
Metabolic Reprogramming in Cancer Cells: Emerging Molecular Mechanisms and Novel Therapeutic Approaches

 ,
,  ,
,  ,
,  , , and
, , and
Abstract
:1. Introduction
2. A Look into the Energy Metabolism of the Normal and Cancerous Cell
3. Metabolic Pathways in Cancer Cells
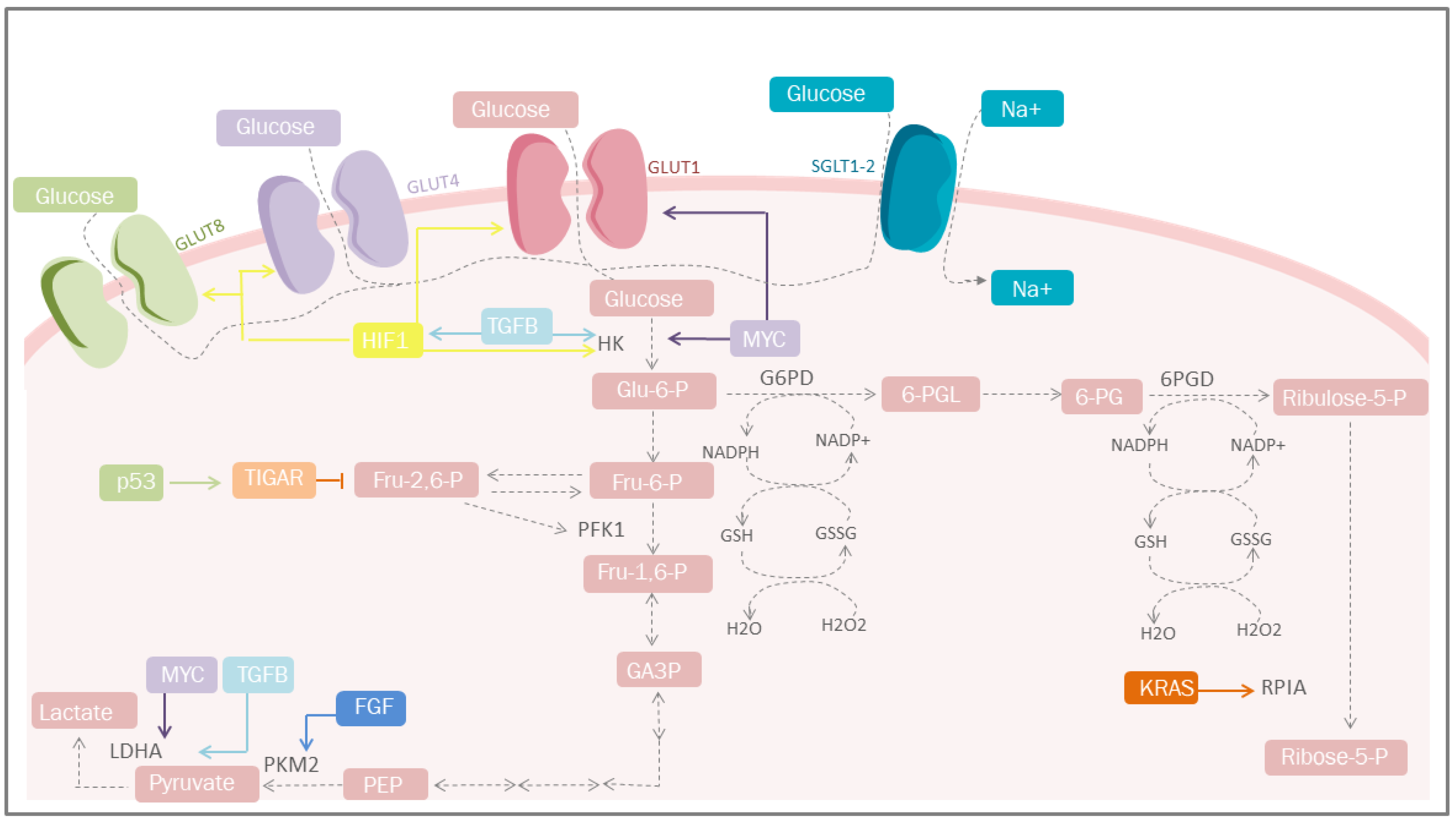
3.1. Carbohydrate Metabolism
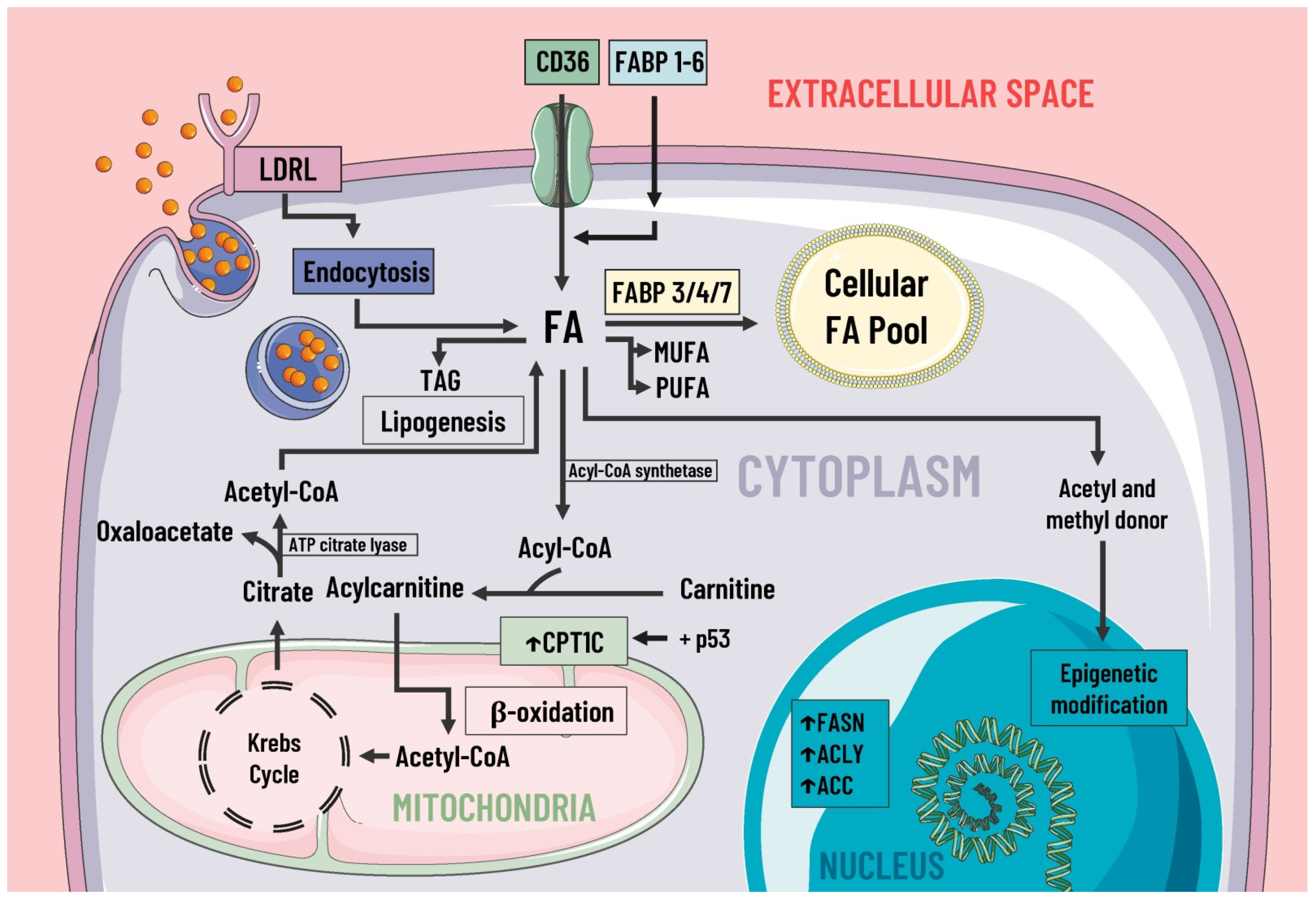
3.2. Lipid Metabolism
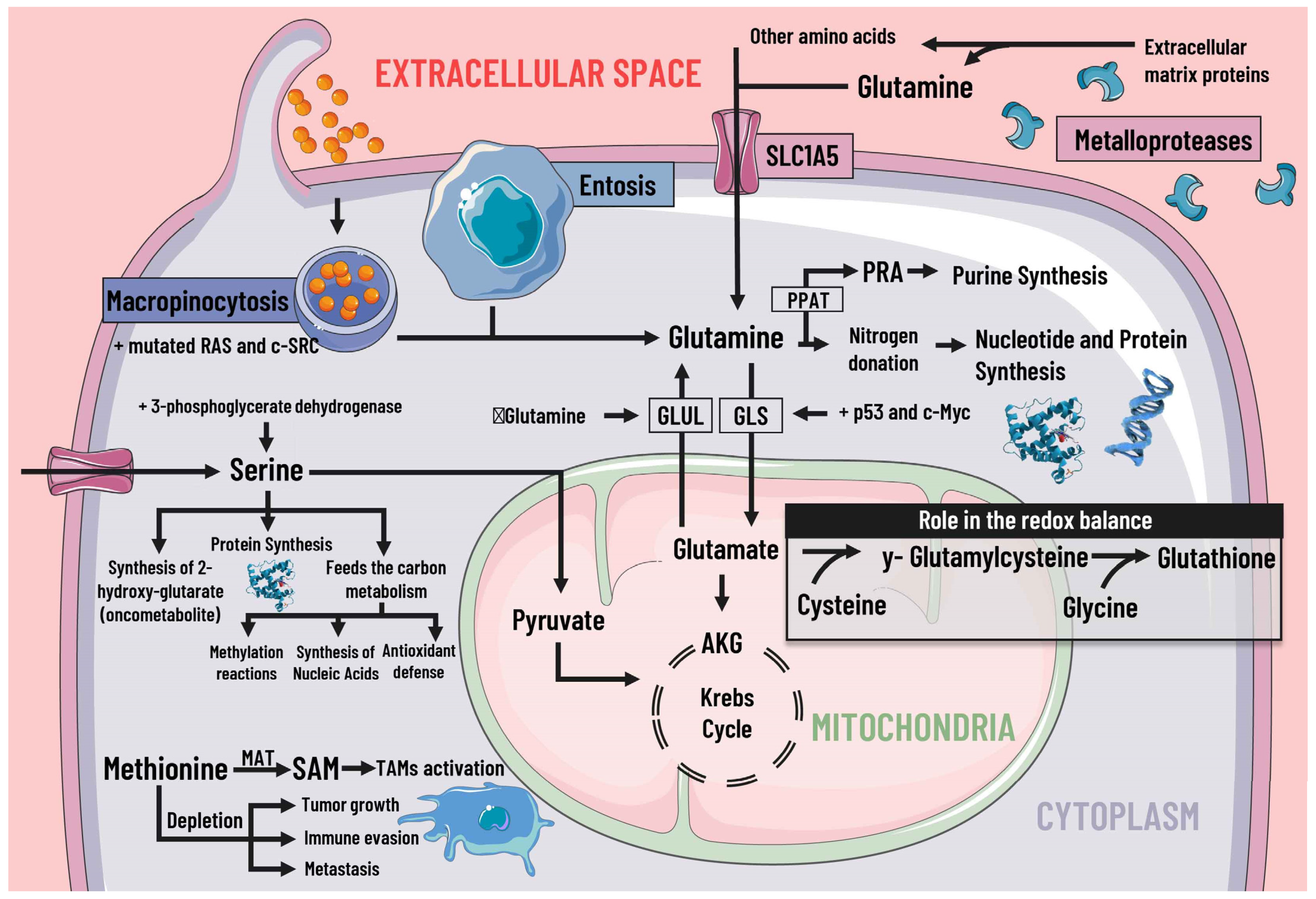
3.3. Protein Metabolism
3.4. Methionine, S-Adenosylmethionine and Cysteine
3.5. Isoenzymes
3.5.1. Fumarate Hydratase (FH)
3.5.2. Isocitrate Dehydrogenase
3.5.3. Glutaminase
3.5.4. Lactate Dehydrogenases
4. Mediators of Metabolic Reprogramming
4.1. Growth Factors
4.2. Hypoxia-Inducible Factors
4.3. Tumor Suppressor Genes
4.3.1. P53
4.3.2. Phosphatase and Tensin Homolog
4.3.3. Liver Kinase B1
4.3.4. Tuberin
4.4. Oncogenes
5. A Brief Analysis of Therapies Modulating Cell Metabolism
5.1. mTOR Inhibitors
Rapalogs
5.2. ATP-Competitive mTOR Inhibitors
Dual PI3K/mTOR Inhibitors
5.3. Glutaminase Inhibition
5.4. Biguanides
5.5. HER2 Inhibitor
5.6. HIF-1 Inhibitor
5.7. Ion Channels as Drug Targets
5.8. Others Therapies
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- da Silva-Diz, V.; Lorenzo-Sanz, L.; Bernat-Peguera, A.; Lopez-Cerda, M.; Muñoz, P. Cancer Cell Plasticity: Impact on Tumor Progression and Therapy Response. Semin. Cancer Biol. 2018, 53, 48–58. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the Basics of Cancer Metabolism: Developing Antitumor Strategies by Exploiting the Differences between Normal and Cancer Cell Metabolism. Redox Biol. 2017, 12, 833–842. [Google Scholar] [CrossRef]
- Liu, C.-L.; Hsu, Y.-C.; Lee, J.-J.; Chen, M.-J.; Lin, C.-H.; Huang, S.-Y.; Cheng, S.-P. Targeting the Pentose Phosphate Pathway Increases Reactive Oxygen Species and Induces Apoptosis in Thyroid Cancer Cells. Mol. Cell. Endocrinol. 2020, 499, 110595. [Google Scholar] [CrossRef]
- Sun, H.; Chen, L.; Cao, S.; Liang, Y.; Xu, Y. Warburg Effects in Cancer and Normal Proliferating Cells: Two Tales of the Same Name. Genom. Proteom. Bioinform. 2019, 17, 273–286. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of Fatty Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yang, C. Oncometabolites in Cancer: Current Understanding and Challenges. Cancer Res. 2021, 81, 2820–2823. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benny, S.; Mishra, R.; Manojkumar, M.K.; Aneesh, T.P. From Warburg Effect to Reverse Warburg Effect; the New Horizons of Anti-Cancer Therapy. Med. Hypotheses 2020, 144, 110216. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Srivastava, S.; Zhang, J. Starve Cancer Cells of Glutamine: Break the Spell or Make a Hungry Monster? Cancers 2019, 11, 804. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine Reliance in Cell Metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Fernández, L.P.; Gómez de Cedrón, M.; Ramírez de Molina, A. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10, 577420. [Google Scholar] [CrossRef]
- Castelli, S.; De Falco, P.; Ciccarone, F.; Desideri, E.; Ciriolo, M.R. Lipid Catabolism and ROS in Cancer: A Bidirectional Liaison. Cancers 2021, 13, 5484. [Google Scholar] [CrossRef]
- Lue, H.; Podolak, J.; Kolahi, K.; Cheng, L.; Rao, S.; Garg, D.; Xue, C.-H.; Rantala, J.K.; Tyner, J.W.; Thornburg, K.L.; et al. Metabolic Reprogramming Ensures Cancer Cell Survival despite Oncogenic Signaling Blockade. Genes Dev. 2017, 31, 2067–2084. [Google Scholar] [CrossRef]
- Sun, L.; Suo, C.; Li, S.-T.; Zhang, H.; Gao, P. Metabolic Reprogramming for Cancer Cells and Their Microenvironment: Beyond the Warburg Effect. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 51–66. [Google Scholar] [CrossRef]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The Cancer Metabolic Reprogramming and Immune Response. Mol. Cancer 2021, 20, 28. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Tan, X.; Yang, R.; Miao, Y.; Zhang, M.; Xi, Y.; Guo, R.; Zheng, M.; Li, B. Discovery of Novel Glyceraldehyde-3-Phosphate Dehydrogenase Inhibitor via Docking-Based Virtual Screening. Bioorg. Chem. 2020, 96, 103620. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari Movahed, Z.; Rastegari-Pouyani, M.; Mohammadi, M.H.; Mansouri, K. Cancer Cells Change Their Glucose Metabolism to Overcome Increased ROS: One Step from Cancer Cell to Cancer Stem Cell? Biomed. Pharmacother. 2019, 112, 108690. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.C.; Dukhande, V.V.; Zhou, Z.; Young, L.E.A.; Emanuelle, S.; Brainson, C.F.; Gentry, M.S. Nuclear Glycogenolysis Modulates Histone Acetylation in Human Non-Small Cell Lung Cancers. Cell Metab. 2019, 30, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Grasmann, G.; Smolle, E.; Olschewski, H.; Leithner, K. Gluconeogenesis in Cancer Cells—Repurposing of a Starvation-Induced Metabolic Pathway? Biochim. Biophys. Acta BBA Rev. Cancer 2019, 1872, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Vincent, E.E.; Sergushichev, A.; Griss, T.; Gingras, M.-C.; Samborska, B.; Ntimbane, T.; Coelho, P.P.; Blagih, J.; Raissi, T.C.; Choinière, L.; et al. Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth. Mol. Cell 2015, 60, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Luo, M.; Cheng, L.-J.; Li, R.-N.; Liu, B.; Linghu, H. Glutamine-Fructose-6-Phosphate Transaminase 2 (GFPT2) Promotes the EMT of Serous Ovarian Cancer by Activating the Hexosamine Biosynthetic Pathway to Increase the Nuclear Location of β-Catenin. Pathol. Res. Pract. 2019, 215, 152681. [Google Scholar] [CrossRef]
- Bu, P.; Chen, K.Y.; Xiang, K.; Johnson, C.; Crown, S.B.; Rakhilin, N.; Ai, Y.; Wang, L.; Xi, R.; Astapova, I.; et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab. 2018, 27, 1249–1262. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.R.V.; Burke, J.E. Novel Roles of Phosphoinositides in Signaling, Lipid Transport, and Disease. Curr. Opin. Cell Biol. 2020, 63, 57–67. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y. CD36 Tango in Cancer: Signaling Pathways and Functions. Theranostics 2019, 9, 4893–4908. [Google Scholar] [CrossRef] [PubMed]
- Tanase, C.; Enciu, A.M.; Codrici, E.; Popescu, I.D.; Dudau, M.; Dobri, A.M.; Pop, S.; Mihai, S.; Gheorghișan-Gălățeanu, A.-A.; Hinescu, M.E. Fatty Acids, CD36, Thrombospondin-1, and CD47 in Glioblastoma: Together and/or Separately? Int. J. Mol. Sci. 2022, 23, 604. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, L.; Chen, W.; Shen, L.; Jiang, J.; Sun, S.; Chen, Z. Role of Intra- and Extracellular Lipid Signals in Cancer Stemness and Potential Therapeutic Strategy. Front. Pharmacol. 2021, 12, 730751. [Google Scholar] [CrossRef] [PubMed]
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935. [Google Scholar] [CrossRef]
