
Semi-Automated Therapeutic Drug Monitoring as a Pillar toward Personalized Medicine for Tuberculosis Management

 , ,
, , 
Abstract
:1. Introduction
2. Overview of Therapeutic Drug Monitoring in TB Treatment
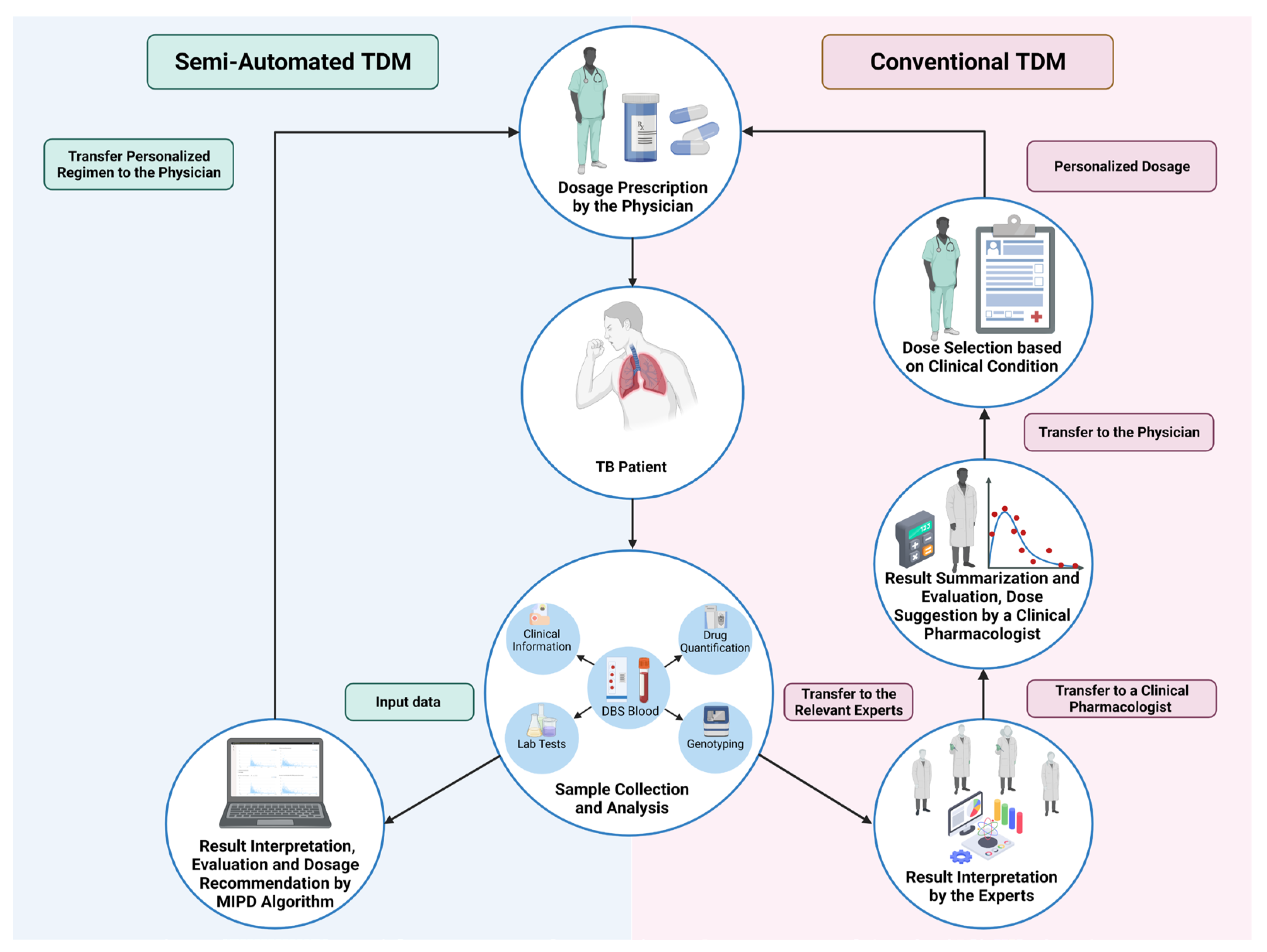
2.1. Conventional TDM
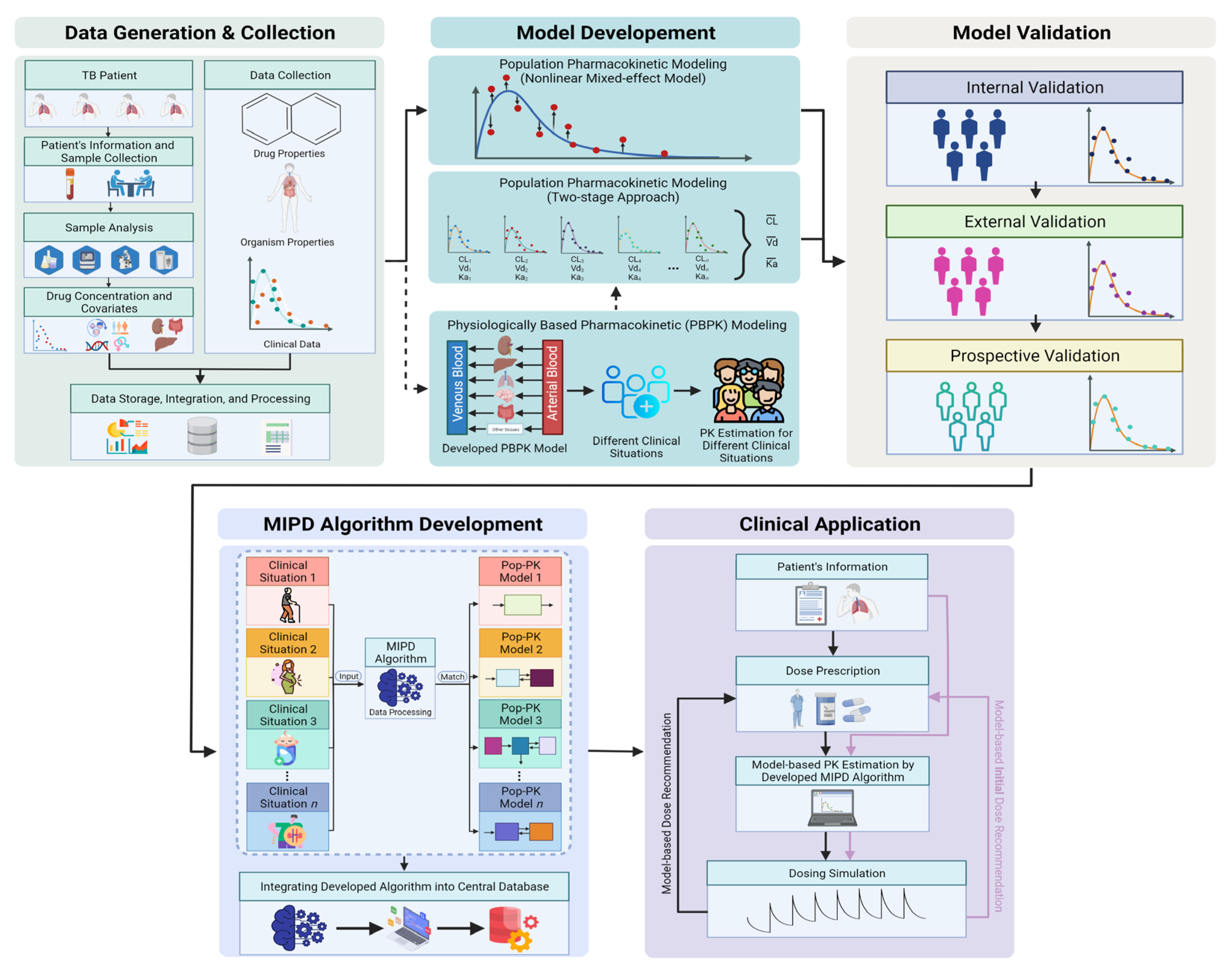
2.2. Model-Informed Precision-Dosing-Based TDM
3. TDM Implementation in Clinical Practice
- Defining the case;
- Obtaining the blood samples;
- Measuring drug concentrations;
- Interpreting the results.
