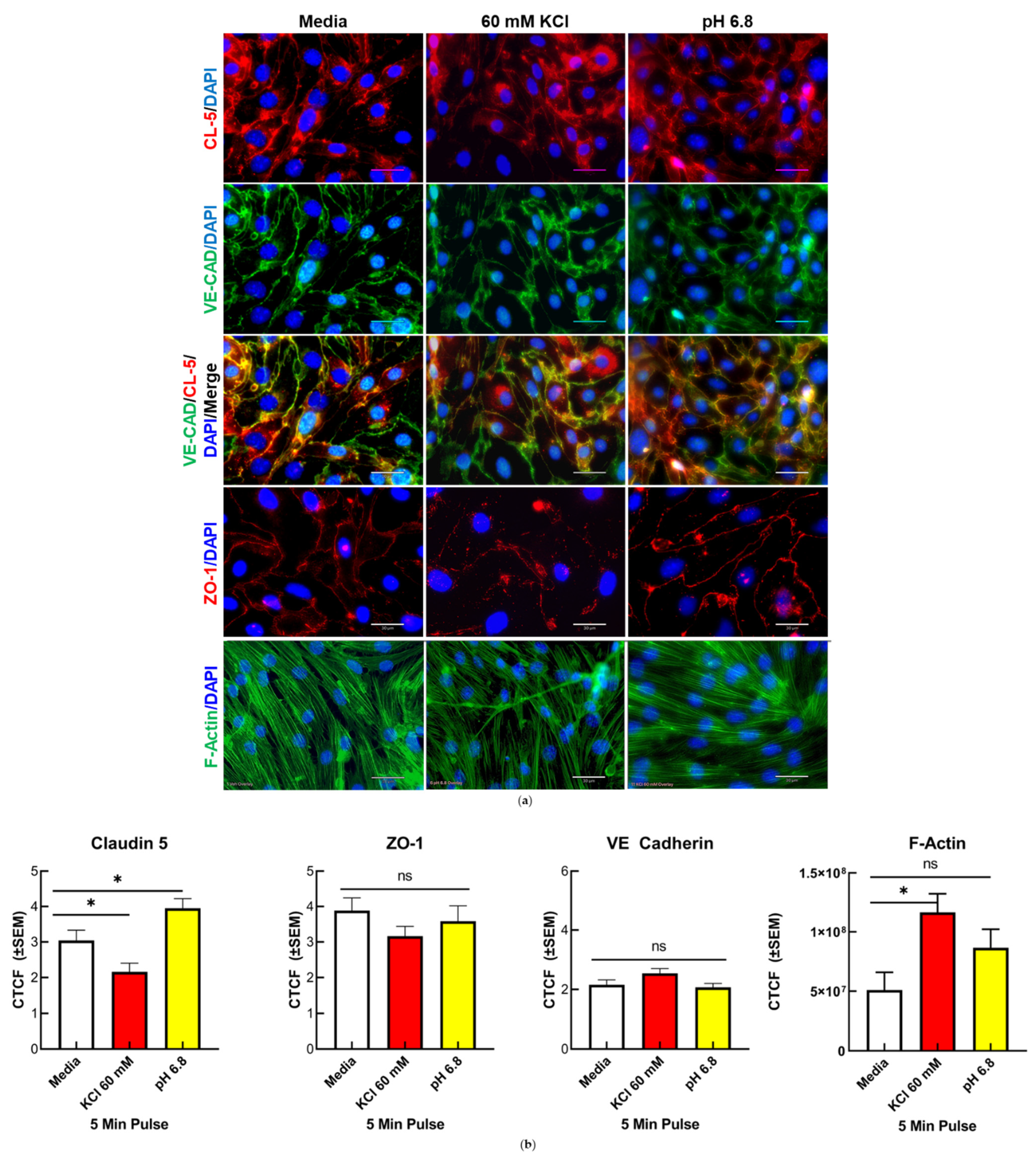
3.2. Changes in TJ Localization, but Not Total Expression, Are Induced by Mediators Released during CSD
3.2.1. Confocal Immunofluorescence and Quantification of CL5, but Not ZO-1 and VE-CAD, Demonstrated a Significant Alteration to Localization Following KCl and Acidified pH Treatment
3.2.2. Confocal Imaging and Quantification of f-Actin Filaments Demonstrate Increase in f-Actin Stress Fibers Following KCl Insult
3.3. Dynamics of CL-5 Trafficking and Localization within Cellular Components
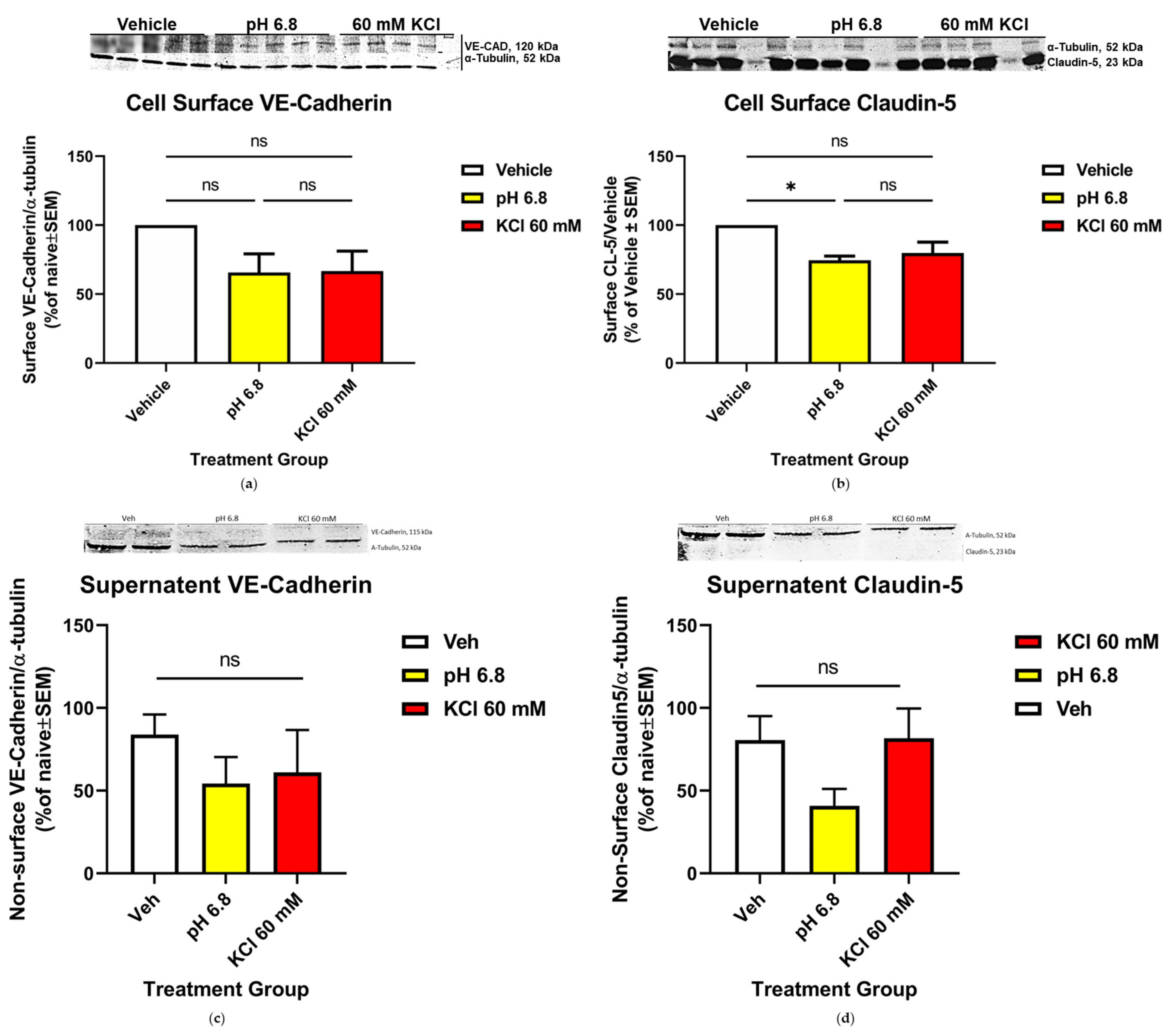
Endothelial Cell Surface Localization of CL-5 Was Significantly Increased Following Acidified pH Treatment, While VE-CAD Surface Detection Was Unaffected
3.4. Mechanisms of CL-5 Reorganization
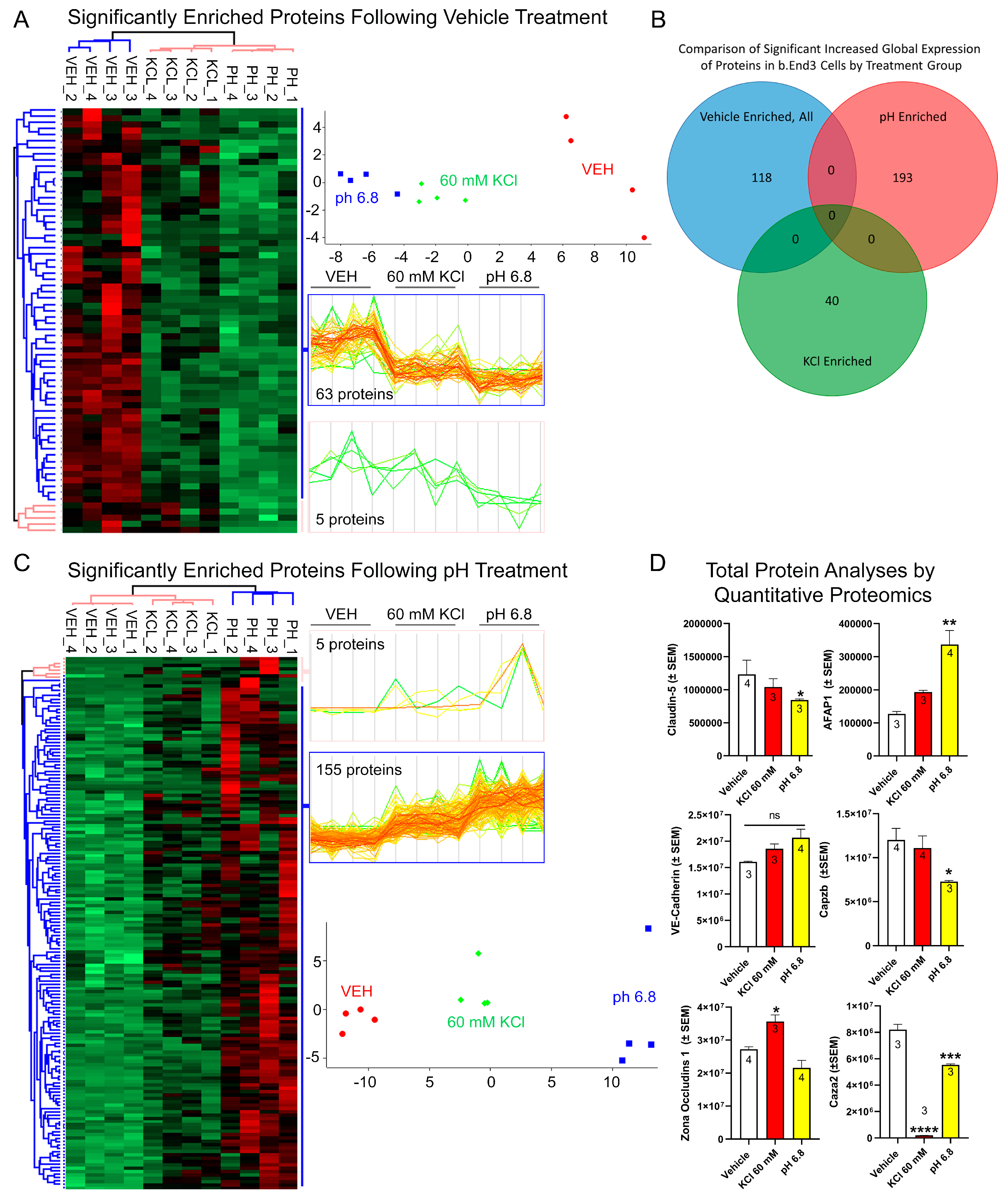
3.4.1. Global Quantitative Unlabeled Proteome Analysis Demonstrated Significant, Differential Changes to Total Enrichment of TJ and Cytoskeletal Associated Proteins Following KCl and Acidified pH Exposure
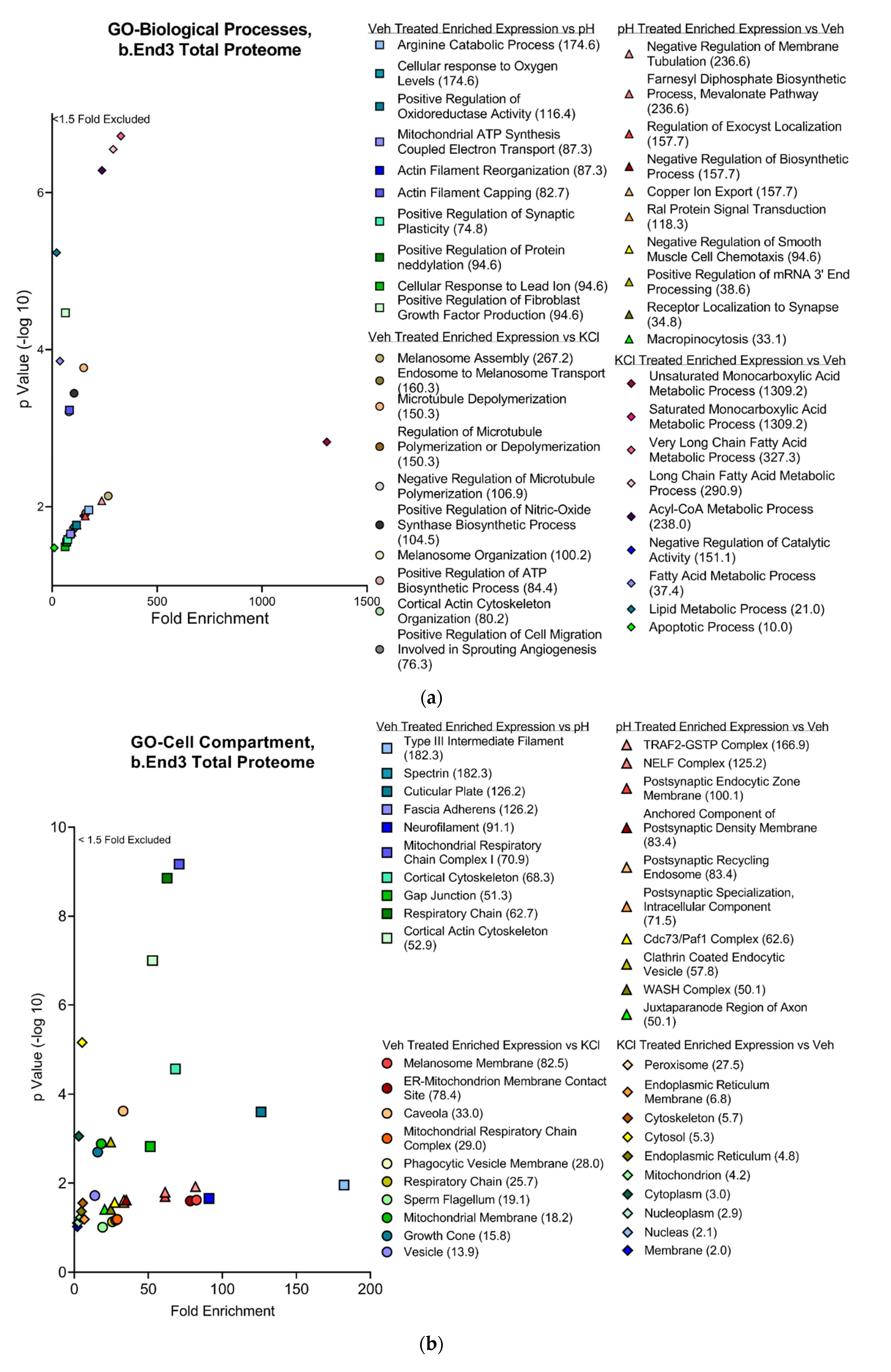
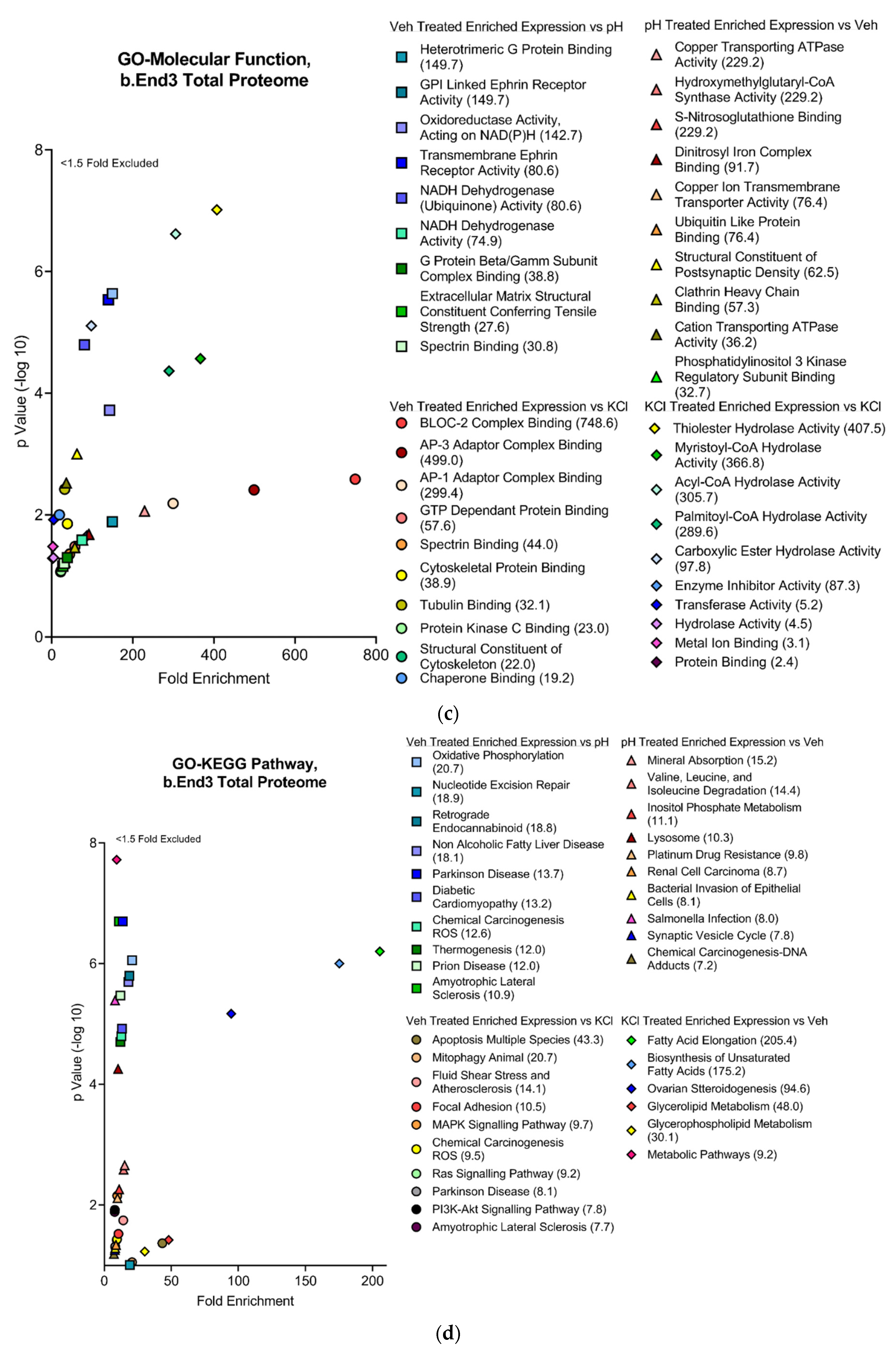
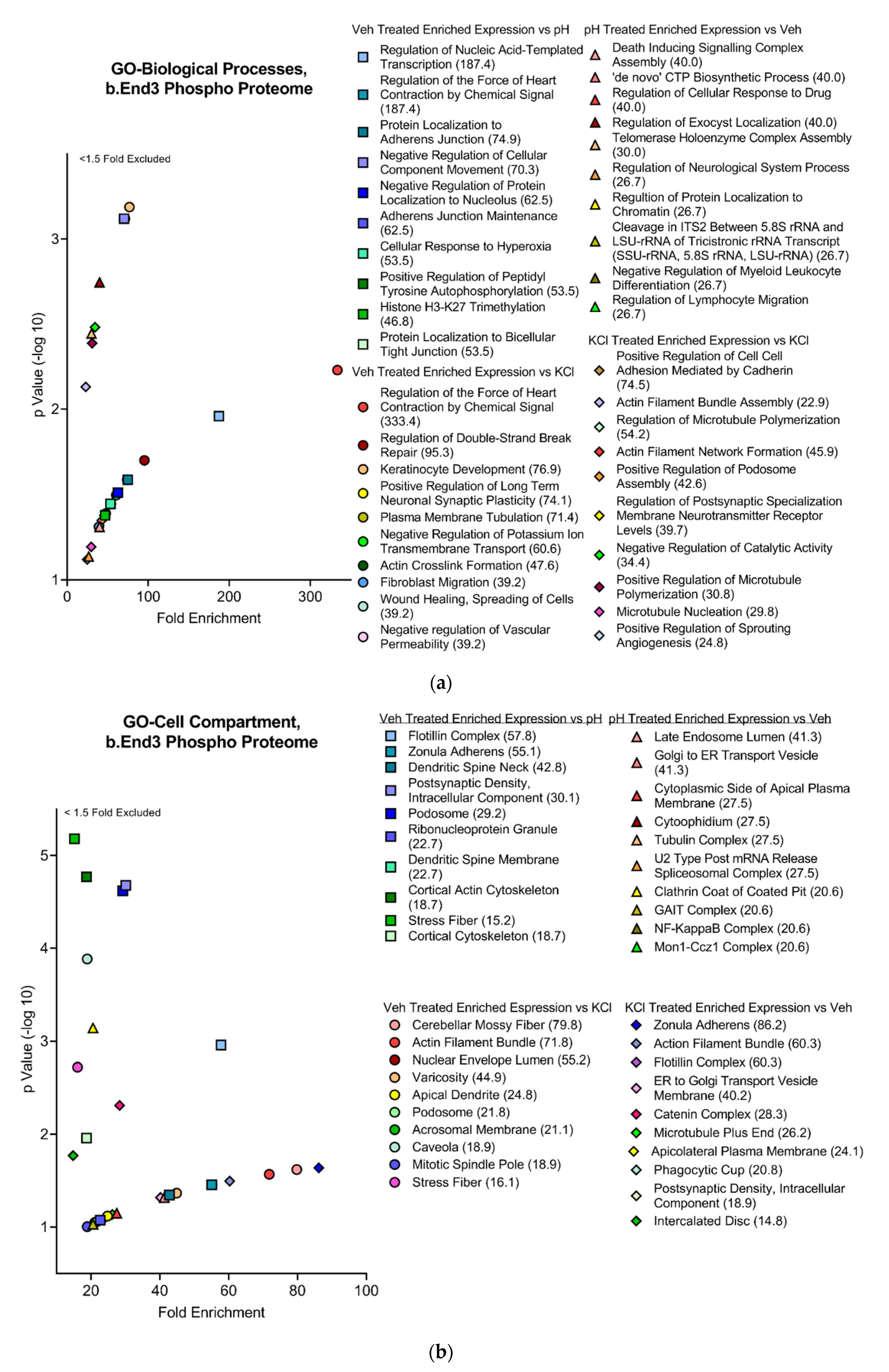
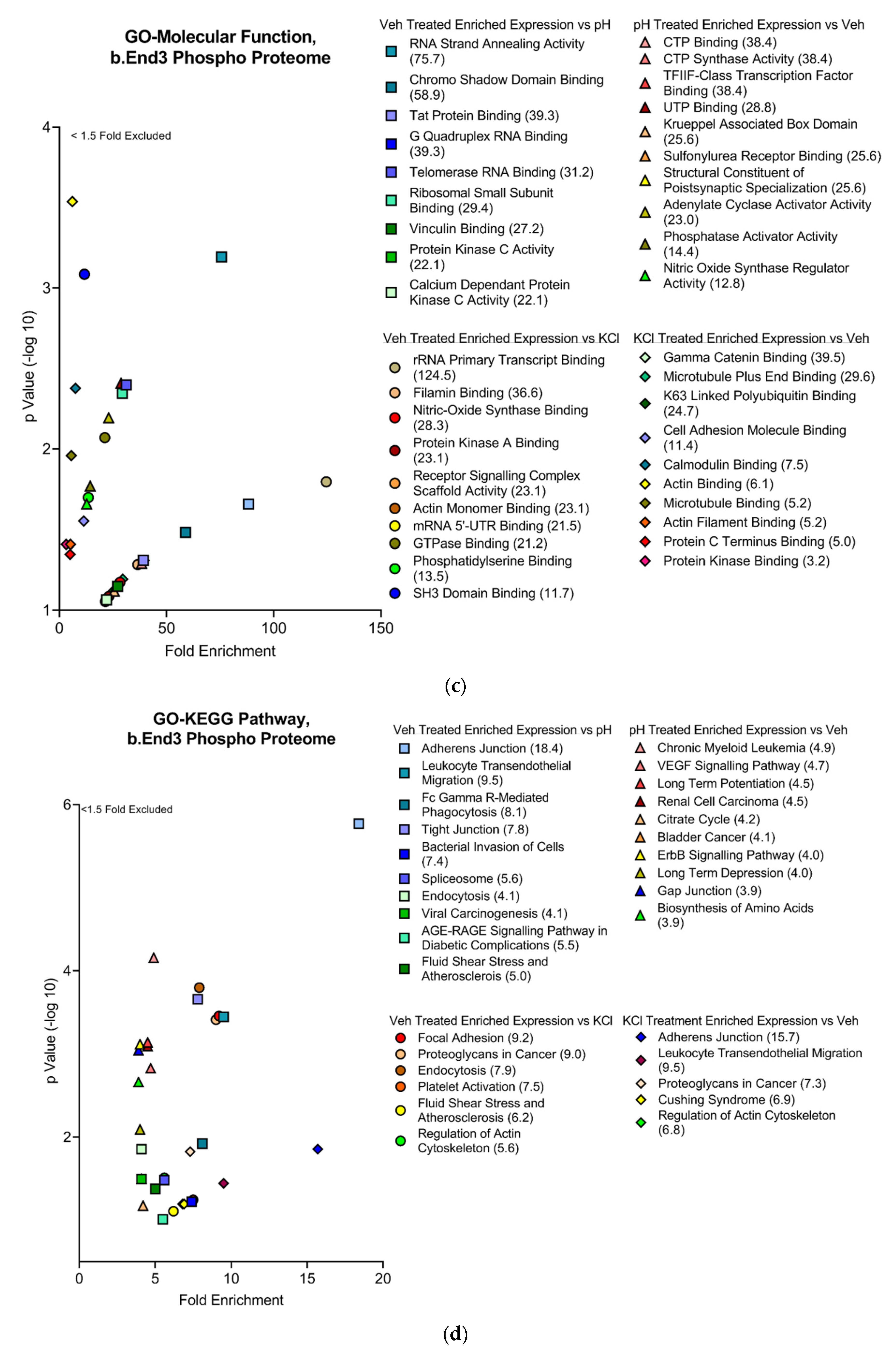
3.4.2. Functional and Pathway Analyses of Global b.End3 Proteome with GO and KEGG Bioinformatic Databases
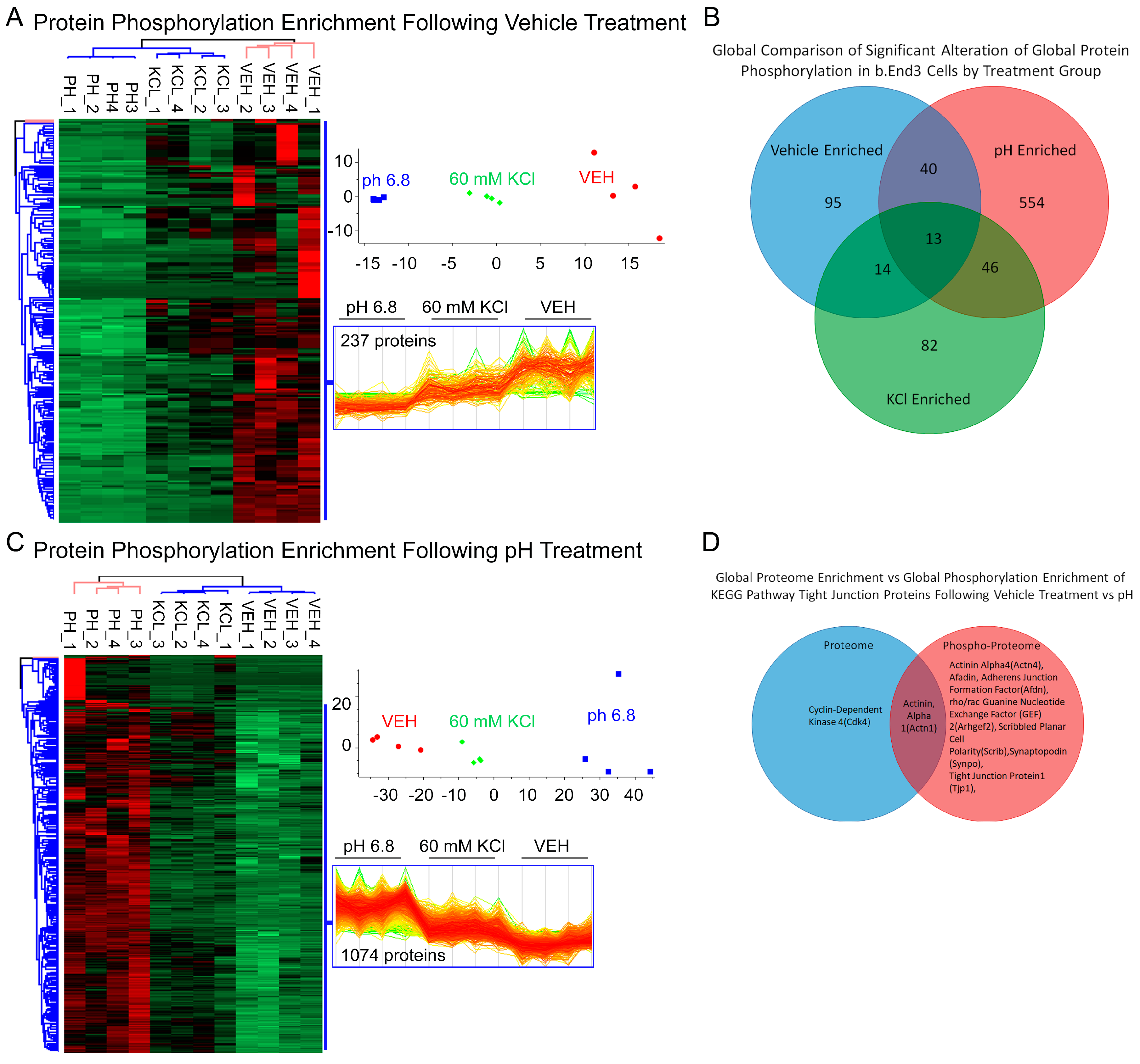
3.4.3. Global b.End3 Phospho-Proteomic and Bioinformatic Analyses Identified Significant and Highly Variable Enrichment of Phosphorylation Sites on TJ and Cytoskeletal Proteins Uniquely Associated with Treatment
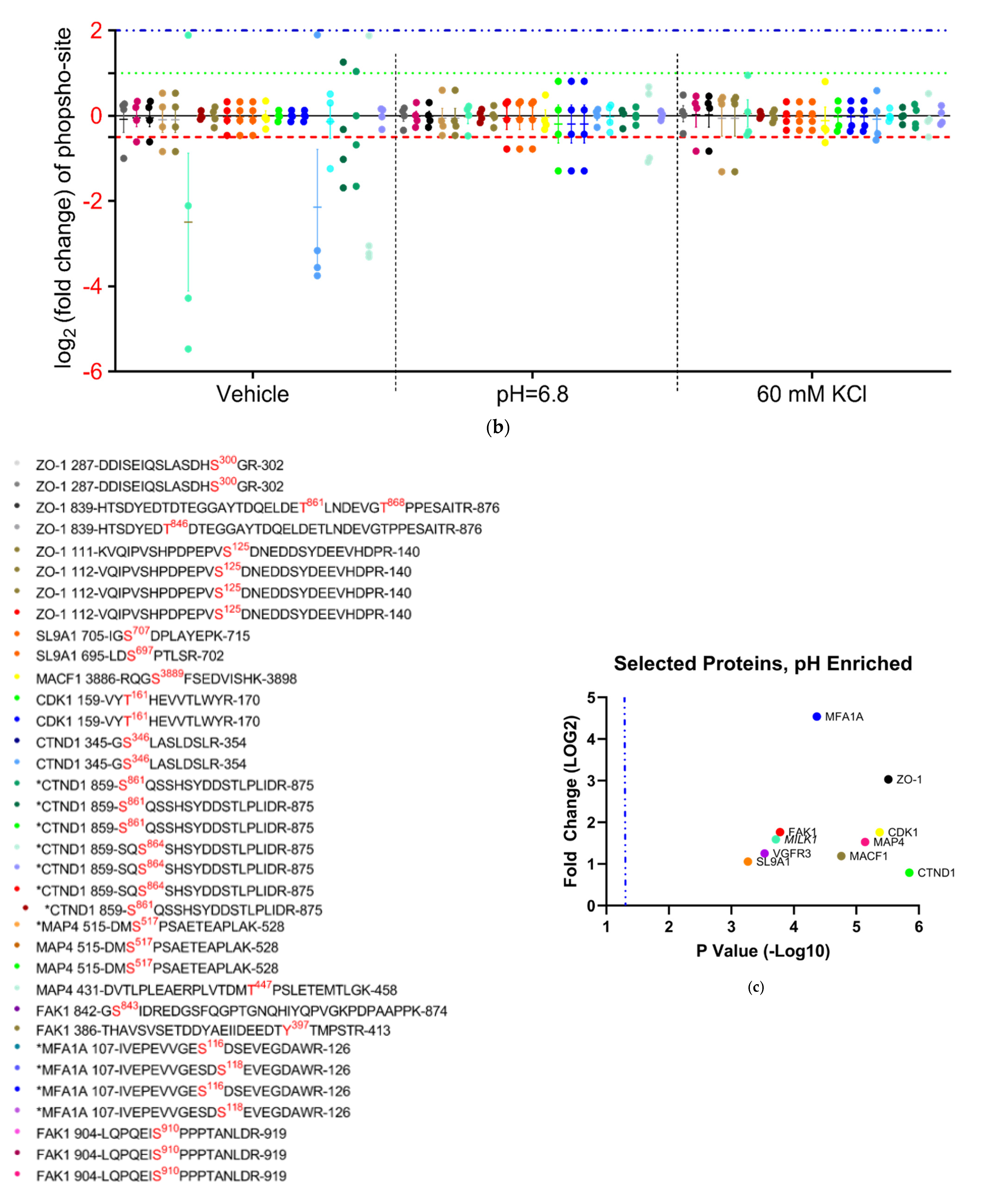
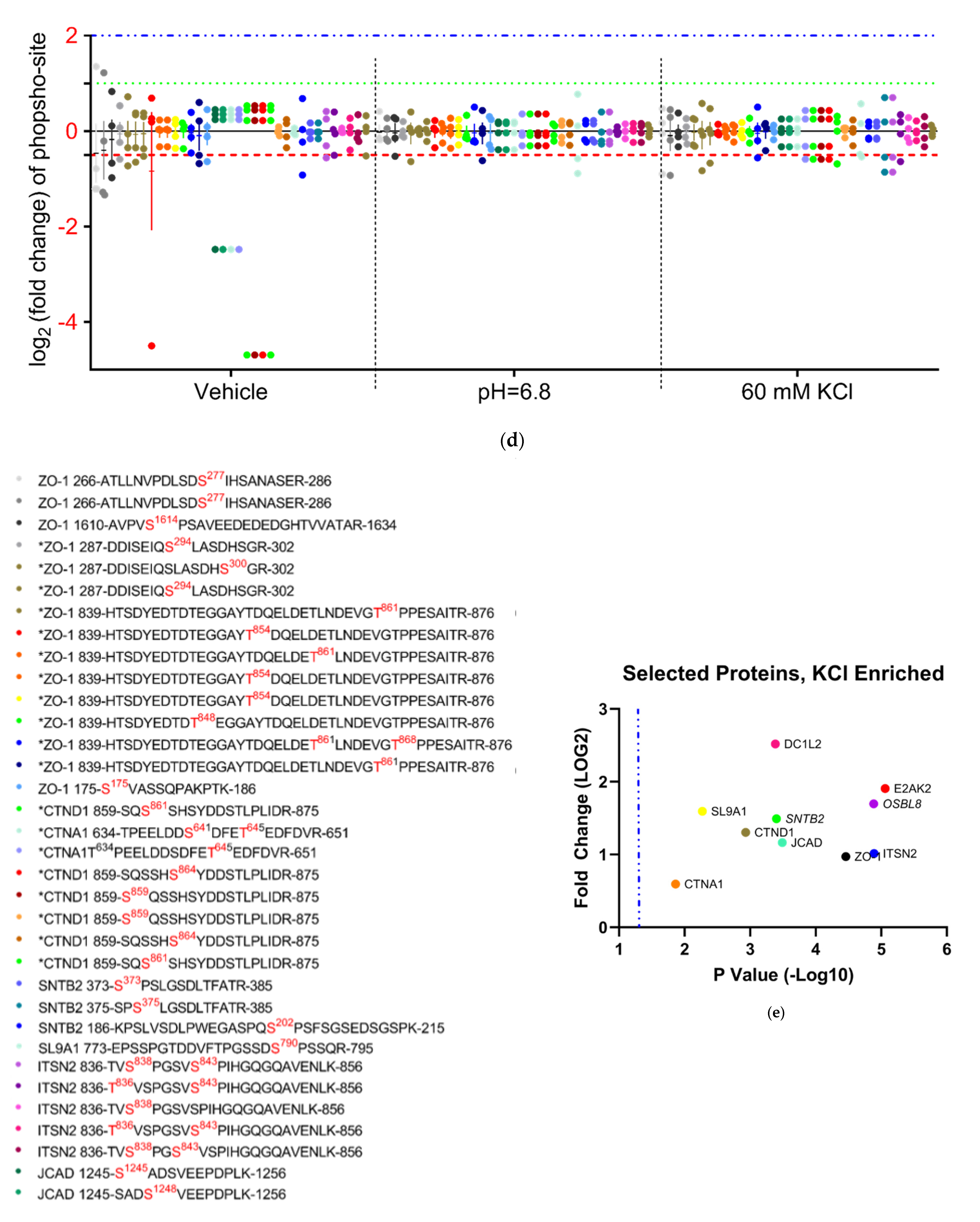
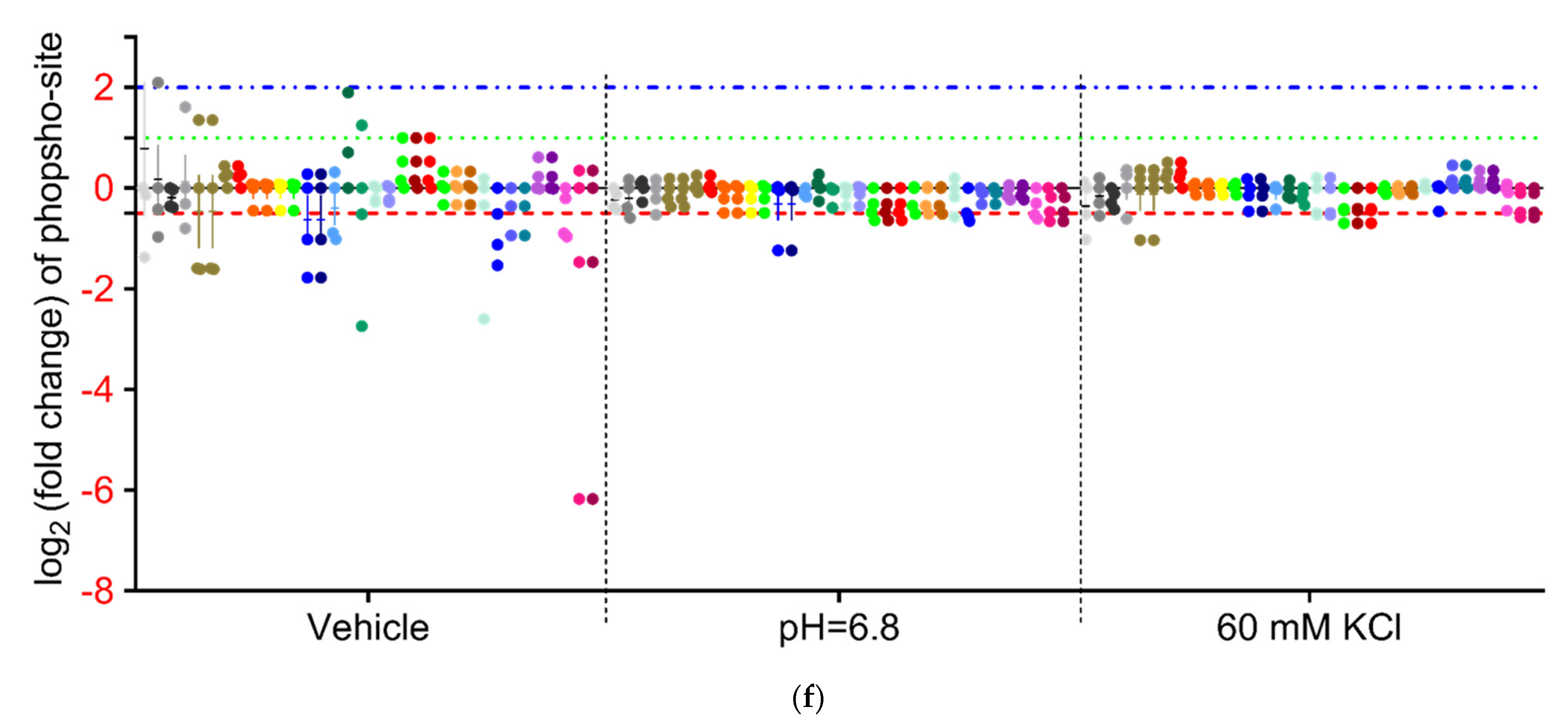
3.4.4. Analysis of Phospho-Peptide Enrichment Demonstrates Unique Enrichment of ZO-1 by Treatment Group