- Bartolacci, C.; Andreani, C.; El-Gammal, Y.; Scaglioni, P.P. Lipid Metabolism Regulates Oxidative Stress and Ferroptosis in RAS-Driven Cancers: A Perspective on Cancer Progression and Therapy. Front. Mol. Biosci. 2021, 8, 706650. [Google Scholar] [CrossRef] [PubMed]
- Casals, N.; Zammit, V.; Herrero, L.; Fadó, R.; Rodríguez-Rodríguez, R.; Serra, D. Carnitine Palmitoyltransferase 1C: From Cognition to Cancer. Prog. Lipid Res. 2016, 61, 134–148. [Google Scholar] [CrossRef] [Green Version]
- Parrales, A.; Iwakuma, T. P53 as a Regulator of Lipid Metabolism in Cancer. Int. J. Mol. Sci. 2016, 17, 2074. [Google Scholar] [CrossRef]
- Bott, A.J.; Shen, J.; Tonelli, C.; Zhan, L.; Sivaram, N.; Jiang, Y.-P.; Yu, X.; Bhatt, V.; Chiles, E.; Zhong, H.; et al. Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism. Cell Rep. 2019, 29, 1287–1298. [Google Scholar] [CrossRef] [Green Version]
- Vettore, L.; Westbrook, R.L.; Tennant, D.A. New Aspects of Amino Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 150–156. [Google Scholar] [CrossRef]
- Hosios, A.M.; Hecht, V.C.; Danai, L.V.; Johnson, M.O.; Rathmell, J.C.; Steinhauser, M.L.; Manalis, S.R.; Vander Heiden, M.G. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev. Cell 2016, 36, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Kurbegovic, A.; Trudel, M. The Master Regulators Myc and P53 Cellular Signaling and Functions in Polycystic Kidney Disease. Cell. Signal. 2020, 71, 109594. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Yoon, H.; Ringel, A.E.; Jeanfavre, S.; Clish, C.B.; Haigis, M.C. Metabolic Recycling of Ammonia via Glutamate Dehydrogenase Supports Breast Cancer Biomass. Science 2017, 358, 941–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukey, M.J.; Katt, W.P.; Cerione, R.A. Targeting Amino Acid Metabolism for Cancer Therapy. Drug Discov. Today 2017, 22, 796–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corchado-Cobos, R.; García-Sancha, N.; Mendiburu-Eliçabe, M.; Gómez-Vecino, A.; Jiménez-Navas, A.; Pérez-Baena, M.J.; Holgado-Madruga, M.; Mao, J.-H.; Cañueto, J.; Castillo-Lluva, S.; et al. Pathophysiological Integration of Metabolic Reprogramming in Breast Cancer. Cancers 2022, 14, 322. [Google Scholar] [CrossRef] [PubMed]
- Durgan, J.; Florey, O. Cancer Cell Cannibalism: Multiple Triggers Emerge for Entosis. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 831–841. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, H.; Zhao, J.; Wan, P.; Hu, Y.; Lv, K.; Hu, Y.; Yang, X.; Ma, M. Activation of MAT2A-RIP1 Signaling Axis Reprograms Monocytes in Gastric Cancer. J. Immunother. Cancer 2021, 9, e001364. [Google Scholar] [CrossRef]
- Zhang, H.-F.; Klein Geltink, R.I.; Parker, S.J.; Sorensen, P.H. Transsulfuration, Minor Player or Crucial for Cysteine Homeostasis in Cancer. Trends Cell Biol. 2022, S0962-8924(22)000605. [Google Scholar] [CrossRef]
- Tyrakis, P.A.; Yurkovich, M.E.; Sciacovelli, M.; Papachristou, E.K.; Bridges, H.R.; Gaude, E.; Schreiner, A.; D’Santos, C.; Hirst, J.; Hernandez-Fernaud, J.; et al. Fumarate Hydratase Loss Causes Combined Respiratory Chain Defects. Cell Rep. 2017, 21, 1036–1047. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, E.; Sciacovelli, M.; Costa, A.S.H.; Tran, M.G.B.; Johnson, T.I.; Machado, D.; Frezza, C.; Saez-Rodriguez, J. Post-Translational Regulation of Metabolism in Fumarate Hydratase Deficient Cancer Cells. Metab. Eng. 2018, 45, 149–157. [Google Scholar] [CrossRef]
- Schmidt, S.; Gay, D.; Uthe, F.W.; Denk, S.; Paauwe, M.; Matthes, N.; Diefenbacher, M.E.; Bryson, S.; Warrander, F.C.; Erhard, F.; et al. A MYC–GCN2–EIF2α Negative Feedback Loop Limits Protein Synthesis to Prevent MYC-Dependent Apoptosis in Colorectal Cancer. Nat. Cell Biol. 2019, 21, 1413–1424. [Google Scholar] [CrossRef]
- Masui, K.; Onizuka, H.; Cavenee, W.K.; Mischel, P.S.; Shibata, N. Metabolic Reprogramming in the Pathogenesis of Glioma: Update. Neuropathol. Off. J. Jpn. Soc. Neuropathol. 2019, 39, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelman, S.J.; Naser, F.; Mahieu, N.G.; McKenzie, L.D.; Dunn, G.P.; Chheda, M.G.; Patti, G.J. Consumption of NADPH for 2-HG Synthesis Increases Pentose Phosphate Pathway Flux and Sensitizes Cells to Oxidative Stress. Cell Rep. 2018, 22, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo-Garcia, J.L.; Viswanath, P.; Eriksson, P.; Cai, L.; Radoul, M.; Chaumeil, M.M.; Blough, M.; Luchman, H.A.; Weiss, S.; Cairncross, J.G.; et al. IDH1 Mutation Induces Reprogramming of Pyruvate Metabolism. Cancer Res. 2015, 75, 2999–3009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, S.F.; Lambden, S.; Macias, D.; Puthucheary, Z.; Pietsch, S.; Mendil, L.; McPhail, M.J.W.; Johnson, R.S. 2-Hydroxyglutarate Metabolism Is Altered in an In Vivo Model of LPS Induced Endotoxemia. Front. Physiol. 2020, 11, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masisi, B.K.; El Ansari, R.; Alfarsi, L.; Rakha, E.A.; Green, A.R.; Craze, M.L. The Role of Glutaminase in Cancer. Histopathology 2020, 76, 498–508. [Google Scholar] [CrossRef]
- Katt, W.P.; Lukey, M.J.; Cerione, R.A. A Tale of Two Glutaminases: Homologous Enzymes with Distinct Roles in Tumorigenesis. Future Med. Chem. 2017, 9, 223–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matés, J.M.; Campos-Sandoval, J.A.; Márquez, J. Glutaminase Isoenzymes in the Metabolic Therapy of Cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2018, 1870, 158–164. [Google Scholar] [CrossRef]
- Gouirand, V.; Guillaumond, F.; Vasseur, S. Influence of the Tumor Microenvironment on Cancer Cells Metabolic Reprogramming. Front. Oncol. 2018, 8, 117. [Google Scholar] [CrossRef]
- Robey, R.B.; Weisz, J.; Kuemmerle, N.; Salzberg, A.C.; Berg, A.; Brown, D.G.; Kubik, L.; Palorini, R.; Al-Mulla, F.; Al-Temaimi, R.; et al. Metabolic Reprogramming and Dysregulated Metabolism: Cause, Consequence and/or Enabler of Environmental Carcinogenesis? Carcinogenesis 2015, 36, S203–S231. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. CB 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Lamouille, S.; Derynck, R. Cell Size and Invasion in TGF-β–Induced Epithelial to Mesenchymal Transition Is Regulated by Activation of the MTOR Pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samarelli, A.V.; Masciale, V.; Aramini, B.; Coló, G.P.; Tonelli, R.; Marchioni, A.; Bruzzi, G.; Gozzi, F.; Andrisani, D.; Castaniere, I.; et al. Molecular Mechanisms and Cellular Contribution from Lung Fibrosis to Lung Cancer Development. Int. J. Mol. Sci. 2021, 22, 12179. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.R.; Santos, L.d.S.; Dias, R.B.; Quadros, C.A.; Bezerra, D.P. Emerging Agents That Target Signaling Pathways to Eradicate Colorectal Cancer Stem Cells. Cancer Commun. 2021, 41, 1275–1313. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Flinck, M.; Pardo, L.A. The Interplay between Dysregulated Ion Transport and Mitochondrial Architecture as a Dangerous Liaison in Cancer. Int. J. Mol. Sci. 2021, 22, 5209. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Fan, M.; Wang, J.; Ullah, A.; Ghauri, M.A.; Dai, B.; Zhan, Y.; Zhang, D.; Zhang, Y. Sanguinarine Inhibits Epithelial-Mesenchymal Transition via Targeting HIF-1α/TGF-β Feed-Forward Loop in Hepatocellular Carcinoma. Cell Death Dis. 2019, 10, 939. [Google Scholar] [CrossRef]