3.1. Defining the Case
- Poor response to tuberculosis treatment despite adherence and fully drug-susceptible Mycobacterium tuberculosis strain;
- Severe gastrointestinal abnormalities: severe gastroparesis, short bowel syndrome, chronic diarrhea with malabsorption;
- Drug–drug interactions;
- Impaired renal clearance: renal insufficiency, peritoneal dialysis, critically ill patients on continuous renal replacement;
- HIV infection;
- Diabetes mellitus;
- Treatment using second-line drugs.
3.2. Obtaining the Blood Samples
3.3. Measuring Drug Concentrations
3.4. Interpreting the Results
4. Evidence of Benefits
5. Barriers of Implementation in Clinical Practice
5.1. Sampling Strategy
5.2. Logistic and Storage
5.3. Bioanalysis Process
5.4. Human Resources
6. Semi-Automated TDM Process
7. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Migliori, G.B.; Thong, P.M.; Akkerman, O.; Alffenaar, J.W.; Alvarez-Navascues, F.; Assao-Neino, M.M.; Bernard, P.V.; Biala, J.S.; Blanc, F.X.; Bogorodskaya, E.M.; et al. Worldwide effects of coronavirus disease pandemic on tuberculosis services, January-April 2020. Emerg. Infect. Dis. 2020, 26, 2709–2712. [Google Scholar] [CrossRef]
- Polasek, T.M.; Rayner, C.R.; Peck, R.W.; Rowland, A.; Kimko, H.; Rostami-Hodjegan, A. Toward dynamic prescribing information: Codevelopment of companion model-informed precision dosing tools in drug development. Clin. Pharmacol. Drug Dev. 2019, 8, 418–425. [Google Scholar] [CrossRef]
- Polasek, T.M.; Rostami-Hodjegan, A.; Yim, D.S.; Jamei, M.; Lee, H.; Kimko, H.; Kim, J.K.; Nguyen, P.T.T.; Darwich, A.S.; Shin, J.G. What does it take to make model-informed precision dosing common practice? Report from the 1st Asian Symposium on Precision Dosing. AAPS J 2019, 21, 17. [Google Scholar] [CrossRef]
- Visvikis-Siest, S.; Theodoridou, D.; Kontoe, M.S.; Kumar, S.; Marschler, M. Milestones in personalized medicine: From the ancient time to nowadays-the provocation of COVID-19. Front. Genet. 2020, 11, 569175. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Mkrtchian, S.; Zhou, Y.; Lauschke, V.M. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum. Genom. 2018, 12, 26. [Google Scholar] [CrossRef]
- Goetz, L.H.; Schork, N.J. Personalized medicine: Motivation, challenges, and progress. Fertil. Steril. 2018, 109, 952–963. [Google Scholar] [CrossRef]
- Peloquin, C. The role of therapeutic drug monitoring in mycobacterial infections. Microbiol. Spectr. 2017, 5, TNMI7–TNMI0029. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Ulbricht, E.; Ahn, Y.K.; Gillooly, I.S.; Lee, K.J.; Lieu, J.; Nguyen, W.; Young, S.; Cho, J.G.; Alffenaar, J.W. Therapeutic drug monitoring practice in patients with active tuberculosis: Assessment of opportunities. Eur. Respir. J. 2021, 57, 2002349. [Google Scholar] [CrossRef]
- Sturkenboom, M.G.G.; Martson, A.G.; Svensson, E.M.; Sloan, D.J.; Dooley, K.E.; van den Elsen, S.H.J.; Denti, P.; Peloquin, C.A.; Aarnoutse, R.E.; Alffenaar, J.C. Population pharmacokinetics and bayesian dose adjustment to advance TDM of Anti-TB drugs. Clin. Pharmacokinet. 2021, 60, 685–710. [Google Scholar] [CrossRef]
- van Beek, S.W.; Ter Heine, R.; Keizer, R.J.; Magis-Escurra, C.; Aarnoutse, R.E.; Svensson, E.M. Personalized tuberculosis treatment through model-informed dosing of rifampicin. Clin. Pharmacokinet. 2019, 58, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Zuur, M.A.; Bolhuis, M.S.; Anthony, R.; den Hertog, A.; van der Laan, T.; Wilffert, B.; de Lange, W.; van Soolingen, D.; Alffenaar, J.W. Current status and opportunities for therapeutic drug monitoring in the treatment of tuberculosis. Expert Opin. Drug. Metab. Toxicol. 2016, 12, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Polasek, T.M.; Kirkpatrick, C.M.J.; Rostami-Hodjegan, A. Precision dosing to avoid adverse drug reactions. Ther. Adv. Drug Saf. 2019, 10, 2042098619894147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, C.; Aarnoutse, R.; Chesov, D.; van Crevel, R.; Gillespie, S.H.; Grobbel, H.P.; Kalsdorf, B.; Kontsevaya, I.; van Laarhoven, A.; Nishiguchi, T.; et al. Perspective for precision medicine for tuberculosis. Front. Immunol. 2020, 11, 566608. [Google Scholar] [CrossRef]
- Martson, A.G.; Burch, G.; Ghimire, S.; Alffenaar, J.C.; Peloquin, C.A. Therapeutic drug monitoring in patients with tuberculosis and concurrent medical problems. Expert Opin. Drug Metab. Toxicol. 2021, 17, 23–39. [Google Scholar] [CrossRef]
- Alffenaar, J.C.; Gumbo, T.; Dooley, K.E.; Peloquin, C.A.; McIlleron, H.; Zagorski, A.; Cirillo, D.M.; Heysell, S.K.; Silva, D.R.; Migliori, G.B. Integrating pharmacokinetics and pharmacodynamics in operational research to end tuberculosis. Clin. Infect. Dis. 2020, 70, 1774–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsultan, A.; Peloquin, C.A. Therapeutic drug monitoring in the treatment of tuberculosis: An update. Drugs 2014, 74, 839–854. [Google Scholar] [CrossRef] [PubMed]
- Magis-Escurra, C.; van den Boogaard, J.; Ijdema, D.; Boeree, M.; Aarnoutse, R. Therapeutic drug monitoring in the treatment of tuberculosis patients. Pulm. Pharmacol. Ther. 2012, 25, 83–86. [Google Scholar] [CrossRef]
- Alffenaar, J.C.; Akkerman, O.W.; Kim, H.Y.; Tiberi, S.; Migliori, G.B. Precision and personalized medicine and anti-TB treatment: Is TDM feasible for programmatic use? Int. J. Infect. Dis. 2020, 92S, S5–S9. [Google Scholar] [CrossRef] [Green Version]
- Wicha, S.G.; Martson, A.G.; Nielsen, E.I.; Koch, B.C.P.; Friberg, L.E.; Alffenaar, J.W.; Minichmayr, I.K.; International Society of Anti-Infective Pharmacology. From therapeutic drug monitoring to model-informed precision dosing for antibiotics. Clin. Pharmacol. Ther. 2021, 109, 928–941. [Google Scholar] [CrossRef]
