
Toxicity Assessment of Mesoporous Silica Nanoparticles upon Intravenous Injection in Mice: Implications for Drug Delivery

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
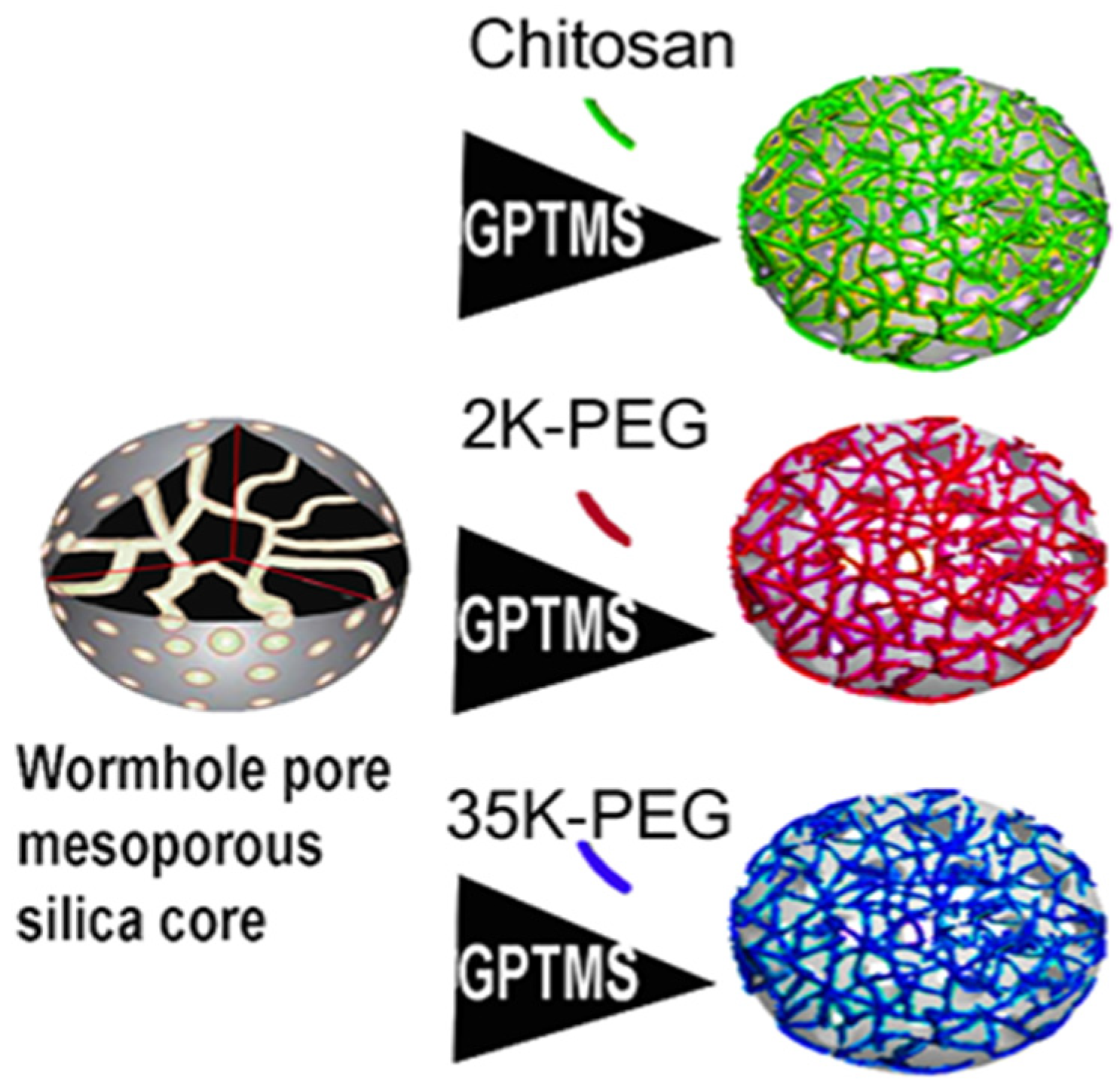
2.2. Nanoparticle Synthesis
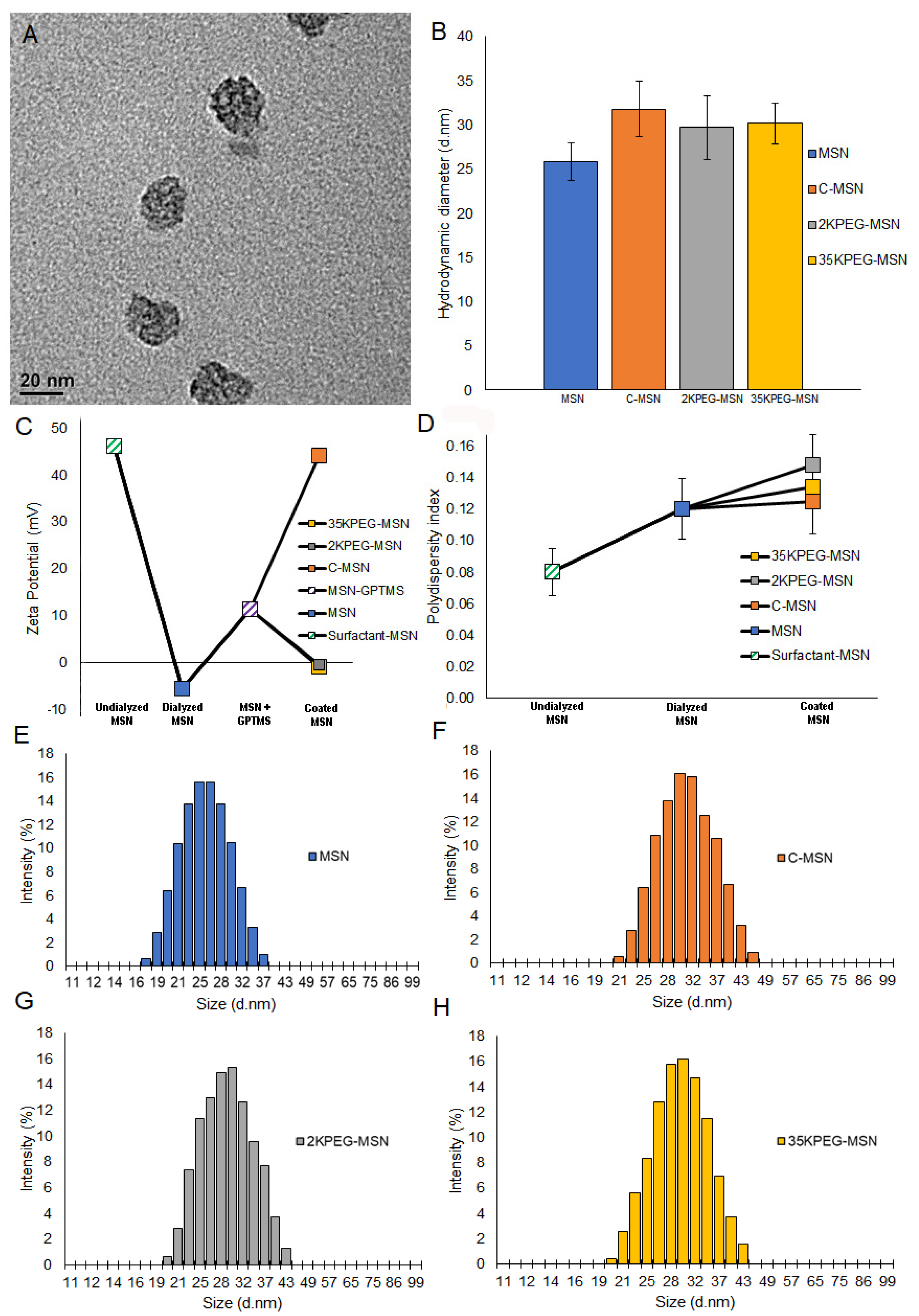
2.3. Nanoparticle Characterization
2.4. Nanoparticle Functionalization
2.5. Functionalized Nanoparticle Characterization
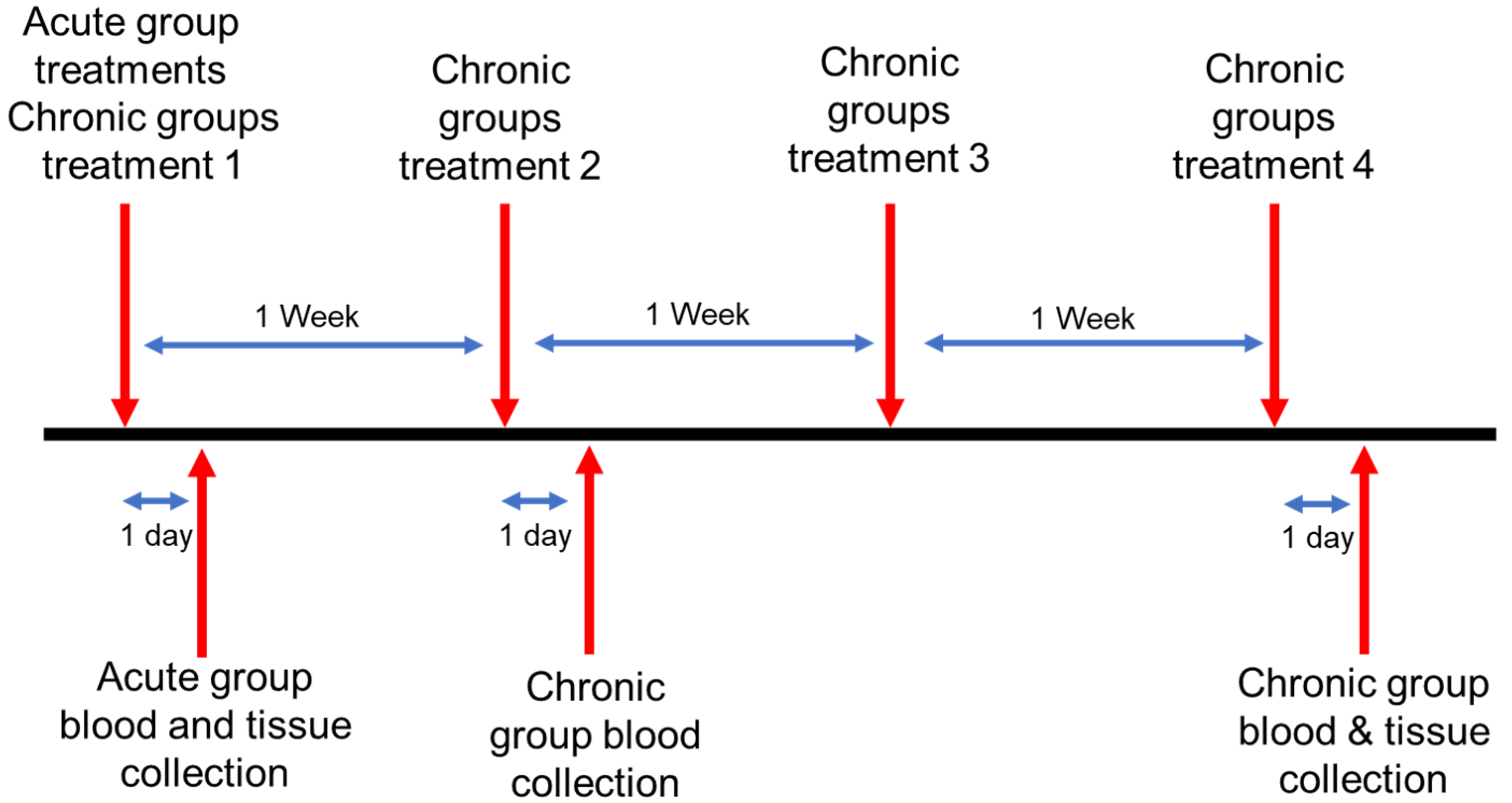
2.6. In Vivo Nanoparticle Toxicity
- Controls (C) n = 3.
- Mesoporous silica nanoparticles with no coating (MSNs) n = 6.
- Mesoporous silica nanoparticles coated with chitosan (C-MSNs) n = 6.
- Mesoporous silica nanoparticles coated with 2 kDa polypropylene glycol (2KPEG-MSNs) n = 6.
- Mesoporous silica nanoparticles coated with 35 kDa polypropylene glycol (35KPEG-MSNs) n = 6.
2.7. Complete Blood Count and Chemistry Panels
2.8. Statistics
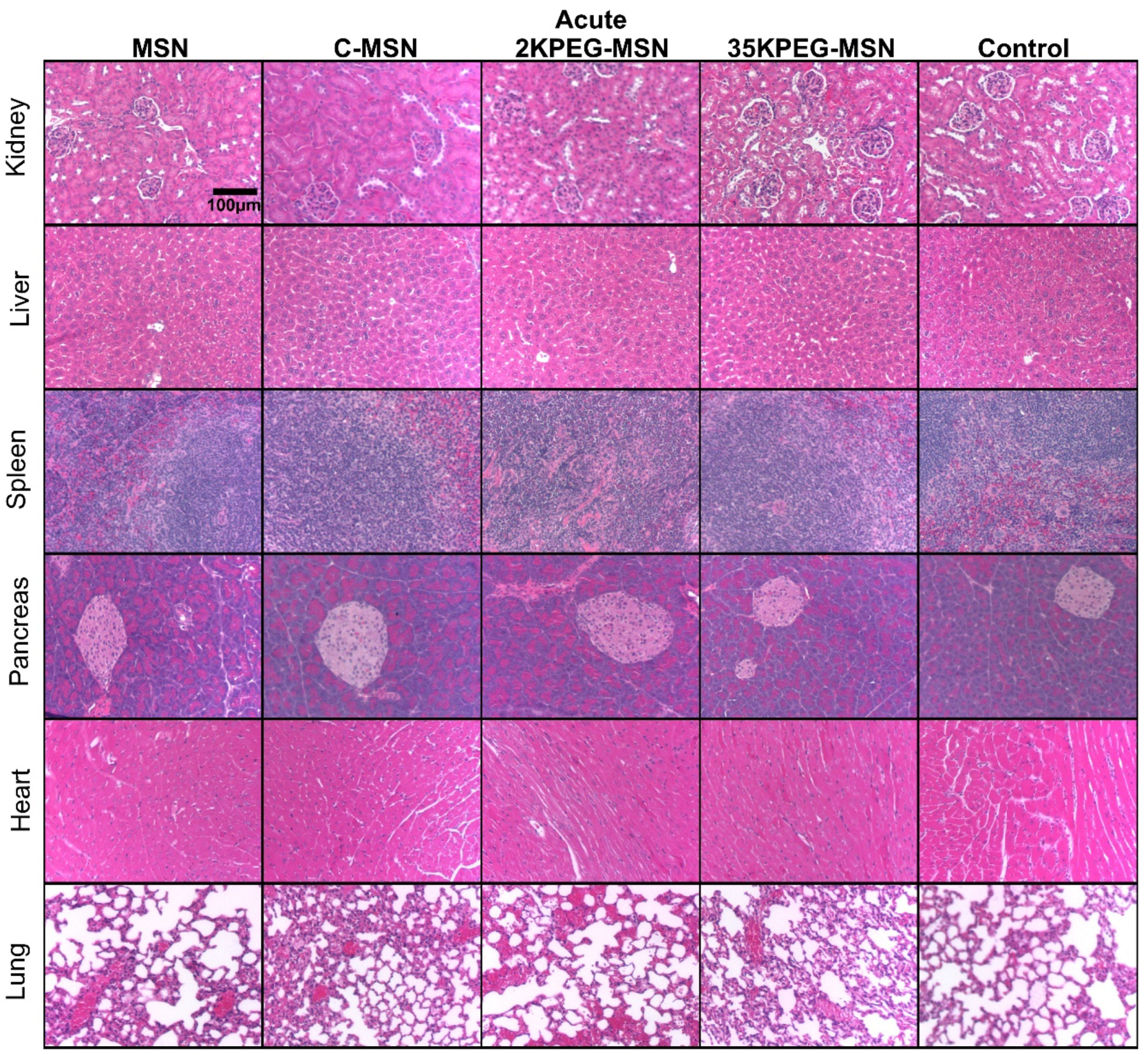
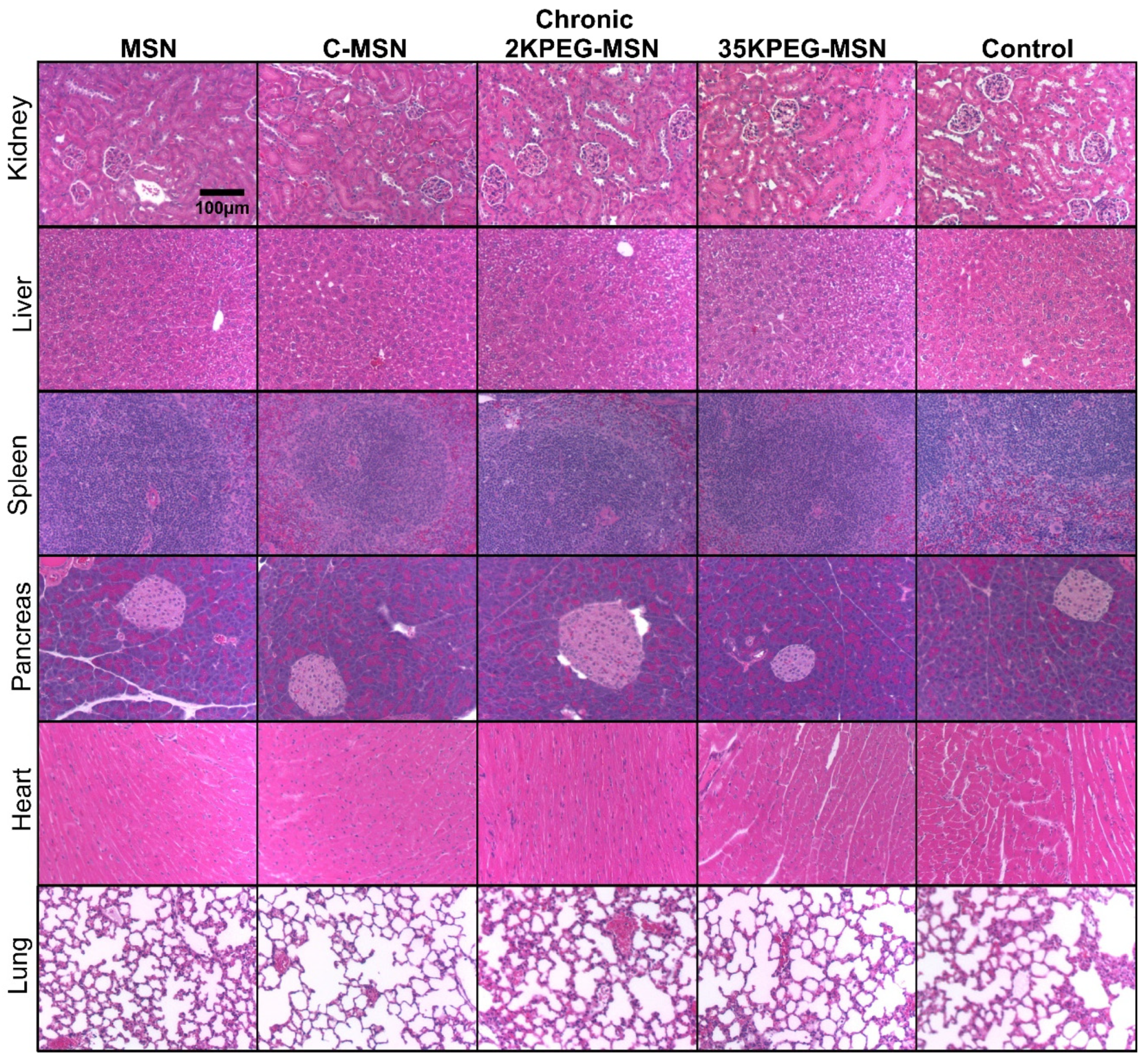
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pelaz, B.; Alexiou, C.; Alvarez-Puebla, R.A.; Alves, F.; Andrews, A.M.; Ashraf, S.; Balogh, L.P.; Ballerini, L.; Bestetti, A.; Brendel, C. Diverse applications of nanomedicine. ACS Nano 2017, 11, 2313–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzano, M.; Vallet-Regí, M. Mesoporous silica nanoparticles for drug delivery. Adv. Funct. Mater. 2020, 30, 1902634. [Google Scholar] [CrossRef]
- Jeelani, P.G.; Mulay, P.; Venkat, R.; Ramalingam, C. Multifaceted application of silica nanoparticles. A review. Silicon 2020, 12, 1337–1354. [Google Scholar] [CrossRef]
- Carniato, F.; Tei, L.; Botta, M. Gd-based mesoporous silica nanoparticles as MRI probes. Eur. J. Inorg. Chem. 2018, 2018, 4936–4954. [Google Scholar] [CrossRef]
- Nakamura, T.; Sugihara, F.; Matsushita, H.; Yoshioka, Y.; Mizukami, S.; Kikuchi, K. Mesoporous silica nanoparticles for 19 F magnetic resonance imaging, fluorescence imaging, and drug delivery. Chem. Sci. 2015, 6, 1986–1990. [Google Scholar] [CrossRef] [Green Version]