- Jonasch, E.; McCutcheon, I.E.; Gombos, D.S.; Ahrar, K.; Perrier, N.D.; Liu, D.; Robichaux, C.C.; Villarreal, M.F.; Weldon, J.A.; Woodson, A.H.; et al. Pazopanib in Patients with von Hippel-Lindau Disease: A Single-Arm, Single-Centre, Phase 2 Trial. Lancet Oncol. 2018, 19, 1351–1359. [Google Scholar] [CrossRef]
- Zhu, W.; Li, Y.; Zhao, D.; Li, H.; Zhang, W.; Xu, J.; Hou, J.; Feng, X.; Wang, H. Dihydroartemisinin Suppresses Glycolysis of LNCaP Cells by Inhibiting PI3K/AKT Pathway and Downregulating HIF-1α Expression. Life Sci. 2019, 233, 116730. [Google Scholar] [CrossRef]
- Hao, L.-S.; Liu, Q.; Tian, C.; Zhang, D.-X.; Wang, B.; Zhou, D.-X.; Li, Z.-P.; Yuan, Z.-X. Correlation and Expression Analysis of Hypoxia-inducible Factor 1α, Glucose Transporter 1 and Lactate Dehydrogenase 5 in Human Gastric Cancer. Oncol. Lett. 2019, 18, 1431–1441. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Jiang, H.; Li, Z.; Zhuang, Y.; Liu, Y.; Zhou, S.; Xiao, Y.; Xie, C.; Zhou, F.; Zhou, Y. 2-Methoxyestradiol Enhances Radiosensitivity in Radioresistant Melanoma MDA-MB-435R Cells by Regulating Glycolysis via HIF-1α/PDK1 Axis. Int. J. Oncol. 2017, 50, 1531–1540. [Google Scholar] [CrossRef] [Green Version]
- Ullmann, P.; Qureshi-Baig, K.; Rodriguez, F.; Ginolhac, A.; Nonnenmacher, Y.; Ternes, D.; Weiler, J.; Gäbler, K.; Bahlawane, C.; Hiller, K.; et al. Hypoxia-Responsive MiR-210 Promotes Self-Renewal Capacity of Colon Tumor-Initiating Cells by Repressing ISCU and by Inducing Lactate Production. Oncotarget 2016, 7, 65454–65470. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Yang, F.; Yang, X.-A.; Zhang, L.; Yu, H.; Cheng, X.; Xu, S.; Pan, J.; Wang, K.; Li, P. Mitochondrial Metabolism Is Inhibited by the HIF1α-MYC-PGC-1β Axis in BRAF V600E Thyroid Cancer. FEBS J. 2019, 286, 1420–1436. [Google Scholar] [CrossRef] [PubMed]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid Metabolism in Cancer Cells under Metabolic Stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- van der Mijn, J.C.; Fu, L.; Khani, F.; Zhang, T.; Molina, A.M.; Barbieri, C.E.; Chen, Q.; Gross, S.S.; Gudas, L.J.; Nanus, D.M. Combined Metabolomics and Genome-Wide Transcriptomics Analyses Show Multiple HIF1α-Induced Changes in Lipid Metabolism in Early Stage Clear Cell Renal Cell Carcinoma. Transl. Oncol. 2020, 13, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Melone, M.A.B.; Valentino, A.; Margarucci, S.; Galderisi, U.; Giordano, A.; Peluso, G. The Carnitine System and Cancer Metabolic Plasticity. Cell Death Dis. 2018, 9, 228. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, N.; Nagano, H.; Tanaka, T. The Role of Tumor Suppressor P53 in Metabolism and Energy Regulation, and Its Implication in Cancer and Lifestyle-Related Diseases. Endocr. J. 2019, 66, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, H.; Moriyama, M.; Ozawa, T.; Tsuruta, D.; Iguchi, T.; Tamada, S.; Nakatani, T.; Nakagawa, K.; Hayakawa, T. Notch Signaling Enhances Stemness by Regulating Metabolic Pathways Through Modifying P53, NF-ΚB, and HIF-1α. Stem Cells Dev. 2018, 27, 935–947. [Google Scholar] [CrossRef]
- Nakajima, K.; Kawashima, I.; Koshiisi, M.; Kumagai, T.; Suzuki, M.; Suzuki, J.; Mitsumori, T.; Kirito, K. Glycolytic Enzyme Hexokinase II Is a Putative Therapeutic Target in B-Cell Malignant Lymphoma. Exp. Hematol. 2019, 78, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Lei, R.; Shen, J.; Zhang, S.; Liu, A.; Chen, X.; Wang, Y.; Sun, J.; Dai, S.; Xu, J. Inactivating the Ubiquitin Ligase Parkin Suppresses Cell Proliferation and Induces Apoptosis in Human Keloids. J. Cell. Physiol. 2019, 234, 16601–16608. [Google Scholar] [CrossRef]
- Ramos, H.; Calheiros, J.; Almeida, J.; Barcherini, V.; Santos, S.; Carvalho, A.T.P.; Santos, M.M.M.; Saraiva, L. SLMP53-1 Inhibits Tumor Cell Growth through Regulation of Glucose Metabolism and Angiogenesis in a P53-Dependent Manner. Int. J. Mol. Sci. 2020, 21, 596. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Choi, S.I.; Won, K.Y.; Lim, S.-J. Distinctive Interrelation of P53 with SCO2, COX, and TIGAR in Human Gastric Cancer. Pathol. Res. Pract. 2016, 212, 904–910. [Google Scholar] [CrossRef]
- Liu, M.; Hu, Y.; Lu, S.; Lu, M.; Li, J.; Chang, H.; Jia, H.; Zhou, M.; Ren, F.; Zhong, J. IC261, a Specific Inhibitor of CK1δ/ε, Promotes Aerobic Glycolysis through P53-Dependent Mechanisms in Colon Cancer. Int. J. Biol. Sci. 2020, 16, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.; Das, G.M. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, E89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Xu, Z.; Song, D.; Zhang, H.; Li, B.; Gao, L.; Zhang, Y.; Feng, Q.; Yu, D.; Hu, L.; et al. Antitumor Effects of Rafoxanide in Diffuse Large B Cell Lymphoma via the PTEN/PI3K/Akt and JNK/c-Jun Pathways. Life Sci. 2020, 243, 117249. [Google Scholar] [CrossRef]
- Zhou, X.; Yang, X.; Sun, X.; Xu, X.; Li, X.; Guo, Y.; Wang, J.; Li, X.; Yao, L.; Wang, H.; et al. Effect of PTEN Loss on Metabolic Reprogramming in Prostate Cancer Cells. Oncol. Lett. 2019, 17, 2856–2866. [Google Scholar] [CrossRef] [Green Version]
- Phadngam, S.; Castiglioni, A.; Ferraresi, A.; Morani, F.; Follo, C.; Isidoro, C. PTEN Dephosphorylates AKT to Prevent the Expression of GLUT1 on Plasmamembrane and to Limit Glucose Consumption in Cancer Cells. Oncotarget 2016, 7, 84999–85020. [Google Scholar] [CrossRef] [Green Version]
- Ryu, M.J.; Han, J.; Kim, S.J.; Lee, M.J.; Ju, X.; Lee, Y.L.; Son, J.H.; Cui, J.; Jang, Y.; Chung, W.; et al. PTEN/AKT Signaling Mediates Chemoresistance in Refractory Acute Myeloid Leukemia through Enhanced Glycolysis. Oncol. Rep. 2019, 42, 2149–2158. [Google Scholar] [CrossRef]
- Wu, Q.; Li, Z.; Liu, Q. An Important Role of SREBP-1 in HBV and HCV Co-Replication Inhibition by PTEN. Virology 2018, 520, 94–102. [Google Scholar] [CrossRef]
- Kachaylo, E.; Tschuor, C.; Calo, N.; Borgeaud, N.; Ungethüm, U.; Limani, P.; Piguet, A.-C.; Dufour, J.-F.; Foti, M.; Graf, R.; et al. PTEN Down-Regulation Promotes β-Oxidation to Fuel Hypertrophic Liver Growth After Hepatectomy in Mice. Hepatol. Baltim. Md 2017, 66, 908–921. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef] [Green Version]