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hagihara, H.; Catts, V.S.; Katayama, Y.; Shoji, H.; Takagi, T.; Huang, F.L.; Nakao, A.; Mori, Y.; Huang, K.-P.; Ishii, S.; et al. Decreased Brain pH as a Shared Endophenotype of Psychiatric Disorders. Neuropsychopharmacology 2018, 43, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, Y.; Németh, E.; Schomburg, R.; Chemla, A.; Fülöp, L.; Menger, M.D.; Liu, Y.; Fassbender, K. Decreased pH in the aging brain and Alzheimer’s disease. Neurobiol. Aging 2021, 101, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, M. Pathophysiology of the migraine aura. The spreading depression theory. Brain 1994, 117 Pt 1, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Olsen, T.S. Pathophysiology of the migraine aura. The spreading depression theory. Brain 1995, 118 Pt 1, 307–308. [Google Scholar] [CrossRef] [PubMed]
- Ayata, C.; Lauritzen, M. Spreading Depression, Spreading Depolarizations, and the Cerebral Vasculature. Physiol. Rev. 2015, 95, 953–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottier, K.E.; Galloway, E.A.; Calabrese, E.C.; Tome, M.E.; Liktor-Busa, E.; Kim, J.; Davis, T.P.; Vanderah, T.W.; Largent-Milnes, T.M. Loss of Blood-Brain Barrier Integrity in a KCl-Induced Model of Episodic Headache Enhances CNS Drug Delivery. eNeuro 2018, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, M.; Dreier, J.; Fabricius, M.; Hartings, J.; Graf, R.; Strong, A.J. Clinical relevance of cortical spreading depression in neurological disorders: Migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J. Cereb. Blood Flow Metab. 2011, 31, 17–35. [Google Scholar] [CrossRef]
- Mathew, A.A.; Panonnummal, R. Cortical spreading depression: Culprits and mechanisms. Exp. Brain Res. 2022, 240, 733–749. [Google Scholar] [CrossRef]