- Gurka, M.K.; Pender, D.; Chuong, P.; Fouts, B.L.; Sobelov, A.; McNally, M.W.; Mezera, M.; Woo, S.Y.; McNally, L.R. Identification of pancreatic tumors in vivo with ligand-targeted, pH responsive mesoporous silica nanoparticles by multispectral optoacoustic tomography. J. Control. Release 2016, 231, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Khanal, A.; Ullum, C.; Kimbrough, C.W.; Garbett, N.C.; Burlison, J.A.; McNally, M.W.; Chuong, P.; El-Baz, A.S.; Jasinski, J.B.; McNally, L.R. Tumor targeted mesoporous silica-coated gold nanorods facilitate detection of pancreatic tumors using Multispectral optoacoustic tomography. Nano Res. 2015, 8, 3864–3877. [Google Scholar] [CrossRef]
- Samykutty, A.; Grizzle, W.E.; Fouts, B.L.; McNally, M.W.; Chuong, P.; Thomas, A.; Chiba, A.; Otali, D.; Woloszynska, A.; Said, N.; et al. Optoacoustic imaging identifies ovarian cancer using a microenvironment targeted theranostic wormhole mesoporous silica nanoparticle. Biomaterials 2018, 182, 114–126. [Google Scholar] [CrossRef]
- Rimal, B.; Greenberg, A.K.; Rom, W.N. Basic pathogenetic mechanisms in silicosis: Current understanding. Curr. Opin. Pulm. Med. 2005, 11, 169–173. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Desouky, E.M.; Hozayen, W.G.; Bin-Jumah, M.; El-Nahass, E.-S.; Soliman, H.A.; Farghali, A.A. Mesoporous silica nanoparticles trigger liver and kidney injury and fibrosis via altering TLR4/NF-κB, JAK2/STAT3 and Nrf2/HO-1 signaling in rats. Biomolecules 2019, 9, 528. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Li, Y.; Wang, W.; Jin, M.; Du, Z.; Li, Y.; Duan, J.; Yu, Y.; Sun, Z. Acute toxicity of amorphous silica nanoparticles in intravenously exposed ICR mice. PLoS ONE 2013, 8, e61346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waegeneers, N.; Brasseur, A.; Van Doren, E.; Van der Heyden, S.; Serreyn, P.-J.; Pussemier, L.; Mast, J.; Schneider, Y.-J.; Ruttens, A.; Roels, S. Short-term biodistribution and clearance of intravenously administered silica nanoparticles. Toxicol. Rep. 2018, 5, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Tarn, D.; Ashley, C.E.; Xue, M.; Carnes, E.C.; Zink, J.I.; Brinker, C.J. Mesoporous silica nanoparticle nanocarriers: Biofunctionality and biocompatibility. Acc. Chem. Res. 2013, 46, 792–801. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xie, C.; Xia, H.; Wang, Z. pH and ultrasound dual-responsive polydopamine-coated mesoporous silica nanoparticles for controlled drug delivery. Langmuir 2018, 34, 9974–9981. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Feng, N. Mesoporous silica nanoparticles: Synthesis, classification, drug loading, pharmacokinetics, biocompatibility, and application in drug delivery. Expert Opin. Drug Deliv. 2019, 16, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Yu, X.; Ruan, Z.; Zhu, M.; Zhu, Y.; Hanagata, N. Magnetic mesoporous silica nanoparticles coated with thermo-responsive copolymer for potential chemo-and magnetic hyperthermia therapy. Microporous Mesoporous Mater. 2018, 256, 1–9. [Google Scholar] [CrossRef]
- Amoozgar, Z.; Yeo, Y. Recent advances in stealth coating of nanoparticle drug delivery systems. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2012, 4, 219–233. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Nie, H.; Wang, K.; Tan, W.; Wu, X.; Zhang, P. In vivo study of biodistribution and urinary excretion of surface-modified silica nanoparticles. Anal. Chem. 2008, 80, 9597–9603. [Google Scholar] [CrossRef]
- Cui, J.; Björnmalm, M.; Ju, Y.; Caruso, F. Nanoengineering of poly (ethylene glycol) particles for stealth and targeting. Langmuir 2018, 34, 10817–10827. [Google Scholar] [CrossRef]
- Dai, Q.; Bertleff-Zieschang, N.; Braunger, J.A.; Björnmalm, M.; Cortez-Jugo, C.; Caruso, F. Particle targeting in complex biological media. Adv. Healthc. Mater. 2018, 7, 1700575. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Li, Z.; Sun, J.; Luo, C.; Li, L.; Liu, Y.; Du, Y.; Qiu, S.; Ai, X.; Wu, C. Stealth CD44-targeted hyaluronic acid supramolecular nanoassemblies for doxorubicin delivery: Probing the effect of uncovalent pegylation degree on cellular uptake and blood long circulation. J. Control. Release 2015, 197, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-D.; Di Wu, X.S.; Liu, P.-X.; Yang, N.; Zhao, B.; Zhang, H.; Sun, Y.-M.; Zhang, L.-A.; Fan, F.-Y. Size-dependent in vivo toxicity of PEG-coated gold nanoparticles. Int. J. Nanomed. 2011, 6, 2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerdá, V.J.; Pacheco, R.R.; Witek, J.D.; de la Calle, F.M.M.; de la Sen Fernández, M.L. Immediate hypersensitivity to polyethylene glycols in unrelated products: When standardization in the nomenclature of the components of drugs, cosmetics, and food becomes necessary. Allergy Asthma Clin. Immunol. 2019, 15, 9. [Google Scholar] [CrossRef] [PubMed]