- Saktiawati, A.M.I.; Harkema, M.; Setyawan, A.; Subronto, Y.W.; Sumardi; Stienstra, Y.; Aarnoutse, R.E.; Magis-Escurra, C.; Kosterink, J.G.W.; van der Werf, T.S.; et al. Optimal sampling strategies for therapeutic drug monitoring of first-line tuberculosis drugs in patients with tuberculosis. Clin. Pharmacokinet. 2019, 58, 1445–1454. [Google Scholar] [CrossRef] [Green Version]
- Alshaikheid, M.; Chaabane, A.; Ben Fredj, N.; Ben Brahim, H.; Ben Fadhel, N.; Chadli, Z.; Slama, A.; Boughattas, N.A.; Chakroun, M.; Aouam, K. Limited sampling strategy for predicting isoniazid exposure in patients with extrapulmonary tuberculosis. J. Clin. Pharm. Ther. 2020, 45, 503–512. [Google Scholar] [CrossRef] [PubMed]
- van Beek, S.W.; Ter Heine, R.; Alffenaar, J.C.; Magis-Escurra, C.; Aarnoutse, R.E.; Svensson, E.M.; Isoniazid Precision Dosing, G. A model-informed method for the purpose of precision dosing of isoniazid in pulmonary tuberculosis. Clin. Pharmacokinet. 2021, 60, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Alshaikheid, M.; Ben Fredj, N.; Hannachi, I.; Kolsi, N.; Ben Fadhel, N.; Kerkeni, E.; Ben Romdhane, H.; Chaabane, A.; Koubaa, J.; Chadli, Z.; et al. Development of a population pharmacokinetic model and Bayesian estimators for isoniazid in Tunisian tuberculosis patients. Pharm. J. 2021, 21, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, A.; Prim, D.; Bojescu, E.D.; Segura, J.M.; Pfeifer, M.E. Point-of-care therapeutic drug monitoring for precision dosing of immunosuppressive drugs. J. Appl. Lab. Med. 2020, 5, 738–761. [Google Scholar] [CrossRef] [PubMed]
- Vernon, A.; Fielding, K.; Savic, R.; Dodd, L.; Nahid, P. The importance of adherence in tuberculosis treatment clinical trials and its relevance in explanatory and pragmatic trials. PLoS Med. 2019, 16, e1002884. [Google Scholar] [CrossRef]
- Nahid, P.; Dorman, S.E.; Alipanah, N.; Barry, P.M.; Brozek, J.L.; Cattamanchi, A.; Chaisson, L.H.; Chaisson, R.E.; Daley, C.L.; Grzemska, M.; et al. Official American thoracic society/centers for disease control and prevention/infectious diseases society of America clinical practice guidelines: Treatment of drug-susceptible tuberculosis. Clin. Infect. Dis. 2016, 63, e147–e195. [Google Scholar] [CrossRef]
- Boeree, M.J.; Diacon, A.H.; Dawson, R.; Narunsky, K.; du Bois, J.; Venter, A.; Phillips, P.P.; Gillespie, S.H.; McHugh, T.D.; Hoelscher, M.; et al. A dose-ranging trial to optimize the dose of rifampin in the treatment of tuberculosis. Am. J. Respir. Crit. Care Med. 2015, 191, 1058–1065. [Google Scholar] [CrossRef] [Green Version]
- Perwitasari, D.A.; Darmawan, E.; Mulyani, U.A.; Vlies, P.V.; Alffenaar, J.C.; Atthobar, J.; Wilffert, B. Polymorphisms of NAT2, CYP2E1, GST, and HLA related to drug-induced liver injury in indonesian tuberculosis patients. Int. J. Mycobacteriol. 2018, 7, 380–386. [Google Scholar] [CrossRef]
- Sekaggya-Wiltshire, C.; von Braun, A.; Scherrer, A.U.; Manabe, Y.C.; Buzibye, A.; Muller, D.; Ledergerber, B.; Gutteck, U.; Corti, N.; Kambugu, A.; et al. Anti-TB drug concentrations and drug-associated toxicities among TB/HIV-coinfected patients. J. Antimicrob. Chemother. 2017, 72, 1172–1177. [Google Scholar] [CrossRef] [Green Version]
- Sant´Anna, F.M.; Araújo-Pereira, M.; Schmaltz, C.A.S.; Arriaga, M.B.; de Oliveira, R.V.C.; Andrade, B.B.; Rolla, V.C. Adverse Drug Reactions Related to Treatment of Drug-Susceptible Tuberculosis in Brazil: A Prospective Cohort Study. Front. Trop. Dis. 2022, 2, 748310. [Google Scholar] [CrossRef]
- Srivastava, S.; Pasipanodya, J.G.; Meek, C.; Leff, R.; Gumbo, T. Multidrug-resistant tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J. Infect. Dis. 2011, 204, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Diacon, A.H.; Patientia, R.F.; Venter, A.; van Helden, P.D.; Smith, P.J.; McIlleron, H.; Maritz, J.S.; Donald, P.R. Early bactericidal activity of high-dose rifampin in patients with pulmonary tuberculosis evidenced by positive sputum smears. Antimicrob. Agents Chemother. 2007, 51, 2994–2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burhan, E.; Ruesen, C.; Ruslami, R.; Ginanjar, A.; Mangunnegoro, H.; Ascobat, P.; Donders, R.; van Crevel, R.; Aarnoutse, R. Isoniazid, rifampin, and pyrazinamide plasma concentrations in relation to treatment response in Indonesian pulmonary tuberculosis patients. Antimicrob. Agents Chemother. 2013, 57, 3614–3619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuma, J.; Ohno, M.; Kubota, R.; Yokota, S.; Nagai, T.; Tsuyuguchi, K.; Okuda, Y.; Takashima, T.; Kamimura, S.; Fujio, Y.; et al. NAT2 genotype guided regimen reduces isoniazid-induced liver injury and early treatment failure in the 6-month four-drug standard treatment of tuberculosis: A randomized controlled trial for pharmacogenetics-based therapy. Eur. J. Clin. Pharmacol. 2013, 69, 1091–1101. [Google Scholar] [CrossRef] [Green Version]
- Jing, W.; Zong, Z.; Tang, B.; Wang, J.; Zhang, T.; Wen, S.; Xue, Y.; Chu, N.; Zhao, W.; Huang, H. Population Pharmacokinetic Analysis of Isoniazid among Pulmonary Tuberculosis Patients from China. Antimicrob. Agents Chemother. 2020, 64, e01736-19. [Google Scholar] [CrossRef]
- Yuliwulandari, R.; Prayuni, K.; Razari, I.; Susilowati, R.W.; Zulhamidah, Y.; Soedarsono, S.; Sofro, A.S.M.; Tokunaga, K. Genetic characterization of N-acetyltransferase 2 variants in acquired multidrug-resistant tuberculosis in Indonesia. Pharmacogenomics 2021, 22, 157–163. [Google Scholar] [CrossRef]
- Dong, H.Y.; Xie, J.; Chen, L.H.; Wang, T.T.; Zhao, Y.R.; Dong, Y.L. Therapeutic drug monitoring and receiver operating characteristic curve prediction may reduce the development of linezolid-associated thrombocytopenia in critically ill patients. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1029–1035. [Google Scholar] [CrossRef]