- Momcilovic, M.; Shackelford, D.B. Targeting LKB1 in Cancer—Exposing and Exploiting Vulnerabilities. Br. J. Cancer 2015, 113, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Ciccarese, F.; Zulato, E.; Indraccolo, S. LKB1/AMPK Pathway and Drug Response in Cancer: A Therapeutic Perspective. Oxid. Med. Cell. Longev. 2019, 2019, 8730816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faubert, B.; Vincent, E.E.; Griss, T.; Samborska, B.; Izreig, S.; Svensson, R.U.; Mamer, O.A.; Avizonis, D.; Shackelford, D.B.; Shaw, R.J.; et al. Loss of the Tumor Suppressor LKB1 Promotes Metabolic Reprogramming of Cancer Cells via HIF-1α. Proc. Natl. Acad. Sci. USA 2014, 111, 2554–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, S.J.; Svensson, R.U.; Divakaruni, A.S.; Lefebvre, A.E.; Murphy, A.N.; Shaw, R.J.; Metallo, C.M. LKB1 Promotes Metabolic Flexibility in Response to Energy Stress. Metab. Eng. 2017, 43, 208–217. [Google Scholar] [CrossRef]
- Endo, H.; Owada, S.; Inagaki, Y.; Shida, Y.; Tatemichi, M. Glucose Starvation Induces LKB1-AMPK-Mediated MMP-9 Expression in Cancer Cells. Sci. Rep. 2018, 8, 10122. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Lee, J.; Kim, J.; Kim, S.T.; Lee, S.; Kim, S.Y.; Ha, S.Y.; Park, C.-K.; Lim, H.Y. Loss of Tuberous Sclerosis Complex 2 (TSC2) as a Predictive Biomarker of Response to MTOR Inhibitor Treatment in Patients with Hepatocellular Carcinoma. Transl. Oncol. 2016, 9, 466–471. [Google Scholar] [CrossRef] [Green Version]
- Huynh, H.; Hao, H.-X.; Chan, S.L.; Chen, D.; Ong, R.; Soo, K.C.; Pochanard, P.; Yang, D.; Ruddy, D.; Liu, M.; et al. Loss of Tuberous Sclerosis Complex 2 (TSC2) Is Frequent in Hepatocellular Carcinoma and Predicts Response to MTORC1 Inhibitor Everolimus. Mol. Cancer Ther. 2015, 14, 1224–1235. [Google Scholar] [CrossRef] [Green Version]
- Jan, C.-I.; Tsai, M.-H.; Chiu, C.-F.; Huang, Y.-P.; Liu, C.J.; Chang, N.W. Fenofibrate Suppresses Oral Tumorigenesis via Reprogramming Metabolic Processes: Potential Drug Repurposing for Oral Cancer. Int. J. Biol. Sci. 2016, 12, 786–798. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.-S.; Chen, W.-S.; Chen, L.-L.; Chen, C.-C.; Hsu, Y.-T.; Chua, K.V.; Wang, H.-D.; Huang, T.-S. Osteopontin–Integrin Engagement Induces HIF-1α–TCF12-Mediated Endothelial-Mesenchymal Transition to Exacerbate Colorectal Cancer. Oncotarget 2017, 9, 4998–5015. [Google Scholar] [CrossRef] [Green Version]
- Mossmann, D.; Park, S.; Hall, M.N. MTOR Signalling and Cellular Metabolism Are Mutual Determinants in Cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Chen, J.-F.; Wu, P.; Xia, R.; Yang, J.; Huo, X.-Y.; Gu, D.-Y.; Tang, C.-J.; De, W.; Yang, F. STAT3-Induced LncRNA HAGLROS Overexpression Contributes to the Malignant Progression of Gastric Cancer Cells via MTOR Signal-Mediated Inhibition of Autophagy. Mol. Cancer 2018, 17, 6. [Google Scholar] [CrossRef] [Green Version]
- Mele, L.; la Noce, M.; Paino, F.; Regad, T.; Wagner, S.; Liccardo, D.; Papaccio, G.; Lombardi, A.; Caraglia, M.; Tirino, V.; et al. Glucose-6-Phosphate Dehydrogenase Blockade Potentiates Tyrosine Kinase Inhibitor Effect on Breast Cancer Cells through Autophagy Perturbation. J. Exp. Clin. Cancer Res. CR 2019, 38, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC Transcription Factor Network: Balancing Metabolism, Proliferation and Oncogenesis. Front. Med. 2018, 12, 412–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matés, J.M.; Di Paola, F.J.; Campos-Sandoval, J.A.; Mazurek, S.; Márquez, J. Therapeutic Targeting of Glutaminolysis as an Essential Strategy to Combat Cancer. Semin. Cell Dev. Biol. 2020, 98, 34–43. [Google Scholar] [CrossRef]
- Katayama, R.; Lovly, C.M.; Shaw, A.T. Therapeutic Targeting of Anaplastic Lymphoma Kinase in Lung Cancer: A Paradigm for Precision Cancer Medicine. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2227–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouw, A.M.; Margulis, K.; Liu, N.S.; Raman, S.J.; Mancuso, A.; Toal, G.G.; Tong, L.; Mosley, A.; Hsieh, A.L.; Sullivan, D.K.; et al. The MYC Oncogene Cooperates with Sterol-Regulated Element-Binding Protein to Regulate Lipogenesis Essential for Neoplastic Growth. Cell Metab. 2019, 30, 556–572. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.J.; Schiemann, W.P. Telomerase in Cancer: Function, Regulation, and Clinical Translation. Cancers 2022, 14, 808. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Khattar, E.; Leow, S.C.; Liu, C.Y.; Muller, J.; Ang, W.X.; Li, Y.; Franzoso, G.; Li, S.; Guccione, E.; et al. Telomerase Regulates MYC-Driven Oncogenesis Independent of Its Reverse Transcriptase Activity. J. Clin. Investig. 2015, 125, 2109–2122. [Google Scholar] [CrossRef] [Green Version]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-Driven Metabolic Rewiring Reveals Novel Actionable Targets in Cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef] [Green Version]
- Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic KRAS Supports Pancreatic Cancer through Regulation of Nucleotide Synthesis. Nat. Commun. 2018, 9, 4945. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C. Metabolic Dependencies in RAS-Driven Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1828–1834. [Google Scholar] [CrossRef] [Green Version]
- Frontiers|Macropinocytosis: A Metabolic Adaptation to Nutrient Stress in Cancer|Endocrinology. Available online: https://www.frontiersin.org/articles/10.3389/fendo.2017.00261/full (accessed on 24 January 2022).
- Bernfeld, E.; Foster, D.A. Glutamine as an Essential Amino Acid for KRas-Driven Cancer Cells. Trends Endocrinol. Metab. TEM 2019, 30, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Makinoshima, H.; Takita, M.; Saruwatari, K.; Umemura, S.; Obata, Y.; Ishii, G.; Matsumoto, S.; Sugiyama, E.; Ochiai, A.; Abe, R.; et al. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (MTOR) Axis Is Responsible for Aerobic Glycolysis Mediated by Glucose Transporter in Epidermal Growth Factor Receptor (EGFR)-Mutated Lung Adenocarcinoma. J. Biol. Chem. 2015, 290, 17495–17504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, H.-Y.; Lee, H.-Y. Oncogene-Driven Metabolic Alterations in Cancer. Biomol. Ther. 2018, 26, 45–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Song, F.; Zhao, X.; Jiang, H.; Wu, X.; Wang, B.; Zhou, M.; Tian, M.; Shi, B.; Wang, H.; et al. EGFR Modulates Monounsaturated Fatty Acid Synthesis through Phosphorylation of SCD1 in Lung Cancer. Mol. Cancer 2017, 16, 127. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Yu, C.; Mohamed, E.M.; Shao, H.; Wang, L.; Sundaresan, G.; Zweit, J.; Idowu, M.; Fang, X. A Causal Link from ALK to Hexokinase II Overexpression and Hyperactive Glycolysis in EML4-ALK-Positive Lung Cancer. Oncogene 2016, 35, 6132–6142. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Blake, S.; Kusuma, F.K.; Pearson, R.B.; Kang, J.; Chan, K.T. Oncogene-Induced Senescence: From Biology to Therapy. Mech. Ageing Dev. 2020, 187, 111229. [Google Scholar] [CrossRef]
