
Benchmarking the Solubility Enhancement and Storage Stability of Amorphous Drug–Polyelectrolyte Nanoplex against Co-Amorphous Formulation of the Same Drug


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
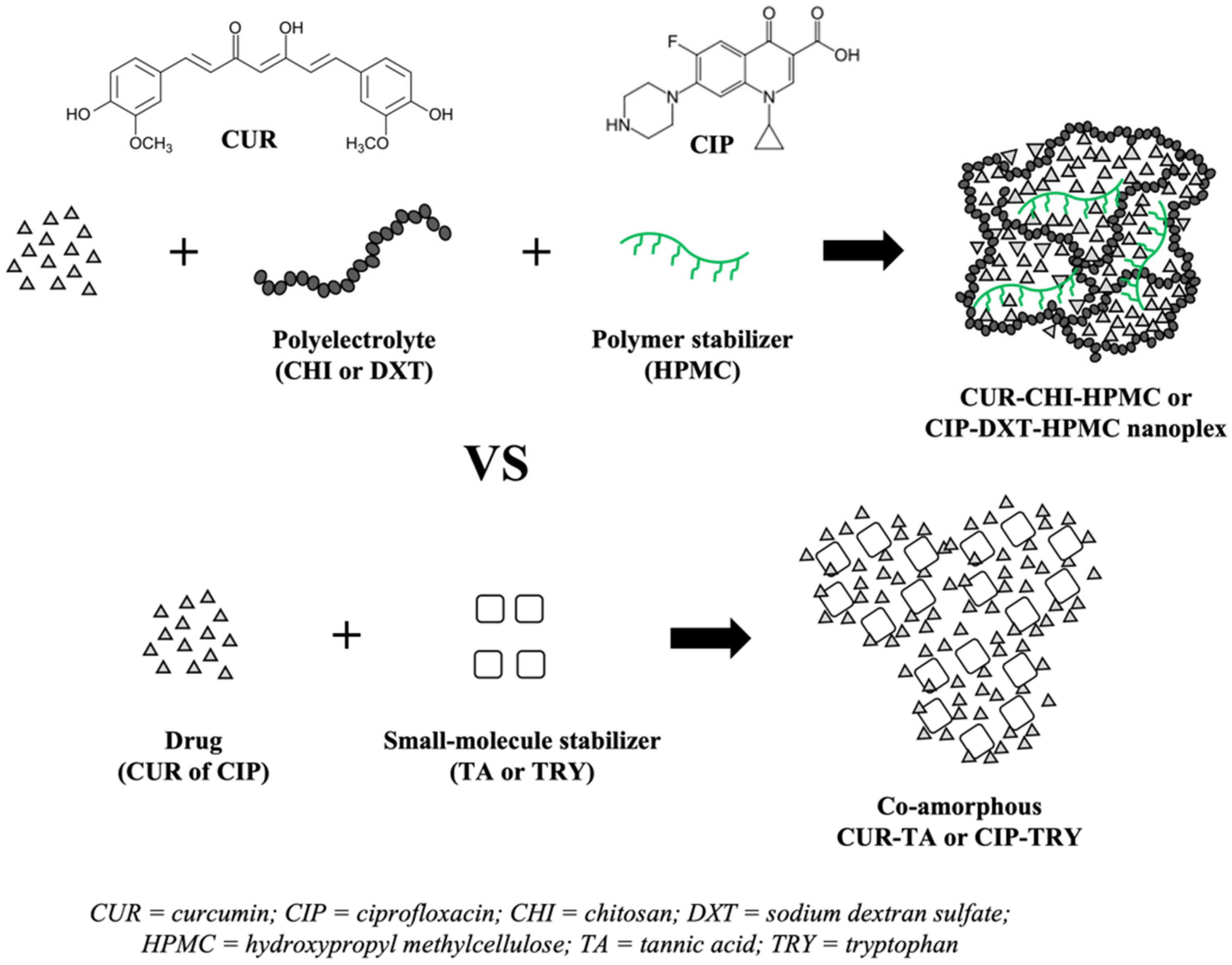
2.2.1. Preparation of CUR-CHI-HMPC and CIP-DXT-HPMC Nanoplexes
2.2.2. Preparation of Co-Amorphous CUR-TA and CIP-TRY
2.2.3. Preparation Efficiency
2.2.4. Physical Characterizations
2.2.5. Dissolution Profile under Sink Condition
2.2.6. Supersaturation Generation
2.2.7. Accelerated Storage Stability
3. Results and Discussion
3.1. Benchmarking Nanoplex against CAM System
3.1.1. Preparation Efficiency
3.1.2. Drug Payload and Morphology
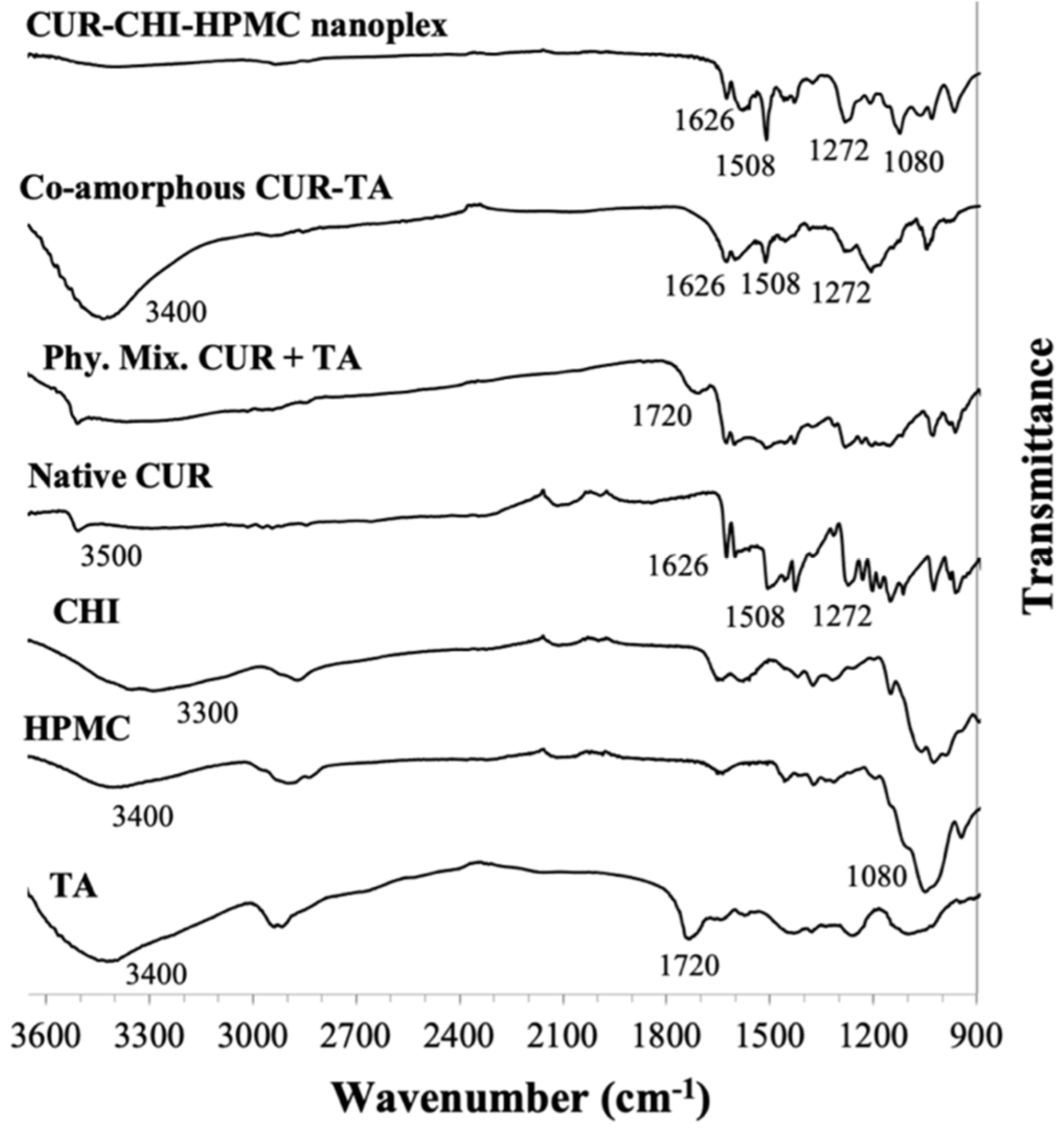
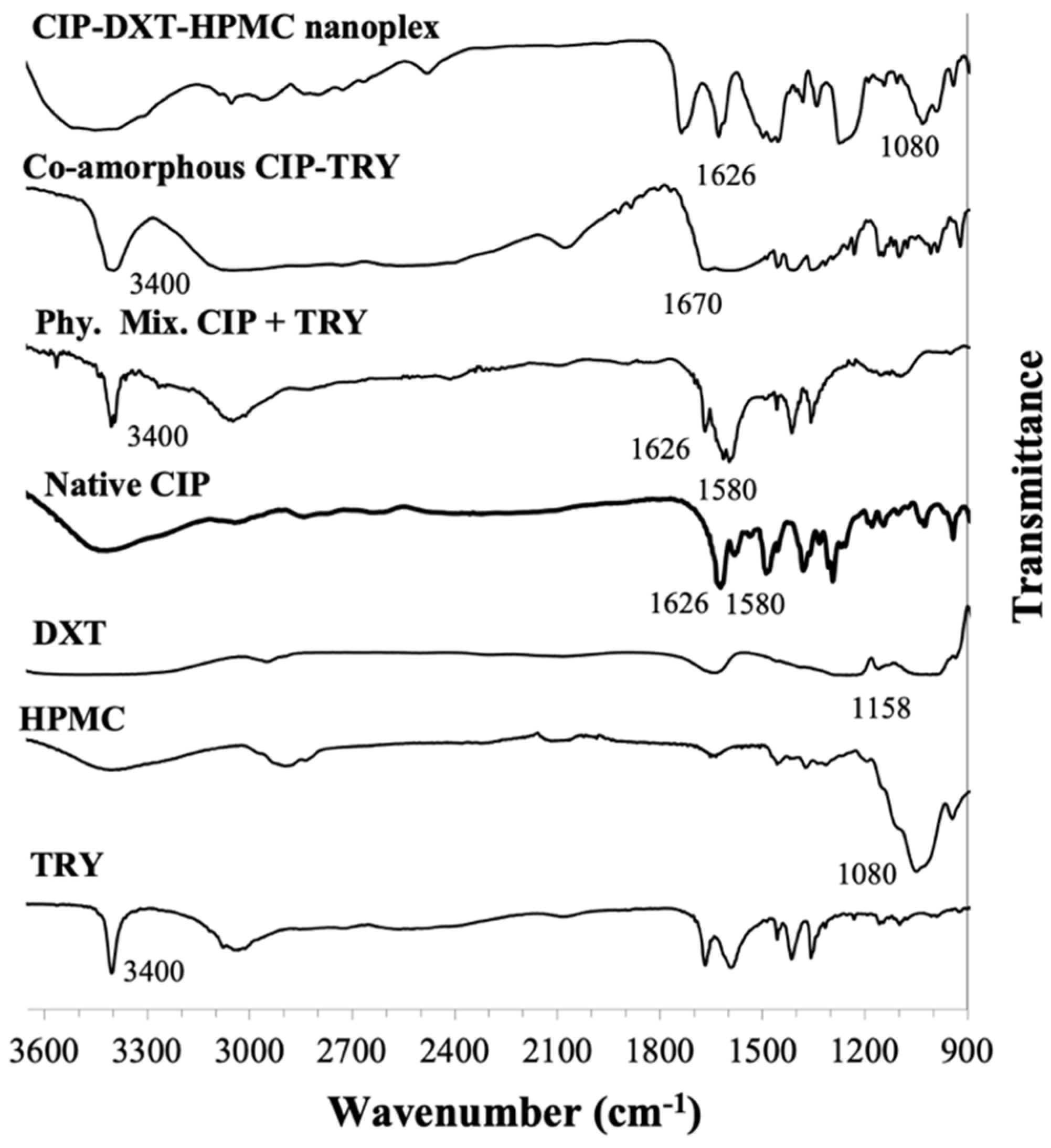
3.1.3. Species Interaction by FTIR
CUR Amorphous Systems
CIP Amorphous Systems
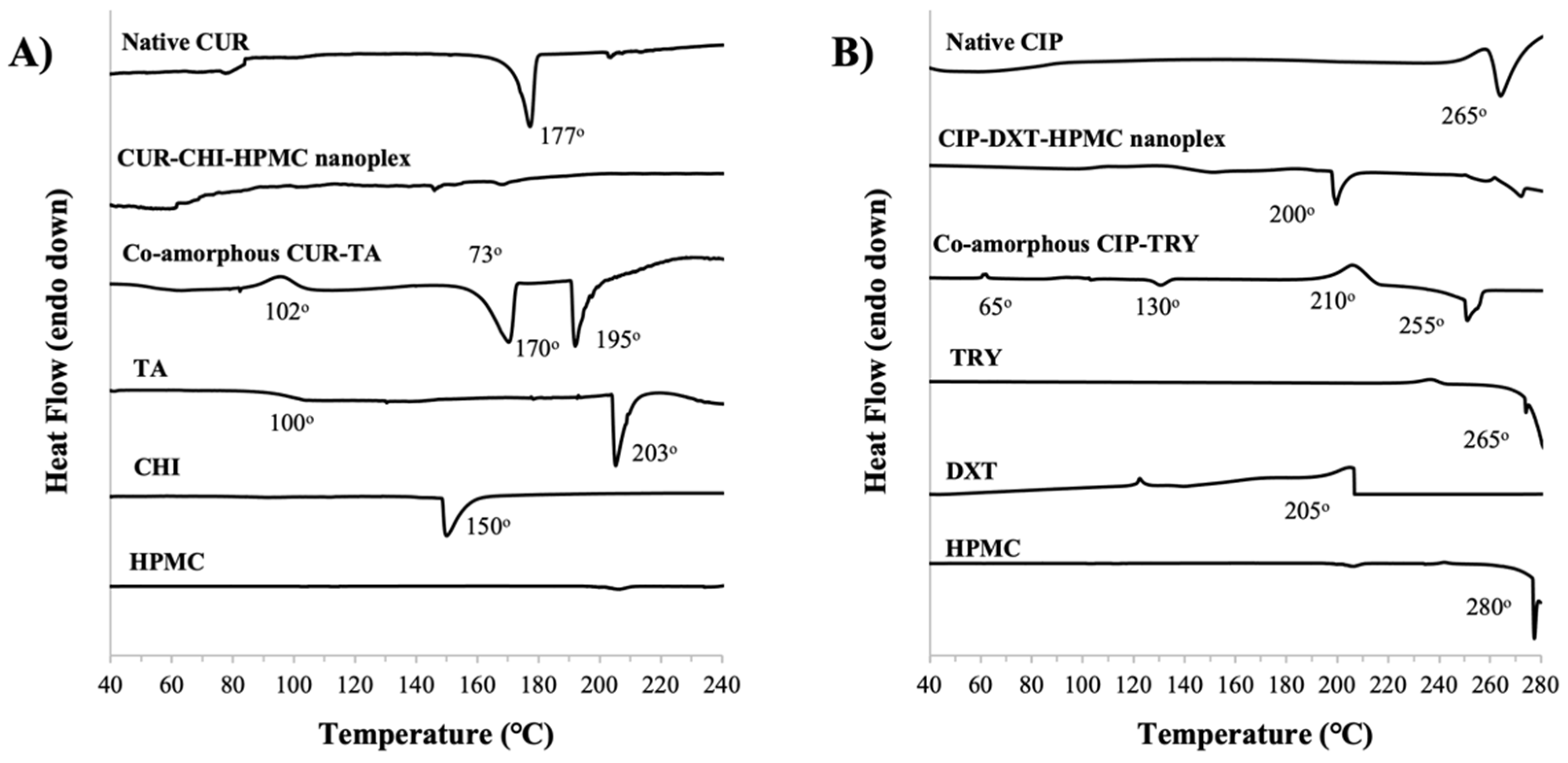
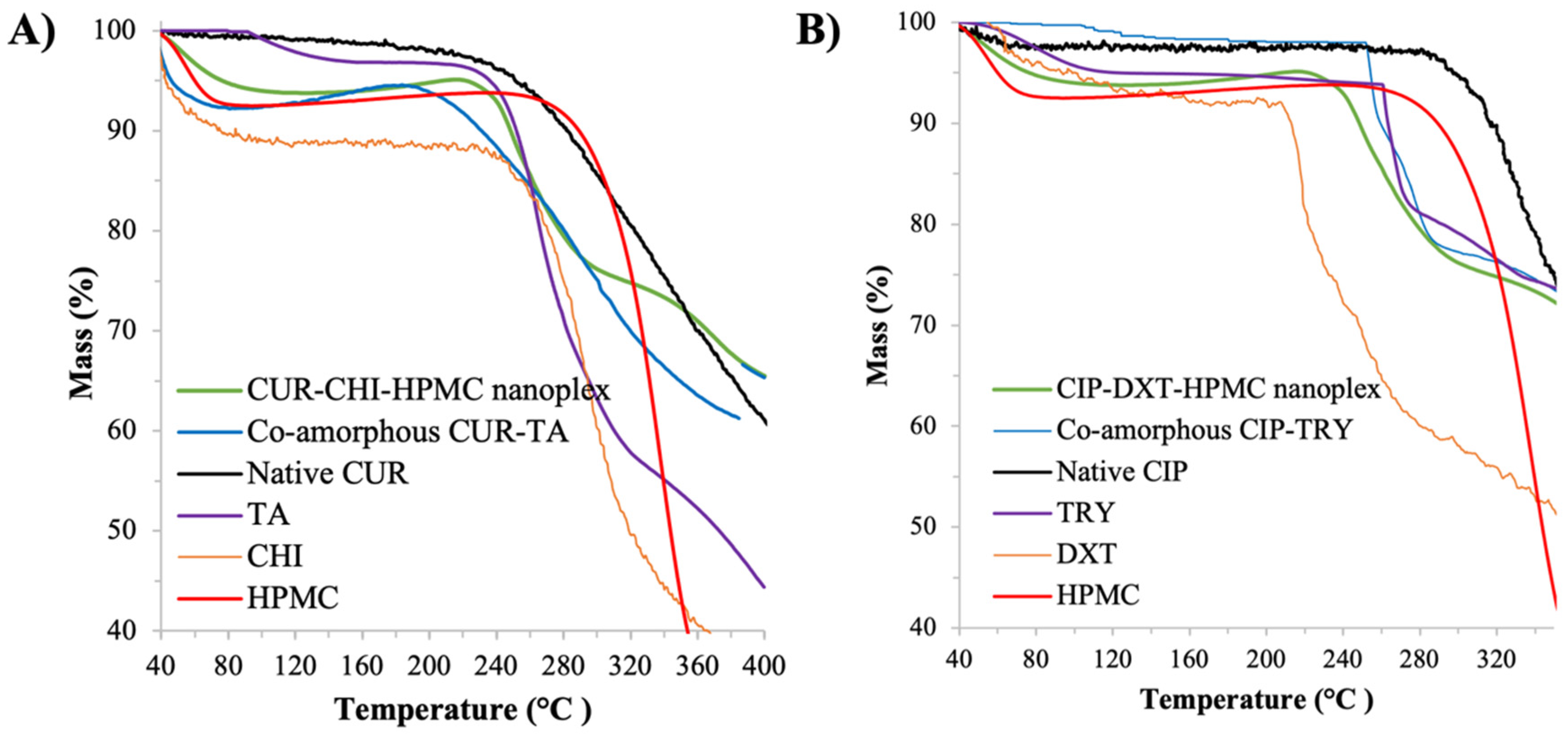
3.1.4. Thermal Stability by TGA and DSC
CUR Amorphous Systems
CIP Amorphous Systems
3.2. Nanoplex vs. CAM’s Solubility Enhancement
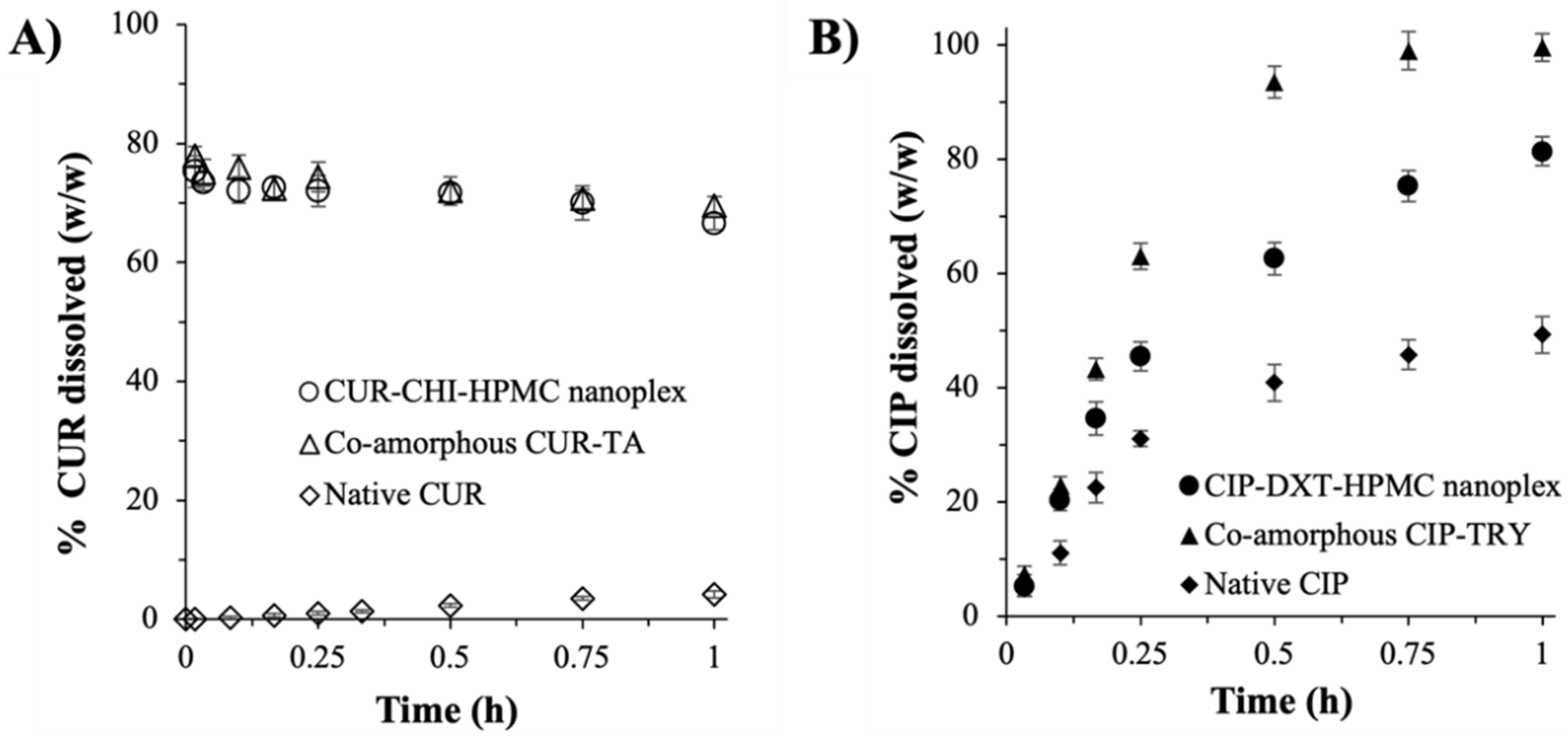
3.2.1. Dissolution Rate
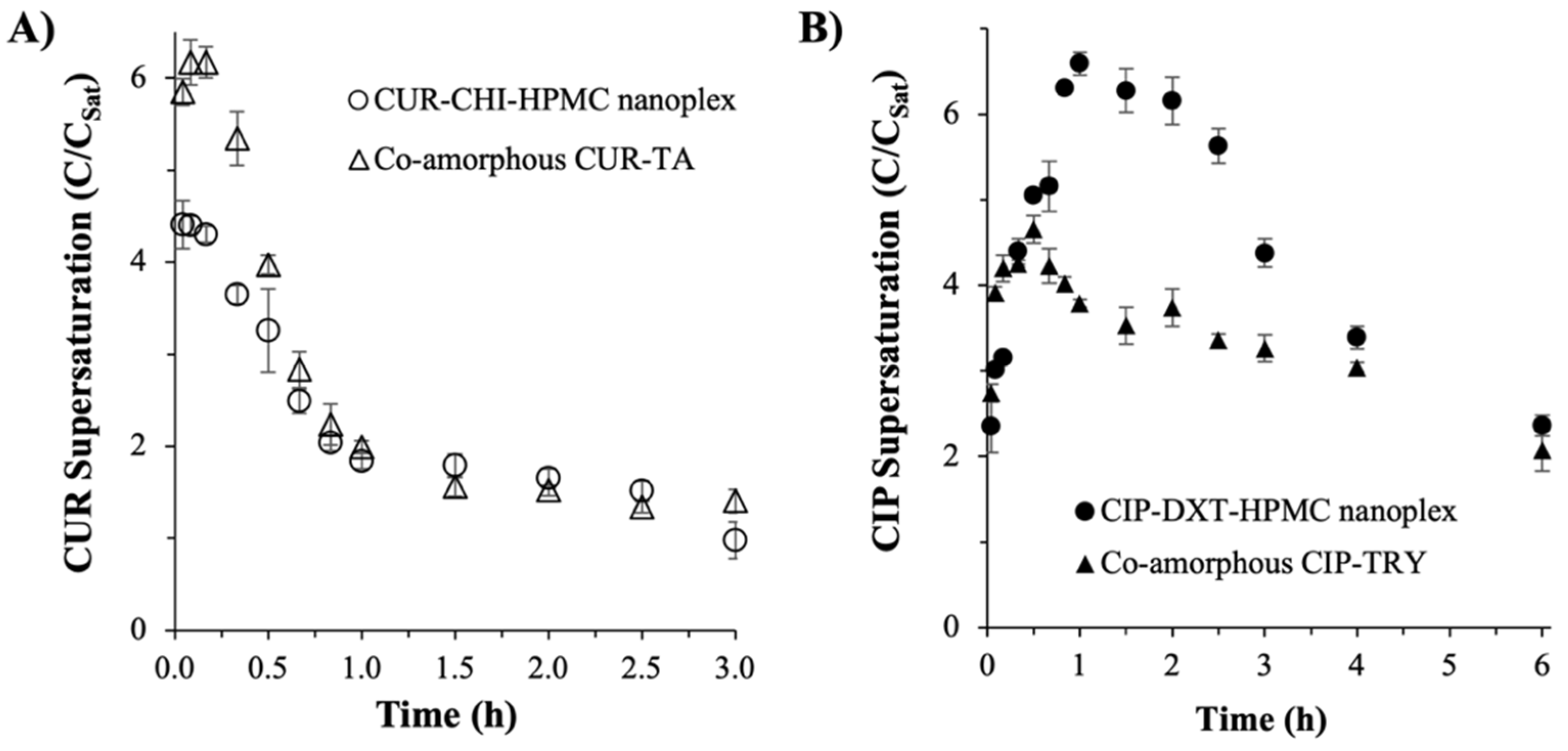
3.2.2. Supersaturation Generation
3.3. Nanoplex vs. CAM’s Storage Stability
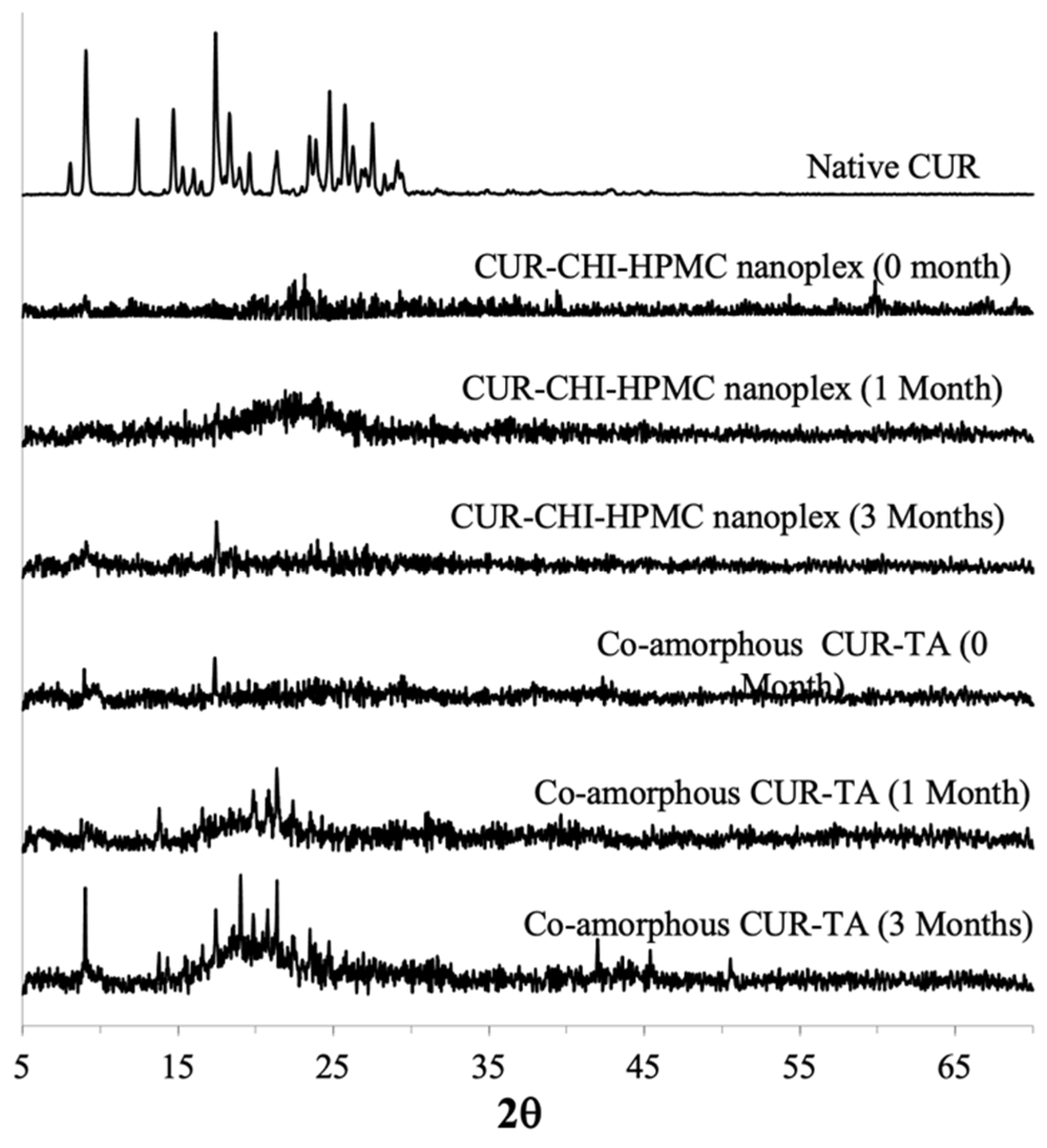
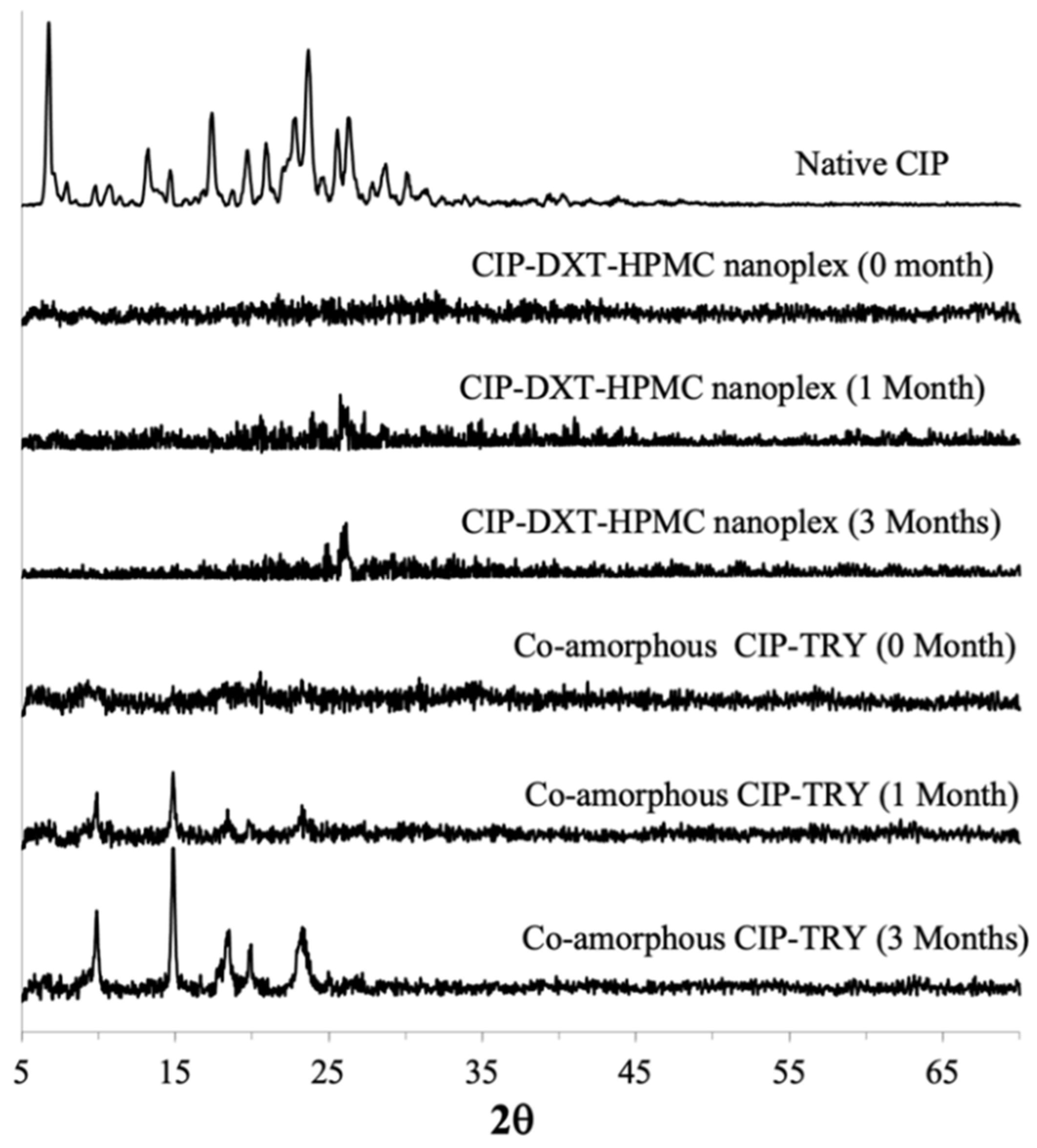
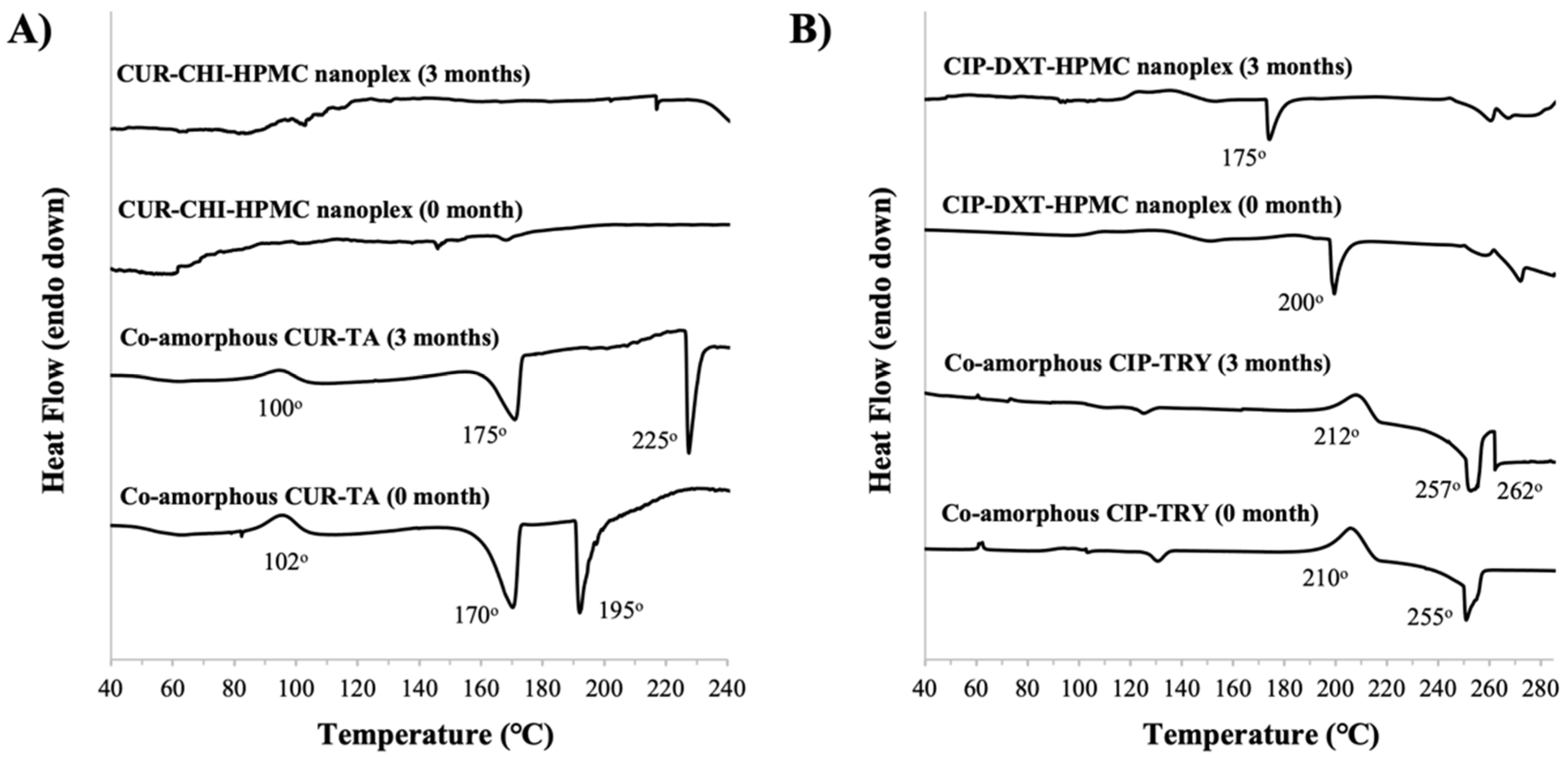
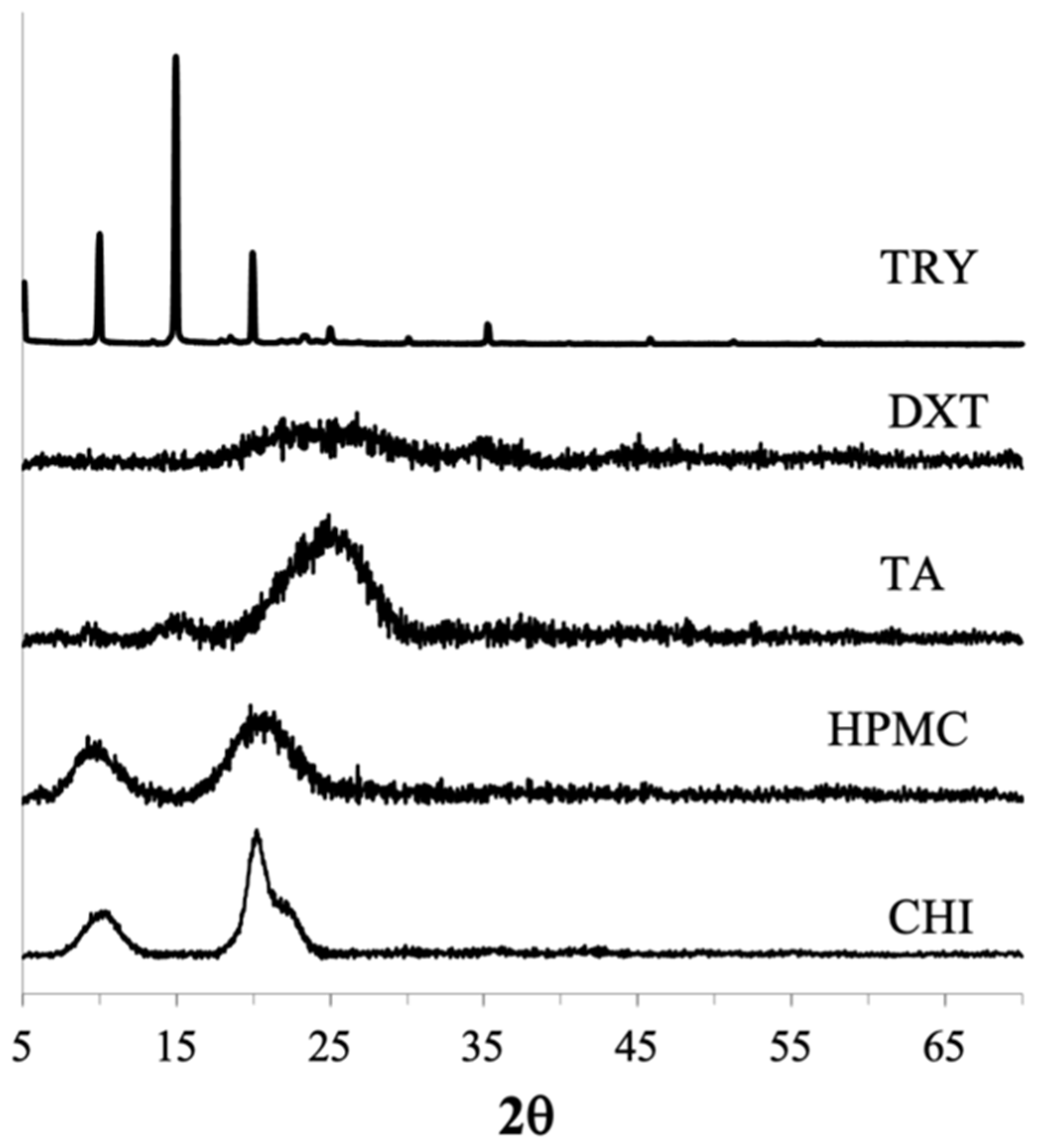
3.3.1. PXRD and DSC
CUR Amorphous Systems
CIP Amorphous Systems
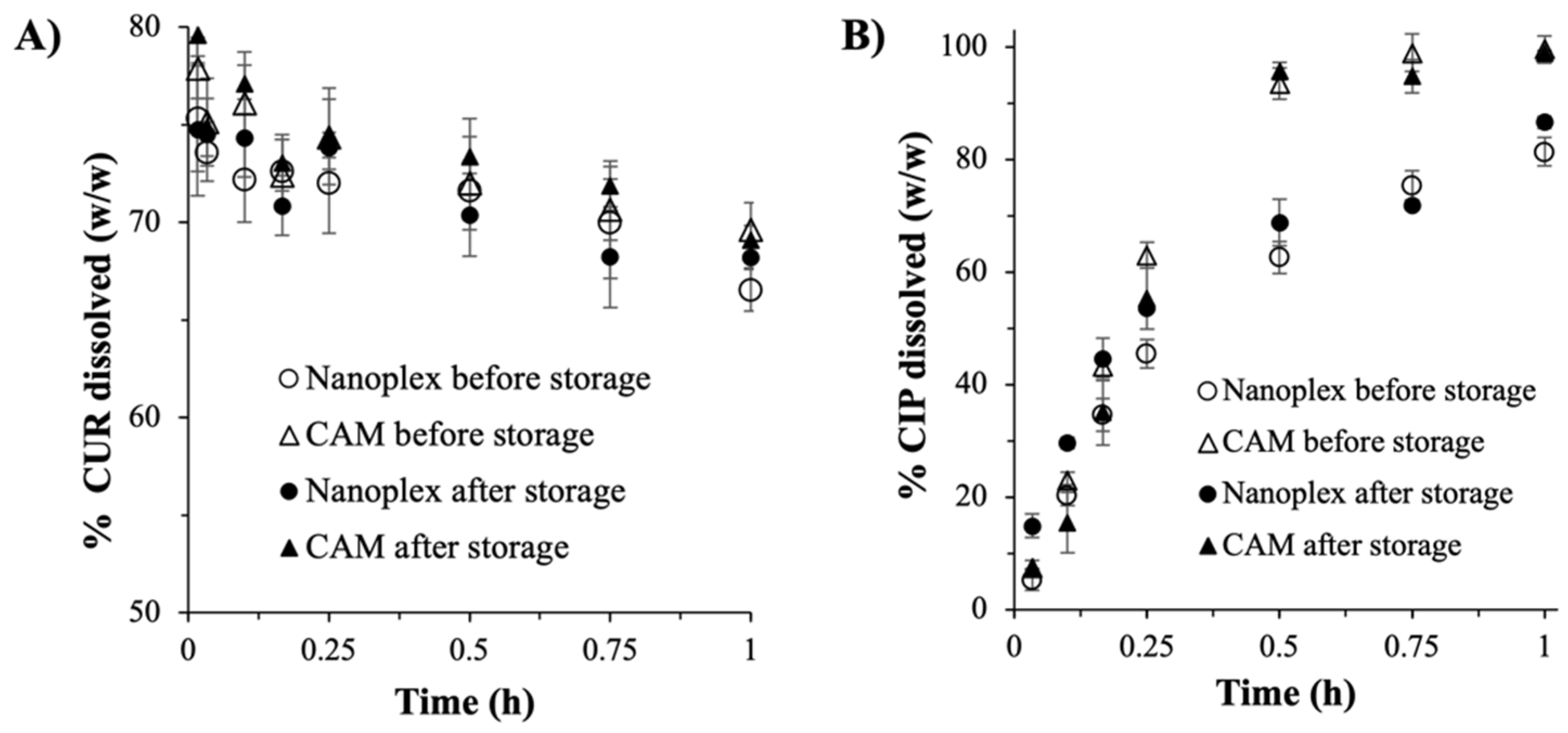
3.3.2. Drug Payload and Dissolution
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A





References
- Di, L.; Fish, P.V.; Mano, T. Bridging solubility between drug discovery and development. Drug Discov. Today 2012, 17, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, Y.; Tin, Y.-Y.; Soe, M.-T.-P.; Ko, B.; Park, S.; Lee, J. Recent Technologies for Amorphization of Poorly Water-Soluble Drugs. Pharmaceutics 2021, 13, 1318. [Google Scholar] [CrossRef] [PubMed]