- Cheng, C.N.; Wu, C.C.; Kuo, C.H.; Wang, C.C.; Wang, J.T.; Lin, Y.T.; Jhang, R.S.; Lin, S.W. Impact of high plasma concentrations of linezolid in Taiwanese adult patients- therapeutic drug monitoring in improving adverse drug reactions. J. Formos. Med. Assoc. 2021, 120, 466–475. [Google Scholar] [CrossRef]
- Ben Salem, C.; Slim, R.; Fathallah, N.; Hmouda, H. Drug-induced hyperuricaemia and gout. Rheumatology 2017, 56, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef]
- Shih, T.Y.; Pai, C.Y.; Yang, P.; Chang, W.L.; Wang, N.C.; Hu, O.Y. A novel mechanism underlies the hepatotoxicity of pyrazinamide. Antimicrob. Agents Chemother. 2013, 57, 1685–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retno, W.S.; Kinasih, P.; Intan, R.; Syukrini, B.; Rika, Y. High frequency of NAT2 slow acetylator alleles in the Malay population of Indonesia: An awareness to the anti-tuberculosis drug induced liver injury and cancer. Med. J. Ind. 2017, 26, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Yuliwulandari, R.; Prayuni, K.; Susilowati, R.W.; AS, M.S.; Tokunaga, K.; Shin, J.G. NAT2 slow acetylator is associated with anti-tuberculosis drug-induced liver injury severity in Indonesian population. Pharmacogenomics 2019, 20, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Holford, N.; Ma, G.; Metz, D. TDM is dead. Long live TCI! Br. J. Clin. Pharmacol. 2020, 88, 1406–1413. [Google Scholar] [CrossRef]
- Holford, N. Pharmacodynamic principles and target concentration intervention. Transl. Clin. Pharmacol. 2018, 26, 150–154. [Google Scholar] [CrossRef] [Green Version]
- Cooney, L.; Loke, Y.K.; Golder, S.; Kirkham, J.; Jorgensen, A.; Sinha, I.; Hawcutt, D. Overview of systematic reviews of therapeutic ranges: Methodologies and recommendations for practice. BMC Med. Res. Methodol. 2017, 17, 84. [Google Scholar] [CrossRef] [Green Version]
- Neely, M.N.; Kato, L.; Youn, G.; Kraler, L.; Bayard, D.; van Guilder, M.; Schumitzky, A.; Yamada, W.; Jones, B.; Minejima, E. Prospective Trial on the Use of Trough Concentration versus Area under the Curve to Determine Therapeutic Vancomycin Dosing. Antimicrob. Agents Chemother. 2018, 62, e02042-17. [Google Scholar] [CrossRef] [Green Version]
- Ates, H.C.; Roberts, J.A.; Lipman, J.; Cass, A.E.G.; Urban, G.A.; Dincer, C. On-site therapeutic drug monitoring. Trends Biotechnol. 2020, 38, 1262–1277. [Google Scholar] [CrossRef]
- Buclin, T.; Thoma, Y.; Widmer, N.; Andre, P.; Guidi, M.; Csajka, C.; Decosterd, L.A. The steps to therapeutic drug monitoring: A structured approach illustrated with imatinib. Front. Pharmacol. 2020, 11, 177. [Google Scholar] [CrossRef]
- Heysell, S.K.; Moore, J.L.; Keller, S.J.; Houpt, E.R. Therapeutic drug monitoring for slow response to tuberculosis treatment in a state control program, Virginia, USA. Emerg. Infect. Dis. 2010, 16, 1546–1553. [Google Scholar] [CrossRef]
- Babalik, A.; Babalik, A.; Mannix, S.; Francis, D.; Menzies, D. Therapeutic drug monitoring in the treatment of active tuberculosis. Can. Respir. J. 2011, 18, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Keller, F. Antibiotic therapy in patients with renal impairment. In Urogenital Infections and Inflammations; Naber, K.G., Ed.; German Medical Science GMS Publishing House: Duesseldorf, Germany, 2018. [Google Scholar]
- Keller, F.; Schroppel, B.; Ludwig, U. Pharmacokinetic and pharmacodynamic considerations of antimicrobial drug therapy in cancer patients with kidney dysfunction. World J. Nephrol. 2015, 4, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Kantasiripitak, W.; Van Daele, R.; Gijsen, M.; Ferrante, M.; Spriet, I.; Dreesen, E. Software tools for model-informed precision dosing: How well do they satisfy the needs? Front. Pharmacol. 2020, 11, 620. [Google Scholar] [CrossRef] [PubMed]
- Drennan, P.; Doogue, M.; van Hal, S.J.; Chin, P. Bayesian therapeutic drug monitoring software: Past, present and future. Int. J. Pharmacokinet. 2018, 3, 109–114. [Google Scholar] [CrossRef]
- Otalvaro, J.D.; Hernandez, A.M.; Rodriguez, C.A.; Zuluaga, A.F. Population pharmacokinetic models of antituberculosis drugs in patients: A systematic critical review. Ther. Drug Monit. 2021, 43, 108–115. [Google Scholar] [CrossRef]
- de Velde, F.; Mouton, J.W.; de Winter, B.C.M.; van Gelder, T.; Koch, B.C.P. Clinical applications of population pharmacokinetic models of antibiotics: Challenges and perspectives. Pharmacol. Res. 2018, 134, 280–288. [Google Scholar] [CrossRef]
- Cho, Y.S.; Jang, T.W.; Kim, H.J.; Oh, J.Y.; Lee, H.K.; Park, H.K.; Ghim, J.L.; Long, N.P.; Park, Y.; Choi, Y.K.; et al. Isoniazid population pharmacokinetics and dose recommendation for Korean patients with tuberculosis based on target attainment analysis. J. Clin. Pharmacol. 2021, 61, 1567–1578. [Google Scholar] [CrossRef]
- Fredj, N.B.; Romdhane, H.B.; Woillard, J.B.; Chickaid, M.; Fadhel, N.B.; Chadly, Z.; Chaabane, A.; Boughattas, N.; Aouam, K. Population pharmacokinetic model of isoniazid in patients with tuberculosis in Tunisia. Int. J. Infect. Dis. 2021, 104, 562–567. [Google Scholar] [CrossRef]
- Svensson, R.J.; Aarnoutse, R.E.; Diacon, A.H.; Dawson, R.; Gillespie, S.H.; Boeree, M.J.; Simonsson, U.S.H. A population pharmacokinetic model incorporating saturable pharmacokinetics and autoinduction for high rifampicin doses. Clin. Pharmacol. Ther. 2018, 103, 674–683. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.A.; Burgard, M.; Stacey, S.; Sandaradura, I.; Lai, T.; Coorey, C.; Cincunegui, M.; Staatz, C.E.; Hennig, S. An evaluation of the user-friendliness of Bayesian forecasting programs in a clinical setting. Br. J. Clin. Pharmacol. 2019, 85, 2436–2441. [Google Scholar] [CrossRef]