- Liu, X.; Ding, J.; Meng, L. Oncogene-Induced Senescence: A Double Edged Sword in Cancer. Acta Pharmacol. Sin. 2018, 39, 1553–1558. [Google Scholar] [CrossRef]
- DeCensi, A.; Puntoni, M.; Goodwin, P.; Cazzaniga, M.; Gennari, A.; Bonanni, B.; Gandini, S. Metformin and Cancer Risk in Diabetic Patients: A Systematic Review and Meta-Analysis. Cancer Prev. Res. 2010, 3, 1451–1461. [Google Scholar] [CrossRef] [Green Version]
- Andronesi, O.C.; Arrillaga-Romany, I.C.; Ly, K.I.; Bogner, W.; Ratai, E.M.; Reitz, K.; Iafrate, A.J.; Dietrich, J.; Gerstner, E.R.; Chi, A.S.; et al. Pharmacodynamics of Mutant-IDH1 Inhibitors in Glioma Patients Probed by in Vivo 3D MRS Imaging of 2-Hydroxyglutarate. Nat. Commun. 2018, 9, 1474. [Google Scholar] [CrossRef]
- Basu, B.; Dean, E.; Puglisi, M.; Greystoke, A.; Ong, M.; Burke, W.; Cavallin, M.; Bigley, G.; Womack, C.; Harrington, E.A.; et al. First-in-Human Pharmacokinetic and Pharmacodynamic Study of the Dual m-TORC 1/2 Inhibitor AZD2014. Clin. Cancer Res. 2015, 21, 3412–3419. [Google Scholar] [CrossRef] [Green Version]
- Graham, L.; Banda, K.; Torres, A.; Carver, B.S.; Chen, Y.; Pisano, K.; Shelkey, G.; Curley, T.; Scher, H.I.; Lotan, T.L.; et al. A Phase II Study of the Dual MTOR Inhibitor MLN0128 in Patients with Metastatic Castration Resistant Prostate Cancer. Investig. New Drugs 2018, 36, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Al-Batran, S.; Jaeger, E.; Schmidt, B.; Bausch, M.; Unger, C.; Sethuraman, N. A Phase IIa Study of PEGylated Glutaminase (PEG-PGA) plus 6-Diazo-5-Oxo-L-Norleucine (DON) in Patients with Advanced Refractory Solid Tumors. J. Clin. Oncol. 2008, 26, 2533. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Lee, R.J.; Carthon, B.C.; Iliopoulos, O.; Mier, J.W.; Patel, M.R.; Tannir, N.M.; Owonikoko, T.K.; Haas, N.B.; Voss, M.H.; et al. CB-839, a Glutaminase Inhibitor, in Combination with Cabozantinib in Patients with Clear Cell and Papillary Metastatic Renal Cell Cancer (MRCC): Results of a Phase I Study. J. Clin. Oncol. 2019, 37, 549. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, Q.; Shang, J.; Lu, L.; Chen, G. Crocin Inhibits the Migration, Invasion, and Epithelial-mesenchymal Transition of Gastric Cancer Cells via MiR-320/KLF5/HIF-1α Signaling. J. Cell. Physiol. 2019, 234, 17876–17885. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhao, L.; Zhang, Y.; Chen, W.; Liu, D.; Hou, H.; Ding, L.; Li, X. Ginsenoside 20(S)-Rg3 Targets HIF-1α to Block Hypoxia-Induced Epithelial-Mesenchymal Transition in Ovarian Cancer Cells. PLoS ONE 2014, 9, e103887. [Google Scholar] [CrossRef] [PubMed]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a Metabolic Gene Regulatory Network Downstream of MTOR Complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT Signaling Pathway and Cancer: An Updated Review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One Drug, Many Effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Su, D.W.; Mita, M.; Mita, A.C. The Clinical Pharmacology and Toxicity Profile of Rapalogs. In mTOR Inhibition for Cancer Therapy: Past, Present and Future; Mita, M., Mita, A., Rowinsky, E.K., Eds.; Springer: Paris, France, 2016; pp. 161–189. ISBN 978-2-8178-0491-0. [Google Scholar]
- Meng, L.; Zheng, X.S. Toward Rapamycin Analog (Rapalog)-Based Precision Cancer Therapy. Acta Pharmacol. Sin. 2015, 36, 1163–1169. [Google Scholar] [CrossRef] [Green Version]
- Kwitkowski, V.E.; Prowell, T.M.; Ibrahim, A.; Farrell, A.T.; Justice, R.; Mitchell, S.S.; Sridhara, R.; Pazdur, R. FDA Approval Summary: Temsirolimus as Treatment for Advanced Renal Cell Carcinoma. Oncologist 2010, 15, 428–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurczak, W.; Ramanathan, S.; Giri, P.; Romano, A.; Mocikova, H.; Clancy, J.; Lechuga, M.; Casey, M.; Boni, J.; Giza, A.; et al. Comparison of Two Doses of Intravenous Temsirolimus in Patients with Relapsed/Refractory Mantle Cell Lymphoma. Leuk. Lymphoma 2018, 59, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Hasskarl, J. Everolimus. In Small Molecules in Oncology; Martens, U.M., Ed.; Recent Results in Cancer Research; Springer International Publishing: Cham, Switzerland, 2018; Volume 211, pp. 101–123. ISBN 978-3-319-91441-1. [Google Scholar]
- Armstrong, A.J.; Shen, T.; Halabi, S.; Kemeny, G.; Bitting, R.L.; Kartcheske, P.; Embree, E.; Morris, K.; Winters, C.; Jaffe, T.; et al. A Phase II Trial of Temsirolimus in Men With Castration-Resistant Metastatic Prostate Cancer. Clin. Genitourin. Cancer 2013, 11, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, A.; Ajani, J.A.; Bai, Y.-X.; Bang, Y.-J.; Chung, H.-C.; Pan, H.-M.; Sahmoud, T.; Shen, L.; Yeh, K.-H.; Chin, K.; et al. Everolimus for Previously Treated Advanced Gastric Cancer: Results of the Randomized, Double-Blind, Phase III GRANITE-1 Study. J. Clin. Oncol. 2013, 31, 3935–3943. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Li, X.; Zhen, L.; Chen, W.; Mu, L.; Zhang, Y.; Song, A. Everolimus Inhibits Breast Cancer Cell Growth through PI3K/AKT/MTOR Signaling Pathway. Mol. Med. Rep. 2018, 17, 7163–7169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chui, M.H.; Kjaer, S.K.; Frederiksen, K.; Hannibal, C.G.; Wang, T.-L.; Vang, R.; Shih, I.-M. BRAF V600E -Mutated Ovarian Serous Borderline Tumors Are at Relatively Low Risk for Progression to Serous Carcinoma. Oncotarget 2019, 10, 6870–6878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, J.C.; Novik, Y.; Stein, S.; Volm, M.; Meyers, M.; Smith, J.; Omene, C.; Speyer, J.; Schneider, R.; Jhaveri, K.; et al. Phase 2 Trial of Everolimus and Carboplatin Combination in Patients with Triple Negative Metastatic Breast Cancer. Breast Cancer Res. 2014, 16, 3389. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Dou, J.; Yu, Y.; Zhao, Y.; Fan, Y.; Cheng, J.; Xu, X.; Liu, W.; Guan, S.; Chen, Z.; et al. MTOR ATP-Competitive Inhibitor INK128 Inhibits Neuroblastoma Growth via Blocking MTORC Signaling. Apoptosis 2015, 20, 50–62. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Song, X.; Cao, D.; Xu, Z.; Fan, B.; Che, L.; Hu, J.; Chen, B.; Dong, M.; Pilo, M.G.; et al. Pan-MTOR Inhibitor MLN0128 Is Effective against Intrahepatic Cholangiocarcinoma in Mice. J. Hepatol. 2017, 67, 1194–1203. [Google Scholar] [CrossRef] [Green Version]