- Cheow, W.S.; Kiew, T.Y.; Hadinoto, K. Combining inkjet printing and amorphous nanonization to prepare personalized dosage forms of poorly-soluble drugs. Eur. J. Pharm. Biopharm. 2015, 96, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Mendonsa, N.; Almutairy, B.; Kallakunta, V.R.; Sarabu, S.; Thipsay, P.; Bandari, S.; Repka, M.A. Manufacturing strategies to develop amorphous solid dispersions: An overview. J. Drug Deliv. Sci. Technol. 2020, 55, 101459. [Google Scholar] [CrossRef]
- Lin, X.; Hu, Y.; Liu, L.; Su, L.; Li, N.; Yu, J.; Tang, B.; Yang, Z. Physical Stability of Amorphous Solid Dispersions: A Physicochemical Perspective with Thermodynamic, Kinetic and Environmental Aspects. Pharm. Res. 2018, 35, 125. [Google Scholar] [CrossRef]
- Ricarte, R.G.; Van Zee, N.J.; Li, Z.; Johnson, L.M.; Lodge, T.P.; Hillmyer, M.A. Recent Advances in Understanding the Micro- and Nanoscale Phenomena of Amorphous Solid Dispersions. Mol. Pharm. 2019, 16, 4089–4103. [Google Scholar] [CrossRef]
- Qiang, W.; Löbmann, K.; McCoy, C.P.; Andrews, G.P.; Zhao, M. Microwave-Induced In Situ Amorphization: A New Strategy for Tackling the Stability Issue of Amorphous Solid Dispersions. Pharmaceutics 2020, 12, 655. [Google Scholar] [CrossRef]
- Bhujbal, S.V.; Mitra, B.; Jain, U.; Gong, Y.; Agrawal, A.; Karki, S.; Taylor, L.S.; Kumar, S.; Zhou, Q. Pharmaceutical amorphous solid dispersion: A review of manufacturing strategies. Acta Pharm. Sin. B 2021, 11, 2505–2536. [Google Scholar] [CrossRef]
- Han, J.; Wei, Y.; Lu, Y.; Wang, R.; Zhang, J.; Gao, Y.; Qian, S. Co-amorphous systems for the delivery of poorly water-soluble drugs: Recent advances and an update. Expert Opin. Drug Deliv. 2020, 17, 1411–1435. [Google Scholar] [CrossRef]
- Liu, J.; Grohganz, H.; Rades, T. Influence of polymer addition on the amorphization, dissolution and physical stability of co-amorphous systems. Int. J. Pharm. 2020, 588, 119768. [Google Scholar] [CrossRef]