- Shabir, O.; Pendry, B.; Lee, L.; Eyre, B.; Sharp, P.S.; Rebollar, M.A.; Drew, D.; Howarth, C.; Heath, P.R.; Wharton, S.B.; et al. Assessment of neurovascular coupling and cortical spreading depression in mixed mouse models of atherosclerosis and Alzheimer’s disease. Elife 2022, 11, e68242. [Google Scholar] [CrossRef]
- Basarsky, T.A.; Feighan, D.; MacVicar, B.A. Glutamate release through volume-activated channels during spreading depression. J. Neurosci. 1999, 19, 6439–6445. [Google Scholar] [CrossRef]
- Fabricius, M.; Jensen, L.H.; Lauritzen, M. Microdialysis of interstitial amino acids during spreading depression and anoxic depolarization in rat neocortex. Brain Res. 1993, 612, 61–69. [Google Scholar] [CrossRef]
- Csiba, L.; Paschen, W.; Mies, G. Regional changes in tissue pH and glucose content during cortical spreading depression in rat brain. Brain Res. 1985, 336, 167–170. [Google Scholar] [CrossRef]
- Gault, L.M.; Lin, C.-W.; LaManna, J.C.; Lust, W.D. Changes in energy metabolites, cGMP and intracellular pH during cortical spreading depression. Brain Res. 1994, 641, 176–180. [Google Scholar] [CrossRef]
- Scheller, D.; Kolb, J.; Tegtmeier, F. Lactate and pH change in close correlation in the extracellular space of the rat brain during cortical spreading depression. Neurosci. Lett. 1992, 135, 83–86. [Google Scholar] [CrossRef]
- Xiong, Z.Q.; Stringer, J.L. Extracellular pH responses in CA1 and the dentate gyrus during electrical stimulation, seizure discharges, and spreading depression. J. Neurophysiol. 2000, 83, 3519–3524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaemi, A.; Alizadeh, L.; Babaei, S.; Jafarian, M.; Ghadiri, M.K.; Meuth, S.G.; Kovac, S.; Gorji, A. Astrocyte-mediated inflammation in cortical spreading depression. Cephalalgia 2018, 38, 626–638. [Google Scholar] [CrossRef] [PubMed]
- van den Maagdenberg, A.M.; Pietrobon, D.; Pizzorusso, T.; Kaja, S.; Broos, L.A.; Cesetti, T.; van de Ven, R.C.; Tottene, A.; van der Kaa, J.; Plomp, J.J.; et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 2004, 41, 701–710. [Google Scholar] [CrossRef]