- Zeiderman, M.R.; Morgan, D.E.; Christein, J.D.; Grizzle, W.E.; McMasters, K.M.; McNally, L.R. Acidic pH-targeted chitosan-capped mesoporous silica coated gold nanorods facilitate detection of pancreatic tumors via multispectral optoacoustic tomography. ACS Biomater. Sci. Eng. 2016, 2, 1108–1120. [Google Scholar] [CrossRef] [Green Version]
- Laramie, M.D.; Fouts, B.L.; MacCuaig, W.M.; Buabeng, E.; Jones, M.A.; Mukherjee, P.; Behkam, B.; McNally, L.R.; Henary, M. Improved pentamethine cyanine nanosensors for optoacoustic imaging of pancreatic cancer. Sci. Rep. 2021, 11, 4366. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.A.; Syeda, J.; Wasan, K.M.; Wasan, E.K. An overview of chitosan nanoparticles and its application in non-parenteral drug delivery. Pharmaceutics 2017, 9, 53. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cai, C.; Li, J.; Li, J.; Li, J.; Sun, T.; Wang, L.; Wu, H.; Yu, G. Chitosan-based nanomaterials for drug delivery. Molecules 2018, 23, 2661. [Google Scholar] [CrossRef] [Green Version]
- Gerweck, L.E.; Seetharaman, K. Cellular pH gradient in tumor versus normal tissue: Potential exploitation for the treatment of cancer. Cancer Res. 1996, 56, 1194–1198. [Google Scholar]
- Garg, U.; Chauhan, S.; Nagaich, U.; Jain, N. Current advances in chitosan nanoparticles based drug delivery and targeting. Adv. Pharm. Bull. 2019, 9, 195. [Google Scholar] [CrossRef]
- Rizeq, B.R.; Younes, N.N.; Rasool, K.; Nasrallah, G.K. Synthesis, bioapplications, and toxicity evaluation of chitosan-based nanoparticles. Int. J. Mol. Sci. 2019, 20, 5776. [Google Scholar] [CrossRef] [Green Version]
- Zubareva, A.; Shagdarova, B.; Varlamov, V.; Kashirina, E.; Svirshchevskaya, E. Penetration and toxicity of chitosan and its derivatives. Eur. Polym. J. 2017, 93, 743–749. [Google Scholar] [CrossRef]