- Voss, M.H.; Gordon, M.S.; Mita, M.; Rini, B.; Makker, V.; Macarulla, T.; Smith, D.C.; Cervantes, A.; Puzanov, I.; Pili, R.; et al. Phase 1 Study of MTORC1/2 Inhibitor Sapanisertib (TAK-228) in Advanced Solid Tumours, with an Expansion Phase in Renal, Endometrial or Bladder Cancer. Br. J. Cancer 2020, 123, 1590–1598. [Google Scholar] [CrossRef]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 Is a Potent, Selective, and Orally Bioavailable ATP-Competitive Mammalian Target of Rapamycin Kinase Inhibitor with In Vitro and In Vivo Antitumor Activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pike, K.G.; Malagu, K.; Hummersone, M.G.; Menear, K.A.; Duggan, H.M.E.; Gomez, S.; Martin, N.M.B.; Ruston, L.; Pass, S.L.; Pass, M. Optimization of Potent and Selective Dual MTORC1 and MTORC2 Inhibitors: The Discovery of AZD8055 and AZD2014. Bioorg. Med. Chem. Lett. 2013, 23, 1212–1216. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin Passes the Torch: A New Generation of MTOR Inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Sparks, C.A.; Guertin, D.A. Targeting MTOR: Prospects for MTOR Complex 2 Inhibitors in Cancer Therapy. Oncogene 2010, 29, 3733–3744. [Google Scholar] [CrossRef] [PubMed]
- Janes, M.R.; Limon, J.J.; So, L.; Chen, J.; Lim, R.J.; Chavez, M.A.; Vu, C.; Lilly, M.B.; Mallya, S.; Ong, S.T.; et al. Effective and Selective Targeting of Leukemia Cells Using a TORC1/2 Kinase Inhibitor. Nat. Med. 2010, 16, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Asahina, H.; Nokihara, H.; Yamamoto, N.; Yamada, Y.; Tamura, Y.; Honda, K.; Seki, Y.; Tanabe, Y.; Shimada, H.; Shi, X.; et al. Safety and Tolerability of AZD8055 in Japanese Patients with Advanced Solid Tumors; a Dose-Finding Phase I Study. Investig. New Drugs 2013, 31, 677–684. [Google Scholar] [CrossRef]
- Eyre, T.A.; Hildyard, C.; Hamblin, A.; Ali, A.S.; Houlton, A.; Hopkins, L.; Royston, D.; Linton, K.M.; Pettitt, A.; Rule, S.; et al. A Phase II Study to Assess the Safety and Efficacy of the Dual MTORC1/2 Inhibitor Vistusertib in Relapsed, Refractory DLBCL. Hematol. Oncol. 2019, 37, 352–359. [Google Scholar] [CrossRef]
- Slotkin, E.K.; Patwardhan, P.P.; Vasudeva, S.D.; de Stanchina, E.; Tap, W.D.; Schwartz, G.K. MLN0128, an ATP-Competitive MTOR Kinase Inhibitor with Potent In Vitro and In Vivo Antitumor Activity, as Potential Therapy for Bone and Soft-Tissue Sarcoma. Mol. Cancer Ther. 2015, 14, 395–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawi, M.; Kim, J.; Dauki, A.; Sutaria, D.; Motiwala, T.; Reyes, R.; Wani, N.; Kolli, S.; Jiang, J.; Coss, C.C.; et al. CD44 Positive and Sorafenib Insensitive Hepatocellular Carcinomas Respond to the ATP-Competitive MTOR Inhibitor INK128. Oncotarget 2018, 9, 26032–26045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.-Y.; Huang, S.-L. Current Development of the Second Generation of MTOR Inhibitors as Anticancer Agents. Chin. J. Cancer 2013, 32, 8–18. [Google Scholar] [CrossRef]
- Bresin, A.; Cristofoletti, C.; Caprini, E.; Cantonetti, M.; Monopoli, A.; Russo, G.; Narducci, M.G. Preclinical Evidence for Targeting PI3K/MTOR Signaling with Dual-Inhibitors as a Therapeutic Strategy against Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2020, 140, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Xia, M.; Wang, J.; Yu, H.; Chai, J.; Zhang, Z.; Sun, Y.; Su, J.; Sun, L. Dual PI3K/MTOR Inhibitor PKI-402 Suppresses the Growth of Ovarian Cancer Cells by Degradation of Mcl-1 through Autophagy. Biomed. Pharmacother. 2020, 129, 110397. [Google Scholar] [CrossRef]
- Wu, Y.-Y.; Wu, H.-C.; Wu, J.-E.; Huang, K.-Y.; Yang, S.-C.; Chen, S.-X.; Tsao, C.-J.; Hsu, K.-F.; Chen, Y.-L.; Hong, T.-M. The Dual PI3K/MTOR Inhibitor BEZ235 Restricts the Growth of Lung Cancer Tumors Regardless of EGFR Status, as a Potent Accompanist in Combined Therapeutic Regimens. J. Exp. Clin. Cancer Res. 2019, 38, 282. [Google Scholar] [CrossRef]
- Rubinstein, M.M.; Hyman, D.M.; Caird, I.; Won, H.; Soldan, K.; Seier, K.; Iasonos, A.; Tew, W.P.; O’Cearbhaill, R.E.; Grisham, R.N.; et al. Phase 2 Study of LY3023414 in Patients with Advanced Endometrial Cancer Harboring Activating Mutations in the PI3K Pathway. Cancer 2020, 126, 1274–1282. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Bell-McGuinn, K.M.; Molina, J.R.; Bendell, J.; Spicer, J.; Kwak, E.L.; Pandya, S.S.; Millham, R.; Borzillo, G.; Pierce, K.J.; et al. First-in-Human Study of PF-05212384 (PKI-587), a Small-Molecule, Intravenous, Dual Inhibitor of PI3K and MTOR in Patients with Advanced Cancer. Clin. Cancer Res. 2015, 21, 1888–1895. [Google Scholar] [CrossRef] [Green Version]
- Bendell, J.C.; Varghese, A.M.; Hyman, D.M.; Bauer, T.M.; Pant, S.; Callies, S.; Lin, J.; Martinez, R.; Wickremsinhe, E.; Fink, A.; et al. A First-in-Human Phase 1 Study of LY3023414, an Oral PI3K/MTOR Dual Inhibitor, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 3253–3262. [Google Scholar] [CrossRef] [Green Version]
- Salazar, R.; Garcia-Carbonero, R.; Libutti, S.K.; Hendifar, A.E.; Custodio, A.; Guimbaud, R.; Lombard-Bohas, C.; Ricci, S.; Klümpen, H.-J.; Capdevila, J.; et al. Phase II Study of BEZ235 versus Everolimus in Patients with Mammalian Target of Rapamycin Inhibitor-Naïve Advanced Pancreatic Neuroendocrine Tumors. Oncologist 2018, 23, 766-e90. [Google Scholar] [CrossRef] [Green Version]
- Khan, K.H.; Wong, M.; Rihawi, K.; Bodla, S.; Morganstein, D.; Banerji, U.; Molife, L.R. Hyperglycemia and Phosphatidylinositol 3-Kinase/Protein Kinase B/Mammalian Target of Rapamycin (PI3K/AKT/MTOR) Inhibitors in Phase I Trials: Incidence, Predictive Factors, and Management. Oncologist 2016, 21, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Meng, G.; Zheng, M.; Zhang, Y.; Chen, A.; Wu, J.; Wei, J. The Glutaminase-1 Inhibitor 968 Enhances Dihydroartemisinin-Mediated Antitumor Efficacy in Hepatocellular Carcinoma Cells. PLoS ONE 2016, 11, e0166423. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef]
- Li, L.; Meng, Y.; Li, Z.; Dai, W.; Xu, X.; Bi, X.; Bian, J. Discovery and Development of Small Molecule Modulators Targeting Glutamine Metabolism. Eur. J. Med. Chem. 2019, 163, 215–242. [Google Scholar] [CrossRef]
- Emadi, A.; Jun, S.A.; Tsukamoto, T.; Fathi, A.T.; Minden, M.D.; Dang, C.V. Inhibition of Glutaminase Selectively Suppresses the Growth of Primary Acute Myeloid Leukemia Cells with IDH Mutations. Exp. Hematol. 2014, 42, 247–251. [Google Scholar] [CrossRef]
- Shukla, K.; Ferraris, D.V.; Thomas, A.G.; Stathis, M.; Duvall, B.; Delahanty, G.; Alt, J.; Rais, R.; Rojas, C.; Gao, P.; et al. Design, Synthesis, and Pharmacological Evaluation of Bis-2-(5-Phenylacetamido-1,2,4-Thiadiazol-2-Yl)Ethyl Sulfide 3 (BPTES) Analogs as Glutaminase Inhibitors. J. Med. Chem. 2012, 55, 10551–10563. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Ruiz-Rodado, V.; Dowdy, T.; Huang, S.; Issaq, S.H.; Beck, J.; Wang, H.; Tran Hoang, C.; Lita, A.; Larion, M.; et al. Glutaminase-1 (GLS1) Inhibition Limits Metastatic Progression in Osteosarcoma. Cancer Metab. 2020, 8, 4. [Google Scholar] [CrossRef]