- Zhao, Z.; Shang, X.; Chen, Y.; Zheng, Y.; Huang, W.; Jiang, H.; Lv, Q.; Kong, D.; Jiang, Y.; Liu, P. Bacteria elevate extracellular adenosine to exploit host signaling for blood-brain barrier disruption. Virulence 2020, 11, 980–994. [Google Scholar] [CrossRef]
- Ebner, T.J.; Chen, G. Spreading acidification and depression in the cerebellar cortex. Neuroscientist 2003, 9, 37–45. [Google Scholar] [CrossRef]
- Dreier, J.P.; Woitzik, J.; Fabricius, M.; Bhatia, R.; Major, S.; Drenckhahn, C.; Lehmann, T.-N.; Sarrafzadeh, A.; Willumsen, L.; Hartings, J.; et al. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain 2006, 129 Pt 12, 3224–3237. [Google Scholar] [CrossRef]
- Gao, H.M.; Li, L.; Zhang, K.-L.; Chen, X.-H.; Tian, S.-Q.; Zhang, Z.-L. Impact of migraine attacks on the blood-brain barrier. Chin. Med. J. 2010, 123, 2559–2561. [Google Scholar] [PubMed]
- Gupta, V.K. CSD, BBB and MMP-9 elevations: Animal experiments versus clinical phenomena in migraine. Expert Rev. Neurother. 2009, 9, 1595–1614. [Google Scholar] [CrossRef] [PubMed]
- Gursoy-Ozdemir, Y.; Qiu, J.; Matsuoka, N.; Bolay, H.; Bermpohl, D.; Jin, H.; Wang, X.; Rosenberg, G.A.; Lo, E.H.; Moskowitz, M.A. Cortical spreading depression activates and upregulates MMP-9. J. Clin. Investig. 2004, 113, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Cermeno, J.; Gobernado, J.M.; Aimeno, A. Transient blood-brain barrier (BBB) damage in migraine. Headache 1986, 26, 437. [Google Scholar] [PubMed]
- Ashina, M.; Tvedskov, J.; Lipka, K.; Bilello, J.; Penkowa, M.; Olesen, J. Matrix metalloproteinases during and outside of migraine attacks without aura. Cephalalgia 2010, 30, 303–310. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Aänismaa, P.; Gatlik-Landwojtowicz, E.; Seelig, A. P-glycoprotein senses its substrates and the lateral membrane packing density: Consequences for the catalytic cycle. Biochemistry 2008, 47, 10197–10207. [Google Scholar] [CrossRef]