- Soedarsono, S.; Jayanti, R.P.; Mertaniasih, N.M.; Kusmiati, T.; Permatasari, A.; Indrawanto, D.W.; Charisma, A.N.; Yuliwulandari, R.; Long, N.P.; Choi, Y.K.; et al. Development of population pharmacokinetics model of isoniazid in indonesian tuberculosis patients. Int. J. Infect. Dis. 2022, 117, 8–14. [Google Scholar] [CrossRef]
- Huang, X.-h.; Wang, K.; Huang, J.-h.; Xu, L.; Li, L.-j.; Sheng, Y.-c.; Zheng, Q.-s. Random sparse sampling strategy using stochastic simulation and estimation for a population pharmacokinetic study. Saudi Pharm. J. 2014, 22, 63–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, J.H.; Tong, D.M.H.; Lucas, S.S.; Faldasz, J.D.; Goswami, S.; Keizer, R.J. Continuous learning in model-informed precision dosing: A case study in pediatric dosing of vancomycin. Clin. Pharmacol. Ther. 2021, 109, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Frymoyer, A.; Schwenk, H.T.; Zorn, Y.; Bio, L.; Moss, J.D.; Chasmawala, B.; Faulkenberry, J.; Goswami, S.; Keizer, R.J.; Ghaskari, S. Model-Informed precision dosing of vancomycin in hospitalized children: Implementation and Adoption at an Academic Children’s Hospital. Front. Pharmacol. 2020, 11, 551. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, S.; Bolhuis, M.S.; Sturkenboom, M.G.; Akkerman, O.W.; de Lange, W.C.; van der Werf, T.S.; Alffenaar, J.W. Incorporating therapeutic drug monitoring into the World Health Organization hierarchy of tuberculosis diagnostics. Eur. Respir. J. 2016, 47, 1867–1869. [Google Scholar] [CrossRef] [Green Version]
- Lange, C.; Alghamdi, W.A.; Al-Shaer, M.H.; Brighenti, S.; Diacon, A.H.; DiNardo, A.R.; Grobbel, H.P.; Groschel, M.I.; von Groote-Bidlingmaier, F.; Hauptmann, M.; et al. Perspectives for personalized therapy for patients with multidrug-resistant tuberculosis. J. Intern. Med. 2018, 284, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, G.; Alffenaar, J.W.; Centis, R.; D’Ambrosio, L.; Spanevello, A.; Piana, A.; Migliori, G.B. Therapeutic drug monitoring: How to improve drug dosage and patient safety in tuberculosis treatment. Int. J. Infect. Dis. 2015, 32, 101–104. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.S.; Lee, M.H. Overview of therapeutic drug monitoring. Korean J. Intern. Med. 2009, 24, 1–10. [Google Scholar] [CrossRef]
- Mota, L.; Al-Efraij, K.; Campbell, J.R.; Cook, V.J.; Marra, F.; Johnston, J. Therapeutic drug monitoring in anti-tuberculosis treatment: A systematic review and meta-analysis. Int. J. Tuberc. Lung Dis. 2016, 20, 819–826. [Google Scholar] [CrossRef]
- Peloquin, C.A. Therapeutic drug monitoring in the treatment of tuberculosis. Drugs 2002, 62, 2169–2183. [Google Scholar] [CrossRef]
- Jang, S.H.; Yan, Z.; Lazor, J.A. Therapeutic drug monitoring: A patient management tool for precision medicine. Clin. Pharmacol. Ther. 2016, 99, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Seo, K.A.; Kim, H.M.; Jeong, E.S.; Ghim, J.L.; Lee, S.H.; Lee, Y.M.; Kim, D.H.; Shin, J.G. Simple and accurate quantitative analysis of 20 anti-tuberculosis drugs in human plasma using liquid chromatography-electrospray ionization-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2015, 102, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Yin, L.; Le, X.; Chen, J.; Zhang, L.; Li, Y.; Lu, H.; Zhang, L. Simultaneous determination of first-line anti-tuberculosis drugs and one metabolite of isoniazid by liquid chromatography/tandem mass spectrometry in patients with human immunodeficiency virus-tuberculosis coinfection. Heliyon 2021, 7, e07532. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.J. Principles and applications of liquid chromatography-mass spectrometry in clinical biochemistry. Clin. Biochem. Rev. 2009, 30, 19–34. [Google Scholar]
- Dusthackeer, A.; Saadhali, S.A.; Thangam, M.; Hassan, S.; Balasubramanian, M.; Balasubramanian, A.; Ramachandran, G.; Kumar, A.K.H.; Thiruvenkadam, K.; Shanmugam, G.; et al. Wild-type MIC distribution for re-evaluating the critical concentration of anti-TB drugs and pharmacodynamics among tuberculosis patients from South India. Front. Microbiol. 2020, 11, 1182. [Google Scholar] [CrossRef]
- Horita, Y.; Alsultan, A.; Kwara, A.; Antwi, S.; Enimil, A.; Ortsin, A.; Dompreh, A.; Yang, H.; Wiesner, L.; Peloquin, C.A. Evaluation of the adequacy of WHO revised dosages of the first-line antituberculosis drugs in children with tuberculosis using population pharmacokinetic modeling and simulations. Antimicrob. Agents Chemother. 2018, 62, e00008-18. [Google Scholar] [CrossRef] [Green Version]
- Rao, P.S.; Moore, C.C.; Mbonde, A.A.; Nuwagira, E.; Orikiriza, P.; Nyehangane, D.; Al-Shaer, M.H.; Peloquin, C.A.; Gratz, J.; Pholwat, S.; et al. Population pharmacokinetics and significant under-dosing of anti-tuberculosis medications in people with HIV and critical illness. Antibiotics 2021, 10, 739. [Google Scholar] [CrossRef]
- Kayhan, S.; Akgüneş, A. Therapeutic monitoring of isoniazid, rifampicin, ethambutol and pyrazinamide serum levels in the treatment of active pulmonary tuberculosis and determinants of their serum concentrations. Afr. J. Pharm. Pharmacol. 2011, 5, 2035–2041. [Google Scholar] [CrossRef]
- Heysell, S.K.; Moore, J.L.; Staley, D.; Dodge, D.; Houpt, E.R. Early therapeutic drug monitoring for isoniazid and rifampin among diabetics with newly diagnosed tuberculosis in Virginia, USA. Tuberc. Res. Treat. 2013, 2013, 129723. [Google Scholar] [CrossRef] [Green Version]