- Moghaddam, S.P.H.; Mohammadpour, R.; Ghandehari, H. In Vitro and in vivo evaluation of degradation, toxicity, biodistribution, and clearance of silica nanoparticles as a function of size, porosity, density, and composition. J. Control. Release 2019, 311, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, D.; Patel, V.; Sawant, K. Systematic investigation of in vitro and in vivo safety, toxicity and degradation of mesoporous silica nanoparticles synthesized using commercial sodium silicate. Microporous Mesoporous Mater. 2019, 284, 343–352. [Google Scholar] [CrossRef]
- Ullman-Culleré, M.H.; Foltz, C.J. Body condition scoring: A rapid and accurate method for assessing health status in mice. Comp. Med. 1999, 49, 319–323. [Google Scholar]
- Lehman, S.E.; Morris, A.S.; Mueller, P.S.; Salem, A.K.; Grassian, V.H.; Larsen, S.C. Silica nanoparticle-generated ROS as a predictor of cellular toxicity: Mechanistic insights and safety by design. Environ. Sci. Nano 2016, 3, 56–66. [Google Scholar] [CrossRef] [Green Version]
- MacCuaig, W.M.; Fouts, B.L.; McNally, M.W.; Grizzle, W.E.; Chuong, P.; Samykutty, A.; Mukherjee, P.; Li, M.; Jasinski, J.B.; Behkam, B. Active targeting significantly outperforms nanoparticle size in facilitating tumor-specific uptake in orthotopic pancreatic cancer. ACS Appl. Mater. Interfaces 2021, 13, 49614–49630. [Google Scholar] [CrossRef]
- Qian, Q.; Zhu, L.; Zhu, X.; Sun, M.; Yan, D. Drug-polymer hybrid macromolecular engineering: Degradable PEG integrated by platinum (IV) for cancer therapy. Matter 2019, 1, 1618–1630. [Google Scholar] [CrossRef] [Green Version]
- Andreani, T.; de Souza, A.L.R.; Kiill, C.P.; Lorenzón, E.N.; Fangueiro, J.F.; Calpena, A.C.; Chaud, M.V.; Garcia, M.L.; Gremião, M.P.D.; Silva, A.M. Preparation and characterization of PEG-coated silica nanoparticles for oral insulin delivery. Int. J. Pharm. 2014, 473, 627–635. [Google Scholar] [CrossRef]
- Xu, H.; Yan, F.; Monson, E.E.; Kopelman, R. Room-temperature preparation and characterization of poly (ethylene glycol)-coated silica nanoparticles for biomedical applications. J. Biomed. Mater. Res. Part A Off. J. Soc. Biomater. Jpn. Soc. Biomater. Aust. Soc. Biomater. Korean Soc. Biomater. 2003, 66, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Berridge, B.R.; Van Vleet, J.F.; Herman, E. Cardiac, vascular, and skeletal muscle systems. In Haschek and Rousseaux’s Handbook of Toxicologic Pathology, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1567–1665. [Google Scholar]