- Aarseth, P.; Barstad, J.A. Blood-brain barrier permeability in various parts of the central nervous system. Arch. Int. Pharmacodyn. Ther. 1968, 176, 434–442. [Google Scholar]
- Blasig, I.E.; Winkler, L.; Lassowski, B.; Mueller, S.L.; Zuleger, N.; Krause, E.; Krause, G.; Gast, K.; Kolbe, M.; Piontek, J. On the self-association potential of transmembrane tight junction proteins. Cell. Mol. Life Sci. 2006, 63, 505–514. [Google Scholar] [CrossRef]
- Andrew, R.D.; Hsieh, Y.T.; Brisson, C.D. Spreading depolarization triggered by elevated potassium is weak or absent in the rodent lower brain. J. Cereb. Blood Flow Metab. 2017, 37, 1735–1747. [Google Scholar] [CrossRef]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.-O.; Deli, M.A.; Förster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef] [PubMed]
- Kruse, R.; Krantz, J.; Barker, N.; Coletta, R.L.; Rafikov, R.; Luo, M.; Højlund, K.; Mandarino, L.J.; Langlais, P.R. Characterization of the CLASP2 Protein Interaction Network Identifies SOGA1 as a Microtubule-Associated Protein. Mol. Cell. Proteom. 2017, 16, 1718–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol. 2018, 1711, 133–148. [Google Scholar] [PubMed] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Colaert, N.; Helsens, K.; Martens, L.; Vandekerckhove, J.; Gevaert, K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 2009, 6, 786–787. [Google Scholar] [CrossRef]
- Colaert, N.; Maddelein, D.; Impens, F.; Van Damme, P.; Plasman, K.; Helsens, K.; Hulstaert, N.; Vandekerckhove, J.; Gevaert, K.; Martens, L. The Online Protein Processing Resource (TOPPR): A database and analysis platform for protein processing events. Nucleic Acids Res. 2013, 41, D333–D337. [Google Scholar] [CrossRef]