- Hanada, M.; Jermain, S.V.; Thompson, S.A.; Furuta, H.; Fukuda, M.; Williams, R.O. Ternary Amorphous Solid Dispersions Containing a High-Viscosity Polymer and Mesoporous Silica Enhance Dissolution Performance. Mol. Pharm. 2021, 18, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Grohganz, H.; Löbmann, K.; Rades, T.; Hempel, N.-J. Co-Amorphous Drug Formulations in Numbers: Recent Advances in Co-Amorphous Drug Formulations with Focus on Co-Formability, Molar Ratio, Preparation Methods, Physical Stability, In Vitro and In Vivo Performance, and New Formulation Strategies. Pharmaceutics 2021, 13, 389. [Google Scholar] [CrossRef] [PubMed]
- Karagianni, A.; Kachrimanis, K.; Nikolakakis, I. Co-Amorphous Solid Dispersions for Solubility and Absorption Improvement of Drugs: Composition, Preparation, Characterization and Formulations for Oral Delivery. Pharmaceutics 2018, 10, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korhonen, O.; Pajula, K.; Laitinen, R. Rational excipient selection for co-amorphous formulations. Expert Opin. Drug Deliv. 2017, 14, 551–569. [Google Scholar] [CrossRef]
- Jog, R.; Burgess, D.J. Pharmaceutical Amorphous Nanoparticles. J. Pharm. Sci. 2017, 106, 39–65. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Teo, J.; Chew, J.W.; Hadinoto, K. Dry powder inhaler formulation of high-payload antibiotic nanoparticle complex intended for bronchiectasis therapy: Spray drying versus spray freeze drying preparation. Int. J. Pharm. 2016, 499, 38–46. [Google Scholar] [CrossRef]
- Dong, B.; Lim, L.M.; Hadinoto, K. Enhancing the physical stability and supersaturation generation of amorphous drug-polyelectrolyte nanoparticle complex via incorporation of crystallization inhibitor at the nanoparticle formation step: A case of HPMC versus PVP. Eur. J. Pharm. Sci. 2019, 138, 105035. [Google Scholar] [CrossRef]