- Babcock, M.B.; Cardell, R.R., Jr. Fine structure of hepatocytes from fasted and fed rats. Am. J. Anat. 1975, 143, 399–437. [Google Scholar] [CrossRef]
- Babcock, M.B.; Cardell, R.R., Jr. Hepatic glycogen patterns in fasted and fed rats. Am. J. Anat. 1974, 140, 299–337. [Google Scholar] [CrossRef] [PubMed]
- Bradley, A.; Mukaratirwa, S.; Petersen-Jones, M. Incidences and range of spontaneous findings in the lymphoid and haemopoietic system of control Charles River CD-1 mice (Crl: CD-1 (ICR) BR) used in chronic toxicity studies. Toxicol. Pathol. 2012, 40, 375–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elangbam, C.S.; Colman, K.A.; Lightfoot, R.M.; Tyler, R.D.; Wall, H.G. Endocardial myxomatous change in harlan sprague-dawley rats (Hsd: S—D) and CD-1 mice: Its microscopic resemblance to drug-induced valvulopathy in humans. Toxicol. Pathol. 2002, 30, 483–491. [Google Scholar] [CrossRef]
- Ruben, Z.; Arceo, R.; Bishop, S.; Elwell, M.; Kerns, W.; Mesfin, G.; Sandusky, G.; Van Vleet, J. Non-proliferative lesions of the heart and vasculature in rats. In Guides for Toxicologic Pathology; STP/ARP/AFIP: Washington, DC, USA, 2000; pp. 1–10. [Google Scholar]
- Berridge, B.R.; Mowat, V.; Nagai, H.; Nyska, A.; Okazaki, Y.; Clements, P.J.; Rinke, M.; Snyder, P.W.; Boyle, M.C.; Wells, M.Y. Non-proliferative and proliferative lesions of the cardiovascular system of the rat and mouse. J. Toxicol. Pathol. 2016, 29, 1S–47S. [Google Scholar] [CrossRef] [Green Version]
- Vervloet, M.G.; Sezer, S.; Massy, Z.A.; Johansson, L.; Cozzolino, M.; Fouque, D. The role of phosphate in kidney disease. Nat. Rev. Nephrol. 2017, 13, 27–38. [Google Scholar] [CrossRef]
- Webster, R.; Elliott, V.; Park, B.K.; Walker, D.; Hankin, M.; Taupin, P. PEG and PEG conjugates toxicity: Towards an understanding of the toxicity of PEG and its relevance to PEGylated biologicals. In PEGylated Protein Drugs: Basic Science and Clinical Applications; Springer: Berlin/Heidelberg, Germany, 2009; pp. 127–146. [Google Scholar]
- Choi, N.; Lee, J.; Chang, Y.; Jung, S.; Kim, Y.; Lee, S.; Lee, J.; Kim, J.; Song, H.; Park, B. Polyethylene glycol bowel preparation does not eliminate the risk of acute renal failure: A population-based case-crossover study. Endoscopy 2013, 45, 208–213. [Google Scholar] [CrossRef]