- Maddelein, D.; Colaert, N.; Buchanan, I.; Hulstaert, N.; Gevaert, K.; Martens, L. The iceLogo web server and SOAP service for determining protein consensus sequences. Nucleic Acids Res. 2015, 43, W543–W546. [Google Scholar] [CrossRef]
- Charles, A.C.; Baca, S.M. Cortical spreading depression and migraine. Nat. Rev. Neurol. 2013, 9, 637–644. [Google Scholar] [CrossRef]
- Charles, A.; Brennan, K. Cortical spreading depression-new insights and persistent questions. Cephalalgia 2009, 29, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Pietrobon, D.; Moskowitz, M.A. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat. Rev. Neurosci. 2014, 15, 379–393. [Google Scholar] [CrossRef]
- Pietrobon, D.; Moskowitz, M.A. Pathophysiology of migraine. Annu. Rev. Physiol. 2013, 75, 365–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskowitz, M.A. Holes in the leaky migraine blood–brain barrier hypothesis? Brain 2017, 140, 1537–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgdorff, P. Arguments against the role of cortical spreading depression in migraine. Neurol. Res. 2018, 40, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, T.; Inagaki, M.; Fujita, Y.; Kimoto, T.; Tanabe-Fujimura, C.; Zou, K.; Liu, J.; Liu, S.; Komano, H. ATP increases the migration of microglia across the brain endothelial cell monolayer. Biosci. Rep. 2016, 36, e00318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldendorf, W.; Braun, L.; Cornford, E. pH dependence of blood-brain barrier permeability to lactate and nicotine. Stroke 1979, 10, 577–581. [Google Scholar] [CrossRef] [Green Version]