- Earhart, R.H.; Amato, D.J.; Chang, Y.-C.A.; Borden, E.C.; Shiraki, M.; Dowd, M.E.; Comis, R.L.; Davis, T.E.; Smith, T.J. Phase II Trial of 6-Diazo-5-Oxo-L-Norleucine versus Aclacinomycin-A in Advanced Sarcomas and Mesotheliomas. Investig. New Drugs 1990, 8, 113–119. [Google Scholar] [CrossRef]
- Calithera Biosciences, Inc Ph1 Study of the Safety, PK, and PDn of Escalating Oral Doses of the Glutaminase Inhibitor CB-839, as a Single Agent and in Combination with Standard Chemotherapy in Patients with Advanced and/or Treatment-Refractory Solid Tumors; clinicaltrials.gov. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT02071862 (accessed on 20 January 2022).
- First-in-Human Study of DRP-104 (Sirpiglenastat) as Single Agent and in Combination with Atezolizumab in Patients with Advanced Solid Tumors—No Study Results Posted—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/results/NCT04471415 (accessed on 24 January 2022).
- Hampp, C.; Borders-Hemphill, V.; Moeny, D.G.; Wysowski, D.K. Use of Antidiabetic Drugs in the U.S., 2003–2012. Diabetes Care 2014, 37, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Torres, W.; Nava, M.; Galbán, N.; Gómez, Y.; Morillo, V.; Rojas, M.; Cano, C.; Chacín, M.; D′Marco, L.; Herazo, Y.; et al. Anti-Aging Effect of Metformin: A Molecular and Therapeutical Perspective. Curr. Pharm. Des. 2020, 26, 4496–4508. [Google Scholar] [CrossRef]
- Shank, J.J.; Yang, K.; Ghannam, J.; Cabrera, L.; Johnston, C.J.; Reynolds, R.K.; Buckanovich, R.J. Metformin Targets Ovarian Cancer Stem Cells In Vitro and In Vivo. Gynecol. Oncol. 2012, 127, 390–397. [Google Scholar] [CrossRef] [Green Version]
- Ugwueze, C.V.; Ogamba, O.J.; Young, E.E.; Onyenekwe, B.M.; Ezeokpo, B.C. Metformin: A Possible Option in Cancer Chemotherapy. Anal. Cell. Pathol. 2020, 2020, 7180923. [Google Scholar] [CrossRef]
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and Reduced Risk of Cancer in Diabetic Patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Zhong, X.; Gao, P.; Shi, J.; Wu, Z.; Guo, Z.; Wang, Z.; Song, Y. The Potential Effect of Metformin on Cancer: An Umbrella Review. Front. Endocrinol. 2019, 10, 617. [Google Scholar] [CrossRef]
- Franciosi, M.; Lucisano, G.; Lapice, E.; Strippoli, G.F.M.; Pellegrini, F.; Nicolucci, A. Metformin Therapy and Risk of Cancer in Patients with Type 2 Diabetes: Systematic Review. PLoS ONE 2013, 8, e71583. [Google Scholar] [CrossRef]
- Kasznicki, J.; Sliwinska, A.; Drzewoski, J. Metformin in Cancer Prevention and Therapy. Ann. Transl. Med. 2014, 2, 57. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, Q.; Zhang, F.; Meng, F.; Liu, S.; Zhou, R.; Wu, Q.; Li, X.; Shen, L.; Huang, J.; et al. HER2 Recruits AKT1 to Disrupt STING Signalling and Suppress Antiviral Defence and Antitumour Immunity. Nat. Cell Biol. 2019, 21, 1027–1040. [Google Scholar] [CrossRef]
- Sung, M.; Tan, X.; Lu, B.; Golas, J.; Hosselet, C.; Wang, F.; Tylaska, L.; King, L.; Zhou, D.; Dushin, R.; et al. Caveolae-Mediated Endocytosis as a Novel Mechanism of Resistance to Trastuzumab Emtansine (T-DM1). Mol. Cancer Ther. 2018, 17, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Yuan, Z.; Xu, D.; Ding, P.; Wang, T.; Zhang, L.; Jiang, Z. Inhibition of Glycolysis by a Novel EGFR/HER2 Inhibitor KU004 Suppresses the Growth of HER2+ Cancer. Exp. Cell Res. 2017, 357, 211–221. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Q.; Bhetuwal, A.; Liu, W. Metabolic Pathways Underlying GATA6 Regulating Trastuzumab Resistance in Gastric Cancer Cells Based on Untargeted Metabolomics. Int. J. Med. Sci. 2020, 17, 3146–3164. [Google Scholar] [CrossRef]
- Su, B.; Huang, T.; Jin, Y.; Yin, H.; Qiu, H.; Yuan, X. Apatinib Exhibits Synergistic Effect with Pyrotinib and Reverses Acquired Pyrotinib Resistance in HER2-Positive Gastric Cancer via Stem Cell Factor/c-Kit Signaling and Its Downstream Pathways. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2021, 24, 352–367. [Google Scholar] [CrossRef]
- Wu, C.-S.; Wei, K.-L.; Chou, J.-L.; Lu, C.-K.; Hsieh, C.-C.; Lin, J.M.J.; Deng, Y.-F.; Hsu, W.-T.; Wang, H.-M.D.; Leung, C.-H.; et al. Aberrant JAK/STAT Signaling Suppresses TFF1 and TFF2 through Epigenetic Silencing of GATA6 in Gastric Cancer. Int. J. Mol. Sci. 2016, 17, E1467. [Google Scholar] [CrossRef]
- Liu, J.; Pan, C.; Guo, L.; Wu, M.; Guo, J.; Peng, S.; Wu, Q.; Zuo, Q. A New Mechanism of Trastuzumab Resistance in Gastric Cancer: MACC1 Promotes the Warburg Effect via Activation of the PI3K/AKT Signaling Pathway. J. Hematol. Oncol. J. Hematol. Oncol. 2016, 9, 76. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Du, Z.; Xu, T.; Wang, X.; Li, W.; Gao, J.; Li, J.; Zhu, H. HIF-1α Is a Rational Target for Future Ovarian Cancer Therapies. Front. Oncol. 2021, 11, 785111. [Google Scholar] [CrossRef]
- Grande, F.; Aiello, F.; Garofalo, A.; Neamati, N. Identification and Preclinical Evaluation of SC144, a Novel Pyrroloquinoxaline Derivative with Broad-Spectrum Anticancer Activity. Mini-Rev. Med. Chem. 2016, 16, 644–650. [Google Scholar] [CrossRef]
- Lu, T.; Tang, J.; Shrestha, B.; Heath, B.R.; Hong, L.; Lei, Y.L.; Ljungman, M.; Neamati, N. Up-Regulation of Hypoxia-Inducible Factor Antisense as a Novel Approach to Treat Ovarian Cancer. Theranostics 2020, 10, 6959–6976. [Google Scholar] [CrossRef]
- Greenberger, L.M.; Horak, I.D.; Filpula, D.; Sapra, P.; Westergaard, M.; Frydenlund, H.F.; Albæk, C.; Schrøder, H.; Ørum, H. A RNA Antagonist of Hypoxia-Inducible Factor-1α, EZN-2968, Inhibits Tumor Cell Growth. Mol. Cancer Ther. 2008, 7, 3598–3608. [Google Scholar] [CrossRef] [Green Version]
- Bin, Y.-L.; Hu, H.-S.; Tian, F.; Wen, Z.-H.; Yang, M.-F.; Wu, B.-H.; Wang, L.-S.; Yao, J.; Li, D.-F. Metabolic Reprogramming in Gastric Cancer: Trojan Horse Effect. Front. Oncol. 2022, 11, 1–14. [Google Scholar] [CrossRef]
- Lu, J.; Chen, H.; He, F.; You, Y.; Feng, Z.; Chen, W.; Li, X.; Zhao, L. Ginsenoside 20(S)-Rg3 Upregulates HIF-1α-targeting MiR-519a-5p to Inhibit the Warburg Effect in Ovarian Cancer Cells. Clin. Exp. Pharmacol. Physiol. 2020, 47, 1455–1463. [Google Scholar] [CrossRef]
- Bortner, C.D.; Cidlowski, J.A. Ion Channels and Apoptosis in Cancer. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2014, 369, 20130104. [Google Scholar] [CrossRef]