- Malta, D.; Petersen, K.S.; Johnson, C.; Trieu, K.; Rae, S.; Jefferson, K.; Santos, J.A.; Wong, M.M.; Raj, T.S.; Webster, J. High sodium intake increases blood pressure and risk of kidney disease. From the Science of Salt: A regularly updated systematic review of salt and health outcomes (August 2016 to March 2017). J. Clin. Hypertens. 2018, 20, 1654–1665. [Google Scholar] [CrossRef] [Green Version]
- Turecek, P.L.; Bossard, M.J.; Schoetens, F.; Ivens, I.A. PEGylation of biopharmaceuticals: A review of chemistry and nonclinical safety information of approved drugs. J. Pharm. Sci. 2016, 105, 460–475. [Google Scholar] [CrossRef] [Green Version]
- Ivens, I.A.; Achanzar, W.; Baumann, A.; Brändli-Baiocco, A.; Cavagnaro, J.; Dempster, M.; Depelchin, B.O.; Irizarry Rovira, A.R.; Dill-Morton, L.; Lane, J.H. PEGylated biopharmaceuticals: Current experience and considerations for nonclinical development. Toxicol. Pathol. 2015, 43, 959–983. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.-Y.; Gößl, D.e.; Haddick, L.; Engelke, H.; Bein, T. Clickable multifunctional large-pore mesoporous silica nanoparticles as nanocarriers. Chem. Mater. 2018, 30, 644–654. [Google Scholar] [CrossRef]
- Wu, S.-H.; Hung, Y.; Mou, C.-Y. Mesoporous silica nanoparticles as nanocarriers. Chem. Commun. 2011, 47, 9972–9985. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.M.; Hung, Y.; Ko, B.S.; Hsu, S.C.; Chen, W.H.; Chien, C.L.; Tsai, C.P.; Kuo, C.T.; Kang, J.C.; Yang, C.S.; et al. Highly efficient cellular labeling of mesoporous nanoparticles in human mesenchymal stem cells: Implication for stem cell tracking. FASEB J. 2005, 19, 2014–2016. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, Y.; Xu, Q.; Liu, Z. Mesoporous silica nanoparticles for stimuli-responsive controlled drug delivery: Advances, challenges, and outlook. Int. J. Nanomed. 2017, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- How, K.N.; Yap, W.H.; Lim, C.L.H.; Goh, B.H.; Lai, Z.W. Hyaluronic acid-mediated drug delivery system targeting for inflammatory skin diseases: A mini review. Front. Pharmacol. 2020, 11, 1105. [Google Scholar] [CrossRef] [PubMed]