- Blawn, K.T.; Kellohen, K.L.; Galloway, E.A.; Wahl, J.; Vivek, A.; Verkhovsky, V.G.; Barker, N.K.; Cottier, K.E.; Vallecillo, T.G.; Langlais, P.R.; et al. Sex hormones regulate NHE1 functional expression and brain endothelial proteome to control paracellular integrity of the blood endothelial barrier. Brain Res. 2021, 1763, 147448. [Google Scholar] [CrossRef]
- Liktor-Busa, E.; Blawn, K.T.; Kellohen, K.L.; Wiese, B.M.; Verkhovsky, V.; Wahl, J.; Vivek, A.; Palomino, S.M.; Davis, T.P.; Vanderah, T.W.; et al. Functional NHE1 expression is critical to blood brain barrier integrity and sumatriptan blood to brain uptake. PLoS ONE 2020, 15, e0227463. [Google Scholar] [CrossRef]
- Kuhlmann, C.R.; Gerigk, M.; Bender, B.; Closhen, D.; Lessmann, V.; Luhmann, H.J. Fluvastatin prevents glutamate-induced blood-brain-barrier disruption in vitro. Life Sci. 2008, 82, 1281–1287. [Google Scholar] [CrossRef]
- Neuhaus, W.; Freidl, M.; Szkokan, P.; Berger, M.; Wirth, M.; Winkler, J.; Gabor, F.; Pifl, C.; Noe, C.R. Effects of NMDA receptor modulators on a blood-brain barrier in vitro model. Brain Res. 2011, 1394, 49–61. [Google Scholar] [CrossRef]
- Fried, N.T.; Maxwell, C.R.; Elliott, M.B.; Oshinsky, M.L. Region-specific disruption of the blood-brain barrier following repeated inflammatory dural stimulation in a rat model of chronic trigeminal allodynia. Cephalalgia 2018, 38, 674–689. [Google Scholar] [CrossRef]
- Sadeghian, H.; Lacoste, B.; Qin, T.; Toussay, X.; Rosa, R.; Oka, F.; Chung, D.; Takizawa, T.; Gu, C.; Ayata, C. Spreading depolarizations trigger caveolin-1-dependent endothelial transcytosis. Ann. Neurol. 2018, 84, 409–423. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Vendor/Catalog Number | Application/Dilution | Lot Number |
|---|---|---|---|
| Claudin-5 Mouse mAb (4C3C2) | Invitrogen | WB: 1:500 | WD |
| 35-2500 | ICC: 1:50 | 327318 | |
| VE-Cadherin Rabbit pAb (CD144) | Invitrogen | WB: 1:500 | UF |
| 36-1900 | ICC: 1:200 | 287723 | |
| Zona Occludens-1 Mouse mAb (ZO1-1A12) | Invitrogen | ICC: 1:200 | WG |