- Mehta, J.B.; Shantaveerapa, H.; Byrd, R.P., Jr.; Morton, S.E.; Fountain, F.; Roy, T.M. Utility of rifampin blood levels in the treatment and follow-up of active pulmonary tuberculosis in patients who were slow to respond to routine directly observed therapy. Chest 2001, 120, 1520–1524. [Google Scholar] [CrossRef] [Green Version]
- Ray, J.; Gardiner, I.; Marriott, D. Managing antituberculosis drug therapy by therapeutic drug monitoring of rifampicin and isoniazid. Intern. Med. J. 2003, 33, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Heysell, S.K.; Moore, J.L.; Peloquin, C.A.; Ashkin, D.; Houpt, E.R. Outcomes and use of therapeutic drug monitoring in multidrug-resistant tuberculosis patients treated in virginia, 2009–2014. Tuberc. Respir. Dis. 2015, 78, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Prahl, J.B.; Johansen, I.S.; Cohen, A.S.; Frimodt-Moller, N.; Andersen, A.B. Clinical significance of 2 h plasma concentrations of first-line anti-tuberculosis drugs: A prospective observational study. J. Antimicrob. Chemother. 2014, 69, 2841–2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammi, S.; El Ataouna, K.; Bouti, K.; Elbouazzi, O.; Laine, M.; Bourkadi, J.E. Importance of therapeutic drug monitoring in the treatment of active tuberculosis—A retrospective study of 4 cases. Integr. J. Med. Sci. 2016, 3, 10–12. [Google Scholar] [CrossRef]
- Srinivas, N.R.; Syed, M. Applicability of a single time point strategy for the prediction of area under the concentration curve of linezolid in patients: Superiority of Ctrough- over Cmax-derived linear regression models. Drugs R D 2016, 16, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.S. Best practice in therapeutic drug monitoring. Br. J. Clin. Pharmacol. 2001, 52 (Suppl. 1), 5S–10S. [Google Scholar] [CrossRef]
- Capiau, S.; Veenhof, H.; Koster, R.A.; Bergqvist, Y.; Boettcher, M.; Halmingh, O.; Keevil, B.G.; Koch, B.C.P.; Linden, R.; Pistos, C.; et al. Official international association for therapeutic drug monitoring and clinical toxicology guideline: Development and validation of dried blood spot-based methods for therapeutic drug monitoring. Ther. Drug Monit. 2019, 41, 409–430. [Google Scholar] [CrossRef]
- Kiang, T.K.L.; Ranamukhaarachchi, S.A.; Ensom, M.H.H. Revolutionizing therapeutic drug monitoring with the use of interstitial fluid and microneedles technology. Pharmaceutics 2017, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- van den Elsen, S.H.J.; Oostenbrink, L.M.; Heysell, S.K.; Hira, D.; Touw, D.J.; Akkerman, O.W.; Bolhuis, M.S.; Alffenaar, J.C. Systematic review of salivary versus blood concentrations of antituberculosis drugs and their potential for salivary therapeutic drug monitoring. Ther. Drug Monit. 2018, 40, 17–37. [Google Scholar] [CrossRef] [Green Version]
- Sayers, J.; Friedman, M. How clinicians use therapeutic drug monitoring. Lab. Med. 1997, 28, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Petersen, E.; Al-Abri, S.; Chakaya, J.; Goletti, D.; Parolina, L.; Wejse, C.; Mucheleng’anga, L.A.; Khalili, S.A.; Yeboah-Manu, D.; Chanda-Kapata, P.; et al. World TB Day 2022: Revamping and reshaping global TB control programs by advancing lessons learnt from the COVID-19 pandemic. Int. J. Infect. Dis. 2022, 4. [Google Scholar] [CrossRef]
- Kim, E.S.; Kwon, B.S.; Park, J.S.; Chung, J.Y.; Seo, S.H.; Park, K.U.; Song, J.; Yoon, S.; Lee, J.H. Relationship among genetic polymorphism of SLCO1B1, rifampicin exposure and clinical outcomes in patients with active pulmonary tuberculosis. Br. J. Clin. Pharmacol. 2021, 87, 3492–3500. [Google Scholar] [CrossRef]
- Ferreira, A.; Lapa, R.; Vale, N. PBPK modeling and simulation and therapeutic drug monitoring: Possible ways for antibiotic dose adjustment. Processes 2021, 9, 2087. [Google Scholar] [CrossRef]
- Perry, C.; Davis, G.; Conner, T.M.; Zhang, T. Utilization of physiologically based pharmacokinetic modeling in clinical pharmacology and therapeutics: An overview. Curr. Pharmacol. Rep. 2020, 6, 71–84. [Google Scholar] [CrossRef]
- Jones, H.; Rowland-Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e63. [Google Scholar] [CrossRef]
- Upton, R.N.; Foster, D.J.; Abuhelwa, A.Y. An introduction to physiologically-based pharmacokinetic models. Paediatr. Anaesth. 2016, 26, 1036–1046. [Google Scholar] [CrossRef]
- Cordes, H.; Thiel, C.; Aschmann, H.E.; Baier, V.; Blank, L.M.; Kuepfer, L. A physiologically based pharmacokinetic model of isoniazid and its application in individualizing tuberculosis chemotherapy. Antimicrob. Agents Chemother. 2016, 60, 6134–6145. [Google Scholar] [CrossRef] [Green Version]
- Wills, K.H.; Behan, S.J.; Nance, M.J.; Dawson, J.L.; Polasek, T.M.; Hopkins, A.M.; van Dyk, M.; Rowland, A. Combining therapeutic drug monitoring and pharmacokinetic modelling deconvolutes physiological and environmental sources of variability in clozapine exposure. Pharmaceutics 2021, 14, 47. [Google Scholar] [CrossRef]
- Zapke, S.E.; Willmann, S.; Grebe, S.O.; Menke, K.; Thurmann, P.A.; Schmiedl, S. Comparing predictions of a PBPK model for cyclosporine with drug levels from therapeutic drug monitoring. Front. Pharmacol. 2021, 12, 630904. [Google Scholar] [CrossRef]
- Broeker, A.; Nardecchia, M.; Klinker, K.P.; Derendorf, H.; Day, R.O.; Marriott, D.J.; Carland, J.E.; Stocker, S.L.; Wicha, S.G. Towards precision dosing of vancomycin: A systematic evaluation of pharmacometric models for Bayesian forecasting. Clin. Microbiol. Infect. 2019, 25, 1286.e1281–1286.e1287. [Google Scholar] [CrossRef]
- de-Madaria, E.; Mira, J.J.; Carrillo, I.; Afif, W.; Ang, D.; Antelo, M.; Bollipo, S.; Castells, A.; Chahal, P.; Heinrich, H.; et al. The present and future of gastroenterology and hepatology: An international SWOT analysis (the GASTROSWOT project). Lancet Gastroenterol. Hepatol. 2022, 7, 485–494. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Relevance | Anti-TB Drug | Evidence |