- Lim, L.M.; Tran, T.T.; Long Wong, J.J.; Wang, D.; Cheow, W.S.; Hadinoto, K. Amorphous ternary nanoparticle complex of curcumin-chitosan-hypromellose exhibiting built-in solubility enhancement and physical stability of curcumin. Colloids Surf. B Biointerfaces 2018, 167, 483–491. [Google Scholar] [CrossRef]
- Pardeshi, C.V.; Belgamwar, V.S. Ropinirole-dextran sulfate nanoplex for nasal administration against Parkinson’s disease: In silico molecular modeling and in vitro-ex vivo evaluation. Artif. Cells Nanomed. Biotechnol. 2017, 45, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Yousefpour, P.; Atyabi, F.; Farahani, E.V.; Sakhtianchi, R.; Dinarvand, R. Polyanionic carbohydrate doxorubicin-dextran nanocomplex as a delivery system for anticancer drugs: In vitro analysis and evaluations. Int. J. Nanomed. 2011, 6, 1487–1496. [Google Scholar]
- Abioye, A.O.; Kola-Mustapha, A. Controlled Electrostatic Self-Assembly of Ibuprofen-Cationic Dextran Nanoconjugates Prepared by low Energy Green Process—A Novel Delivery Tool for Poorly Soluble Drugs. Pharm. Res. 2015, 32, 2110–2131. [Google Scholar] [CrossRef]
- Stoyanova, N.; Ignatova, M.; Manolova, N.; Rashkov, I.; Toshkova, R.; Georgieva, A. Nanoparticles based on complex of berberine chloride and polymethacrylic or polyacrylic acid with antioxidant and in vitro antitumor activities. Int. J. Pharm. 2020, 584, 119426. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Jo, Y.U.; Shin, H.; Lee, J.; Chae, S.U.; Bae, S.K.; Na, K. Anthocyanin-fucoidan nanocomplex for preventing carcinogen induced cancer: Enhanced absorption and stability. Int. J. Pharm. 2020, 586, 119597. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, M.J.; Dehpour, A.R.; Attar, F.; Hasan, A.; Mohammad-Sadeghi, N.; Meratan, A.A.; Aziz, F.M.; Salihi, A.; Shekha, M.S.; Akhtari, K.; et al. Silymarin-albumin nanoplex: Preparation and its potential application as an antioxidant in nervous system in vitro and in vivo. Int. J. Pharm. 2019, 572, 118824. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.H.; Lee, S.E.; Tran, T.T.; Bui, C.B.; Nguyen, T.H.; Vu, N.B.; Tran, T.T.; Nguyen, T.H.; Nguyen, T.T.; Hadinoto, K. A simple strategy to enhance the in vivo wound-healing activity of curcumin in the form of self-assembled nanoparticle complex of curcumin and oligochitosan. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 98, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Hassan, D.; Omolo, C.A.; Gannimani, R.; Waddad, A.Y.; Mocktar, C.; Rambharose, S.; Agrawal, N.; Govender, T. Delivery of novel vancomycin nanoplexes for combating methicillin resistant Staphylococcus aureus (MRSA) infections. Int. J. Pharm. 2019, 558, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.M.; Tran, T.-T.; Cheow, W.S.; Hadinoto, K. Re-evaluating the presumed superiority of amorphous nanoparticles over amorphous microscale solid dispersion in solubility enhancement of poorly soluble drugs. Eur. J. Pharm. Sci. 2017, 109, 455–463. [Google Scholar] [CrossRef]
- Wong, J.J.L.; Yu, H.; Lim, L.M.; Hadinoto, K. A trade-off between solubility enhancement and physical stability upon simultaneous amorphization and nanonization of curcumin in comparison to amorphization alone. Eur. J. Pharm. Sci. 2018, 114, 356–363. [Google Scholar] [CrossRef]
- Cheow, W.S.; Hadinoto, K. Green Amorphous Nanoplex as a New Supersaturating Drug Delivery System. Langmuir 2012, 28, 6265–6275. [Google Scholar] [CrossRef]
- Hou, H.H.; Rajesh, A.; Pandya, K.M.; Lubach, J.W.; Muliadi, A.; Yost, E.; Jia, W.; Nagapudi, K. Impact of Method of Preparation of Amorphous Solid Dispersions on Mechanical Properties: Comparison of Coprecipitation and Spray Drying. J. Pharm. Sci. 2019, 108, 870–879. [Google Scholar] [CrossRef]