| 33-9100 | 329571 | ||
| α-Tubulin (DM1A) Mouse mAb (DM1A) | Cell Signaling Technology | WB: 1:10,000 | 16 |
| 3873 | |||
| Donkey anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa FluorTM 488 | Invitrogen A-21206 | ICC: 1:10,000 | 2156521 |
| Donkey anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa FluorTM 568 | Invitrogen A-10037 | ICC: 1:10,000 | 2156521 |
| IRDye 800CW Donkey anti-Rabbit IgG Secondary Antibody | Li-Cor 926-32213 | WB: 1:10,000 | D10518-05 |
| IRDye 680RD Donkey anti-Rabbit IgG Secondary Antibody | Li-Cor 926-68072 | WB: 1:10,000 | D10728-15 |
| Alexa FluorTM 488 Phalloidin IR Dye 800CW Donkey anti-Rabbit IgG Secondary Antibody | Invitrogen A12379 | WB: 1:10,000 ICC 1:40 | D10518-052219253 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wahl, J.R.; Vivek, A.; Palomino, S.M.; Almuslim, M.; Cottier, K.E.; Langlais, P.R.; Streicher, J.M.; Vanderah, T.W.; Liktor-Busa, E.; Largent-Milnes, T.M. Extracellular Alterations in pH and K+ Modify the Murine Brain Endothelial Cell Total and Phospho-Proteome. Pharmaceutics 2022, 14, 1469. https://doi.org/10.3390/pharmaceutics14071469
Wahl JR, Vivek A, Palomino SM, Almuslim M, Cottier KE, Langlais PR, Streicher JM, Vanderah TW, Liktor-Busa E, Largent-Milnes TM. Extracellular Alterations in pH and K+ Modify the Murine Brain Endothelial Cell Total and Phospho-Proteome. Pharmaceutics. 2022; 14(7):1469. https://doi.org/10.3390/pharmaceutics14071469
Chicago/Turabian StyleWahl, Jared R., Anjali Vivek, Seph M. Palomino, Moyad Almuslim, Karissa E. Cottier, Paul R. Langlais, John M. Streicher, Todd W. Vanderah, Erika Liktor-Busa, and Tally M. Largent-Milnes. 2022. "Extracellular Alterations in pH and K+ Modify the Murine Brain Endothelial Cell Total and Phospho-Proteome" Pharmaceutics 14, no. 7: 1469. https://doi.org/10.3390/pharmaceutics14071469