|---|---|---|
| Inadequate drug levels may lead to a delay in culture conversion and treatment failure | RIF | Current standard dose of RIF has shown inadequate levels of RIF and may contribute to the treatment failure and relapse, high dose RIF has been evaluated and showed promising results for shortening the treatment duration and obtaining early bacterial conversion [28,33]. |
| PZA | Low concentration of PZA with a standard dose was associated with the delayed culture conversion, even though the DOTs had been implemented [34]. | |
| Low drug levels may acquire drug resistance | INH | NAT2 rapid acetylator has a faster clearance rate of INH from the liver, therefore reducing the plasma concentration and exposure of INH and eventually decreased sputum conversion rates and poorer microbiological outcomes [35,36]. Patients with rapid acetylator can mostly be found in patients with drug-resistant TB [37]. |
| RIF | Low exposure of RIF during the initial phase of therapy may put INH under monotherapy, which will eventually emerge as drug resistance [32]. | |
| High drug levels may cause adverse events | LZD | A previous study from China found that Cmin of LZD was significantly higher in the patients with thrombocytopenia (Cmin = 8.81 mg/L, p < 0.0001) [38]. |
| Another study from Taiwan reported that the Cmin and AUC0–24 h of LZD in patients with thrombocytopenia were significantly higher (Cmin = 13 mg/L and AUC0–24 h = 451 mg·h/L) [39]. | ||
| PZA | Pyrazinoic acid, as an active metabolite of PZA, increases serum uric acid based on its trans-stimulatory effect on URAT1, causing the reabsorption of urate from the luminal side into tubular cells and eventually hyperuricemia [40,41]. | |
| The accumulated metabolite concentrations of pyrazinoic acid and 5-hydroxy-pyrazinoic acid have been linked to the PZA-induced liver injury [42]. | ||
| INH | Although it remains arguable, high concentrations of INH also may increase the risk of drug-induced liver injury in slow acetylator patients due to slow clearance rate of INH from liver [43,44]. |
| Author | Country | Study Design | Population Characteristics | Cases (n) | Drugs Measured | TDM Results | Conclusion |
|---|---|---|---|---|---|---|---|
| Heysell et al. (2010) [51] | USA | Retrospective cohort | DS-TB, 42 slow response patients 269 normal patients | 311 | RIF: 600mg INH: 300 mg PZA and EMB: weight based daily dose. | Median C2hr [IQR], μg/mL INH: 1.9 (1.1–3.5) RIF: 7.4 (2.5–11.4) PZA: 2.5 (1.7–3.2) EMB: 28.1 (26.5–33.2) Proportion of patients with low C2hr, (lower limit of therapeutic range, μg/mL): INH: 33% (<3) RIF: 33% (<8) PZA: 0% (<20) EMB: 31% (<2) | Subtherapeutic concentrations of RIF, INH, and EMB were frequently observed, dosage adjustment for INH and RIF from 300 mg and 600 mg daily to 450 mg and 900 mg daily. For intermittent INH interval, the dose was increased from 900 mg to 1200 mg. DM was associated with slow response and low RIF concentrations. Patients with TDM have 2 months shorter therapy. |
| Babalik et al. (2011) [52] | Canada | Retrospective case-control | DS-TB, 20 cases (TDM done) 20 controls (no TDM) 8 with HIV (all cases) | 40 | INH: 5 mg/kg, max 300 mg RIF: 10 mg/kg, max 600 mg PZA: 20 mg/kg EMB: 15 mg/kg RFB: 0.8 ± 0.3 mg/kg | Mean C2hr ± SD, (μg/mL) INH: 2.0 ± 1.3 RIF: 9.1 ± 4 RFB: 0.2 ± 0.1 PZA: 32.9 ± 11.3 Proportion of patients with low C2hr, (lower limit of therapeutic range, μg/mL): INH: 87% (<3) RIF: 67% (<8) RFB: 89% (<0.3) PZA: 15% (<20) | Subtherapeutic concentrations of RIF, RFB, and INH were frequently observed. Mean dosage adjustment ± SD, (mg/kg): INH: 8.1 ± 1.8 RIF: 13.5 ± 1.7 RFB: 2.5 ± 0.9 PZA: 25.5 ± 12.6 Low concentration was found mostly in HIV patients. Subtherapeutic concentrations associated with longer therapy duration. |
| Kayhan et al. (2011) [80] | Turkey | Prospective observational cohort | DS-TB, patients excluded: HIV and DM | 49 | INH: 300 mg RIF: 600 mg PZA: 1500 mg or 2000 mg (weight adjusted) EMB: 1000 or 1500 mg (weight adjusted) | Mean C2hr ± SD, (μg/mL) INH: 3.83 ± 2.09 RIF: 6.13 ± 4.27 PZA: 32.2 ± 16.96 EMB: 3.68 ± 2.41 Proportion of patients with low C2hr, (lower limit of therapeutic range, μg/mL): INH: 29% (<3) RIF: 74% (<8) PZA: 20% (<20) EMB: 18% (<2) | Subtherapeutic concentrations of RIF and INH were frequently observed, dosage adjustment was performed in low serum drug concentrations. |
| Magis-Escurra et al. (2012) [18] | Netherlands | Retrospective case series | Relapse TB, delayed converter | 4 | RIF, INH, PZA, EMB (doses were not described clearly) | Patient 1 C2h, (lower limit of therapeutic range, μg/mL): RIF: 5.6 (<8) INH: <0.025 (<3) PZA: 8.3 (<20) Patient 2 C2h, (lower limit of therapeutic range, μg/mL): RIF: 4.1 (<8) Patient 3 C2h, (lower limit of therapeutic range, μg/mL): RIF: 4.0 (<8) PZA: 10.0 (<20) Patient 4 C2h, (lower limit of therapeutic range, μg/mL): RIF: 2.3 (<8) | Subtherapeutic concentration of RIF associated with delayed conversion, dosage adjustment of: Patient 1: RIF from 600 mg to 1200 mg INH and PZA also were adjusted. Patient 2: RIF from 600 mg to 1200 mg Patient 3: INH from 200 to 250 mg, RIF from 450 to 600 mg, PZA from 1250 to 2000 mg Patient 4: RIF from 600 to 900 mg (not enough to reach the target concentration) then increased to 1200 mg The dose adjustment for all patients improved the treatment outcomes and increased the C2h of RIF to achieve therapeutic target. |