- Wulff, H.; Castle, N.A. Therapeutic Potential of KCa3.1 Blockers: Recent Advances and Promising Trends. Expert Rev. Clin. Pharmacol. 2010, 3, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Sontheimer, H. An Unexpected Role for Ion Channels in Brain Tumor Metastasis. Exp. Biol. Med. Maywood NJ 2008, 233, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Turner, K.L.; Sontheimer, H. Cl- and K+ Channels and Their Role in Primary Brain Tumour Biology. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2014, 369, 20130095. [Google Scholar] [CrossRef] [Green Version]
- Mamelak, A.N.; Jacoby, D.B. Targeted Delivery of Antitumoral Therapy to Glioma and Other Malignancies with Synthetic Chlorotoxin (TM-601). Expert Opin. Drug Deliv. 2007, 4, 175–186. [Google Scholar] [CrossRef]
- Mamelak, A.N.; Rosenfeld, S.; Bucholz, R.; Raubitschek, A.; Nabors, L.B.; Fiveash, J.B.; Shen, S.; Khazaeli, M.B.; Colcher, D.; Liu, A.; et al. Phase I Single-Dose Study of Intracavitary-Administered Iodine-131-TM-601 in Adults with Recurrent High-Grade Glioma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 3644–3650. [Google Scholar] [CrossRef]
- Hockaday, D.C.; Shen, S.; Fiveash, J.; Raubitschek, A.; Colcher, D.; Liu, A.; Alvarez, V.; Mamelak, A.N. Imaging Glioma Extent with 131I-TM-601. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2005, 46, 580–586. [Google Scholar]
- Zuliani, V.; Rapalli, A.; Patel, M.K.; Rivara, M. Sodium Channel Blockers: A Patent Review (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 279–290. [Google Scholar] [CrossRef]
- Djamgoz, M.B.A.; Onkal, R. Persistent Current Blockers of Voltage-Gated Sodium Channels: A Clinical Opportunity for Controlling Metastatic Disease. Recent Patents Anticancer Drug Discov. 2013, 8, 66–84. [Google Scholar] [CrossRef]
- Vandenberg, J.I.; Perry, M.D.; Perrin, M.J.; Mann, S.A.; Ke, Y.; Hill, A.P. HERG K(+) Channels: Structure, Function, and Clinical Significance. Physiol. Rev. 2012, 92, 1393–1478. [Google Scholar] [CrossRef] [Green Version]
- Fnu, G.; Weber, G.F. Alterations of Ion Homeostasis in Cancer Metastasis: Implications for Treatment. Front. Oncol. 2021, 11, 765329. [Google Scholar] [CrossRef]
- Szablewski, L. Glucose Transporters as Markers of Diagnosis and Prognosis in Cancer Diseases. Oncol. Rev. 2022, 16, 561. [Google Scholar] [CrossRef]
- Pliszka, M.; Szablewski, L. Glucose Transporters as a Target for Anticancer Therapy. Cancers 2021, 13, 4184. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors (REF) | Treatment | Inhibitor Target | Methodology | Results |
|---|---|---|---|---|
| DeCensi et al. [119] | Metformin | Insulin-lowering agent. | Meta-analysis combining 11 observational studies and trials (N = 4042) focused on evaluating the effects of metformin on cancer. | A 31% reduction in cancer incidence or mortality was observed in subjects taking metformin compared with other antidiabetics (SRR, 0.69; 95% CI, 0.61–0.79). |
| Andronesi et al. [120] | IDH305 | Mutant-selective allosteric high affinity IDH1 inhibitor. | Phase I clinical trial of IDH305. | A rapid decrease of 2HG levels by 70% (CI 13%, p = 0.019) after 1 week of treatment. Importantly, inhibition of mutant IDH1 may lead to reprogramming of tumor metabolism, suggested by simultaneous changes in glutathione, glutamine, glutamate, and lactate. |
| Basu et al. [121] | AZD2014 | Dual m-TORC 1/2 Inhibitor. | Phase II clinical trial where 56 adults were treated with AZD2014 orally twice daily with doses of 25 to 100 mg. | Two PRs were evidenced in the study. The first was a patient with acinar pancreatic cancer with KRAS, PDGFRA, APC, ERB4, KIT, and FBXW7 mutations. The second to respond was a breast cancer patient with HRAS, NRAS, TP53 and ERBB2 mutations. |
| Graham et al. [122] | MLN0128 | Dual m-TORC 1/2 Inhibitor. | Phase II clinical trial focused on evaluating the efficacy and safety of MLN0128 in mCRPC at a dose of 4 mg for a median time of 11 weeks (range = 3–30) in 9 subjects. | The best response was stable disease. All subjects showed an increase in PSA during treatment (159%) and no decreases in cancer cell counts after treatment. Correlative studies of biopsy samples before and after treatment indicate limited inhibition of AKT phosphorylation, 4EBP1, and eIF4E activity. |
| Mueller et al. [123] | 6-diazo-5-oxo-L-norleucine (DON) | Glutamine antagonist and inhibits irreversibly the purine/pyrimidine synthesis. | Phase IIa multicenter study evaluating the safety and clinical activity of the glutamine-reducing enzyme PEG-PGA in combination with DON in 55 patients treated with 120 IU/m2 of PEG-PGA once a week, and 140 mg/m2 DON twice a week between 1 and 379 days. | After the second infusion, glutamine levels were reduced by 10% and remained reduced in >90% of patients. Among the patients with metastatic colorectal cancer, 1 PR was found, 9 free of progression at 3 months, and 5 free of progression at 5 months. |
| Meric-Bernstam et al. [124] | CB-839 | Glutaminase Inhibitor. | Escalating doses of CB-839 (600–800 mg PO BID) plus Cabo (60 mg PO QD) were evaluated using a 3+3 design. Tumor response was assessed per RECIST 1.1 every 8 weeks. | CB-839 plus Cabo showed encouraging clinical activity and tolerability in heavily pretreated mRCC patients, with response rates (50% ORR, 100% DCR) comparing favorably to historical Cabo monotherapy. |
| Zhou et al. [125] | Crocin | Inhibits the migration, invasion, and epithelial-mesenchymal transition of gastric cancer cells via miR-320/KLF5/HIF-1α signaling. | Preclinical study | Crocin inhibits the EMT, migration, and invasion of gastric cancer cells, and this activity is mediated by miR-320/KLF5/HIF-1α signaling |
| Liu et al. [126] | Ginsenoside 20(S)-Rg3 | Reduces the expression of HIF-1α by activating the ubiquitin-proteasome pathway to promote HIF-1α degradation. | In vivo and in vitro preclinical study | Ginsenoside 20(S)-Rg3 potently blocks hypoxia-induced EMT of ovarian cancer cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navarro, C.; Ortega, Á.; Santeliz, R.; Garrido, B.; Chacín, M.; Galban, N.; Vera, I.; De Sanctis, J.B.; Bermúdez, V. Metabolic Reprogramming in Cancer Cells: Emerging Molecular Mechanisms and Novel Therapeutic Approaches. Pharmaceutics 2022, 14, 1303. https://doi.org/10.3390/pharmaceutics14061303
Navarro C, Ortega Á, Santeliz R, Garrido B, Chacín M, Galban N, Vera I, De Sanctis JB, Bermúdez V. Metabolic Reprogramming in Cancer Cells: Emerging Molecular Mechanisms and Novel Therapeutic Approaches. Pharmaceutics. 2022; 14(6):1303. https://doi.org/10.3390/pharmaceutics14061303
Chicago/Turabian StyleNavarro, Carla, Ángel Ortega, Raquel Santeliz, Bermary Garrido, Maricarmen Chacín, Néstor Galban, Ivana Vera, Juan Bautista De Sanctis, and Valmore Bermúdez. 2022. "Metabolic Reprogramming in Cancer Cells: Emerging Molecular Mechanisms and Novel Therapeutic Approaches" Pharmaceutics 14, no. 6: 1303. https://doi.org/10.3390/pharmaceutics14061303