- Szabó, E.; Záhonyi, P.; Brecska, D.; Galata, D.L.; Mészáros, L.A.; Madarász, L.; Csorba, K.; Vass, P.; Hirsch, E.; Szafraniec-Szczęsny, J.; et al. Comparison of Amorphous Solid Dispersions of Spironolactone Prepared by Spray Drying and Electrospinning: The Influence of the Preparation Method on the Dissolution Properties. Mol. Pharm. 2021, 18, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Craye, G.; Löbmann, K.; Grohganz, H.; Rades, T.; Laitinen, R. Characterization of Amorphous and Co-Amorphous Simvastatin Formulations Prepared by Spray Drying. Molecules 2015, 20, 21532–21548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, X.; Tang, H.; Mao, H.-Q. Effective encapsulation of curcumin in nanoparticles enabled by hydrogen bonding using flash nanocomplexation. Int. J. Pharm. 2019, 564, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Gao, H.; Babu, S.; Garad, S. Co-Amorphous Formation of High-Dose Zwitterionic Compounds with Amino Acids To Improve Solubility and Enable Parenteral Delivery. Mol. Pharm. 2018, 15, 97–107. [Google Scholar] [CrossRef]
- Leung, M.H.M.; Colangelo, H.; Kee, T.W. Encapsulation of curcumin in cationic micelles suppresses alkaline hydrolysis. Langmuir ACS J. Surf. Colloids 2008, 24 11, 5672–5675. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, L.; Yang, D.; Zhang, N.; He, L.; Du, G.; Lu, Y. Salt screening and characterization of ciprofloxacin. Acta Crystallogr B Struct Sci Cryst Eng Mater. 2016, 72 Pt 1, 20–28. [Google Scholar] [CrossRef]
- Naksuriya, O.; van Steenbergen, M.J.; Torano, J.S.; Okonogi, S.; Hennink, W.E. A Kinetic Degradation Study of Curcumin in Its Free Form and Loaded in Polymeric Micelles. Aaps J. 2016, 18, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Grimm, W. Extension of the International Conference on Harmonization Tripartite Guideline for Stability Testing of New Drug Substances and Products to countries of climatic zones III and IV. Drug Dev. Ind Pharm. 1998, 24, 313–325. [Google Scholar] [CrossRef]
- Que, C.; Lou, X.; Zemlyanov, D.Y.; Mo, H.; Indulkar, A.S.; Gao, Y.; Zhang, G.G.Z.; Taylor, L.S. Insights into the Dissolution Behavior of Ledipasvir–Copovidone Amorphous Solid Dispersions: Role of Drug Loading and Intermolecular Interactions. Mol. Pharm. 2019, 16, 5054–5067. [Google Scholar] [CrossRef]