| Heysell et al. (2013) [81] | USA | Retrospective cohort | TB-DM: 21 patients TB-slow responders: 14 patients | 35 | RIF: 600 mg INH: 300 mg INH (intermittent): 900 mg | Mean C2hr ± SD, (μg/mL): Daily INHDM: 2.0 ± 1.3 INHslow: 3.1 ± 1.1 RIFDM: 6.6 ± 4.3 RIFslow: 8.2 ± 6.2 Intermittent INHDM: 6.0 ± 3.0 INHslow: 11.3 ± 2.5 Proportion of patients with low C2hr, (lower limit of therapeutic range, μg/mL): Daily INHDM: 65% (<3) INHslow: 63% (<3) RIFDM: 60% (<8) RIFslow: 41% (<8) Intermittent INHDM: 75% (<9) INHslow: 17% (<9) | Subtherapeutic concentrations of RIF and INH were frequently observed, dosage adjustment for INH and RIF from 300 mg and 600 mg daily to 450 mg and 900 mg daily. For intermittent INH dose from 900 mg was increased to 1200 mg. DM is associated with subtherapeutic concentration of RIF and INH. Early dose correction using TDM decreased the number of slow responders. |
| Mehta et al. (2001) [82] | USA | Retrospective case series | DS-TB, slow response to treatment HIV: 1 patient | 6 | RIF: 600 mg INH: 300 mg PZA: 25 mg/kg EMB: 25 mg/kg | Patient 1 C2h, (lower limit of therapeutic range, μg/mL): RIF: 1.5 (<8) Patient 2 C2h, (lower limit of therapeutic range, μg/mL): RIF: 5.9 (<8) Patient 3 C2h, (lower limit of therapeutic range, μg/mL): RIF: <1.0 (<8) Patient 4 C2h, (lower limit of therapeutic range, μg/mL): RIF: <1.0 (<8) Patient 5 C2h, (lower limit of therapeutic range, μg/mL): RIF: <1.0 (<8) Patient 6 C2h, (lower limit of therapeutic range, μg/mL): RIF: 3.54 (<8) | Subtherapeutic concentrations of RIF were observed in all patients, dosage adjustment was performed from 600 mg to 900 mg, one patient adjusted to 1500 mg (Patient 4). Dose adjustment improved the response of the patients. |
| Ray et al. (2003) [83] | Australia | Prospective cohort | DS-TB | 90 | RIF: 150, 300, 450, 600 and 750 mg, daily or 3 times weekly INH: 150, 200, 300, 350, 400, 450, 500, 600, and 750 mg daily or 3 divided-dose, weekly | Mean C2hr ± SD, (μmol/L) INH: 11.1 ± 7 RIF: 28.5 ± 20.4 Proportion of patients with low C2hr, (lower limit of therapeutic range, μmol/L): INH: 46% (<22) RIF: 48% (<10) Proportion of patients with high C2hr, (upper limit of therapeutic range, μmol/L): INH: 29% (>37) RIF: 2% (>29) | High concentration of INH related to ADR and low concentration related to therapeutic failure. A case report of a slow converter that needed dosage adjustments of INH and RIF from 300 mg and 450 mg to 400 mg and 600 mg was presented. Sputum smear was improved after dose adjustment. |
| Heysell et al. (2015) [84] | USA | Retrospective cohort | MDR-TB, DM: 1 patient HIV: 1 patient | 10 | CAP: 15 mg/kg dose (maximum 1 g) MFX: 400 mg daily CS: 250 mg daily LZD: 400–600 mg AMK, PAS, EMB, PZA, ETA (doses were not described clearly for these drugs) | Mean C2hr ± SD, (μg/mL): Daily CAP: 21.5 ± 14.0 AMK: 35.3 ± 3.7 MFX: 3.2 ± 1.5 CS: 16.6 ± 10.2 PAS: 65.0 ± 29.1 (C6hr) LZD: 11.4 ± 4.1 EMB: 1.8 ± 1.85 PZA: 39.9 ± 1.8 ETA: 1.5 Proportion of patients with low C2hr, (lower limit of therapeutic range, μg/mL): CAP: 60% (<35) AMK: 50% (< 35) MFX: 20% (<3) CS: 57% (<20) PAS: 0 LZD: 33% (<12) EMB: 33% (<2) PZA: 0% (<20) ETA: 0% (<1) | Subtherapeutic concentrations were frequently observed in CAP, AMK, and CS. The doses were adjusted in CAP, MFX, CS, LZD, EMB (increased), and PZA (decreased). The outcome resulted in patients being cured or clinically improved. |
| Prahl et al. (2014) [85] | Denmark | Prospective observational study | DS-TB, HIV: 2 patients | 32 | INH: 5 mg/kg, max 300 mg RIF: 10 mg/kg, max 600 mg PZA: 30 mg/kg, max 2000 mg EMB: 20 mg/kg, max 1200 mg | Median C2hr (range), μg/mL INH: 2.1 (0.5–12.1) RIF: 6.5 (0–31) PZA: 31.3 (14.9–110.2) EMB: 2.2 (0.5–5.9) Proportion of patients with low C2hr, (lower limit of therapeutic range, μg/mL): INH: 71% (<3) RIF: 58% (<8) PZA: 10% (<20) EMB: 46% (<2) | Subtherapeutic concentrations of RIF and INH were frequently observed, dosage adjustment for the low concentration drugs. Low INH and RIF C2hr associated with poor outcome. |
| Hammi et al. (2016) [86] | Morocco | Retrospective case series | DS-TB, Delayed converter | 4 | Patient 1: RIF: 600 mg INH: 300 mg PZA: 1600 mg EMB: 1100 mg Patient 2: RIF: 450 mg INH: 225 mg PZA: 1200 mg EMB: 825 mg Patient 3: RIF: 450 mg INH: 225 mg PZA: 1200 mg EMB: 825 mg Patient 4: Unknown doses of RIF/INH/PZA/EMB | Patient 1 C2h, (lower limit of therapeutic range, μg/mL): RIF: 2.9 (<8) Patient 2 C2h, (lower limit of therapeutic range, μg/mL): RIF: 4.8 (<8) Patient 3 C2h, (lower limit of therapeutic range, μg/mL): RIF: 3.85 (<8) INH: 0.59 (<3) Patient 4 C2h, (lower limit of therapeutic range, μg/mL): RIF: 1.79 (<8) | Subtherapeutic concentration of RIF associated with delayed conversion, dosage adjustment of: Patient 1: RIF from 600 mg to 855 mg. Patient 2: RIF from 450 mg to 750 mg. Patient 3: INH from 225 to 300 mg, RIF from 450 to 600 mg, Patient 4: RIF dose was increased. The dose adjustment for all patients improved the treatment outcomes and increased C2h of RIF to achieve therapeutic target. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jayanti, R.P.; Long, N.P.; Phat, N.K.; Cho, Y.-S.; Shin, J.-G. Semi-Automated Therapeutic Drug Monitoring as a Pillar toward Personalized Medicine for Tuberculosis Management. Pharmaceutics 2022, 14, 990. https://doi.org/10.3390/pharmaceutics14050990
Jayanti RP, Long NP, Phat NK, Cho Y-S, Shin J-G. Semi-Automated Therapeutic Drug Monitoring as a Pillar toward Personalized Medicine for Tuberculosis Management. Pharmaceutics. 2022; 14(5):990. https://doi.org/10.3390/pharmaceutics14050990
Chicago/Turabian StyleJayanti, Rannissa Puspita, Nguyen Phuoc Long, Nguyen Ky Phat, Yong-Soon Cho, and Jae-Gook Shin. 2022. "Semi-Automated Therapeutic Drug Monitoring as a Pillar toward Personalized Medicine for Tuberculosis Management" Pharmaceutics 14, no. 5: 990. https://doi.org/10.3390/pharmaceutics14050990