- Oćwieja, M.; Adamczyk, Z.; Morga, M. Adsorption of tannic acid on polyelectrolyte monolayers determined in situ by streaming potential measurements. J. Colloid Interface Sci. 2015, 438, 249–258. [Google Scholar] [CrossRef]
- Kharat, M.; Du, Z.; Zhang, G.; McClements, D.J. Physical and Chemical Stability of Curcumin in Aqueous Solutions and Emulsions: Impact of pH, Temperature, and Molecular Environment. J. Agric. Food Chem. 2017, 65, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Faiz Afzal, M.A.; Lehmkemper, K.; Sobich, E.; Hughes, T.F.; Giesen, D.J.; Zhang, T.; Krauter, C.M.; Winget, P.; Degenhardt, M.; Kyeremateng, S.O.; et al. Molecular-Level Examination of Amorphous Solid Dispersion Dissolution. Mol. Pharm. 2021, 18, 3999–4014. [Google Scholar] [CrossRef] [PubMed]
- Yani, Y.; Kanaujia, P.; Chow, P.S.; Tan, R.B.H. Effect of API-Polymer Miscibility and Interaction on the Stabilization of Amorphous Solid Dispersion: A Molecular Simulation Study. Ind. Eng. Chem. Res. 2017, 56, 12698–12707. [Google Scholar] [CrossRef]
- Mugheirbi, N.A.; Mosquera-Giraldo, L.I.; Borca, C.H.; Slipchenko, L.V.; Taylor, L.S. Phase Behavior of Drug-Hydroxypropyl Methylcellulose Amorphous Solid Dispersions Produced from Various Solvent Systems: Mechanistic Understanding of the Role of Polymer using Experimental and Theoretical Methods. Mol. Pharm. 2018, 15, 3236–3251. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.D.; Lee, P.I. Evolution of Supersaturation of Amorphous Pharmaceuticals: The Effect of Rate of Supersaturation Generation. Mol. Pharm. 2013, 10, 4330–4346. [Google Scholar] [CrossRef]
- Moseson, D.E.; Parker, A.S.; Beaudoin, S.P.; Taylor, L.S. Amorphous solid dispersions containing residual crystallinity: Influence of seed properties and polymer adsorption on dissolution performance. Eur. J. Pharm. Sci. 2020, 146, 105276. [Google Scholar] [CrossRef]
- Han, J.; Li, L.; Su, M.; Heng, W.; Wei, Y.; Gao, Y.; Qian, S. Deaggregation and Crystallization Inhibition by Small Amount of Polymer Addition for a Co-Amorphous Curcumin-Magnolol System. Pharmaceutics 2021, 13, 1725. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | CUR–CHI–HPMC Nanoplex | Co-Amorphous CUR-TA | CIP–DXT–HPMC Nanoplex | Co-Amorphous CIP–TRY |
|---|---|---|---|---|
| Drug utilization (% w/w) | 84 ± 2 | 39 ± 4 | 81 ± 5 | 94 ± 1 |
| Yield (% w/w) | 46 ± 1 | 34 ± 3 | 48 ± 3 | 91 ± 3 |
| Drug payload (% w/w) | 55 ± 1 | 49 ± 4 | 59 ± 4 | 51 ± 2 |
| Size (nm) | 326 ± 31 | 331 ± 76 | 298 ± 12 | 1790 ± 430 |
| PDI | 0.22 ± 0.06 | 0.34 ± 0.06 | 0.36 ± 0.03 | NA |
| Zeta potential (mV) | 32.6 ± 1.1 | −36.7 ± 1.4 | −32.1 ± 0.8 | NA |
| Drug Payload (% w/w) | CUR–CHI–HPMC Nanoplex | Co-Amorphous CUR–TA | CIP–DXT–HPMC Nanoplex | Co-Amorphous CIP–TRY |
|---|---|---|---|---|
| Before storage | 55 ± 1 | 49 ± 4 | 59 ± 4 | 51 ± 2 |
| After storage | 56 ± 4 | 53 ± 6 | 56 ± 5 | 47 ± 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, L.M.; Park, J.-W.; Hadinoto, K. Benchmarking the Solubility Enhancement and Storage Stability of Amorphous Drug–Polyelectrolyte Nanoplex against Co-Amorphous Formulation of the Same Drug. Pharmaceutics 2022, 14, 979. https://doi.org/10.3390/pharmaceutics14050979
Lim LM, Park J-W, Hadinoto K. Benchmarking the Solubility Enhancement and Storage Stability of Amorphous Drug–Polyelectrolyte Nanoplex against Co-Amorphous Formulation of the Same Drug. Pharmaceutics. 2022; 14(5):979. https://doi.org/10.3390/pharmaceutics14050979
Chicago/Turabian StyleLim, Li Ming, Jin-Won Park, and Kunn Hadinoto. 2022. "Benchmarking the Solubility Enhancement and Storage Stability of Amorphous Drug–Polyelectrolyte Nanoplex against Co-Amorphous Formulation of the Same Drug" Pharmaceutics 14, no. 5: 979. https://doi.org/10.3390/pharmaceutics14050979