- Castillo, R.R.; Lozano, D.; González, B.; Manzano, M.; Izquierdo-Barba, I.; Vallet-Regí, M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery: An update. Expert Opin. Drug Deliv. 2019, 16, 415–439. [Google Scholar] [CrossRef] [PubMed]
- Frickenstein, A.N.; Hagood, J.M.; Britten, C.N.; Abbott, B.S.; McNally, M.W.; Vopat, C.A.; Patterson, E.G.; MacCuaig, W.M.; Jain, A.; Walters, K.B. Mesoporous silica nanoparticles: Properties and strategies for enhancing clinical effect. Pharmaceutics 2021, 13, 570. [Google Scholar] [CrossRef]
- Zhang, G.; Li, X.; Liao, Q.; Liu, Y.; Xi, K.; Huang, W.; Jia, X. Water-dispersible PEG-curcumin/amine-functionalized covalent organic framework nanocomposites as smart carriers for in vivo drug delivery. Nat. Commun. 2018, 9, 2785. [Google Scholar] [CrossRef] [Green Version]
- Reboredo, C.; González-Navarro, C.J.; Martínez-López, A.L.; Martínez-Ohárriz, C.; Sarmento, B.; Irache, J.M. Zein-based nanoparticles as oral carriers for insulin delivery. Pharmaceutics 2021, 14, 39. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FBS Treatment Characterizations | Diameter by DLS (nm) | Zeta Potential (mV) | ||||
|---|---|---|---|---|---|---|
| MSN Coating | As-Prepared | FBS-Treated | Change | As-Prepared | FBS-Treated | Change |
| MSN | 25.8 | 28 | +2.2 | −5.59 | −2.84 | +2.75 |
| C-MSN | 31.8 | 48.9 | +17.1 | 44.07 | 12.62 | −31.45 |
| 2KPEG-MSN | 29.7 | 29.9 | +0.2 | −0.58 | −1.35 | −0.77 |
| 35KPEG-MSN | 30.1 | 30.3 | +0.2 | −0.91 | −1.72 | −0.81 |
| A | Blood Chemistry-Acute | B | Blood Chemistry-Chronic | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Untreated | MSN | C-MSN | 2KPEG-MSN | 35KPEG-MSN | Untreated | MSN | C-MSN | 2KPEG-MSN | 35KPEG-MSN | ||
| ALB | 4.5 | 4.37 | 4.57 | 4.57 | 4.4 | ALB | 4.53 | 4.53 | 4.67 | 4.63 | 4.57 |
| ALP | 110.3 | 88.7 | 104.3 | 118 | 110.3 | ALP | 82.3 | 95.7 | 91.7 | 92.7 | 80 |
| ALT | 38 | 34 | 49 | 51.3 | 28.3 | ALT | 38 | 30.7 | 36.7 | 23.7 | 35 |
| AMY | 1189 | 1163 | 1067 | 986 | 1083 | AMY | 1095 | 1066 | 1085 | 1174 | 1067 |
| TBIL | 0.33 | 0.4 | 0.4 | 0.4 | 0.3 | TBIL | 0.47 | 0.23 | 0.3 | 0.27 | 0.3 |
| BUN | 20 | 21.7 | 21.3 | 18.7 | 19.3 | BUN | 20.3 | 23.7 | 22.3 | 18.7 | 20.7 |
| CA | 11.8 | 11.3 | 11 | 11.8 | 11.1 | CA | 11.3 | 11 | 11.1 | 11.3 | 11.2 |
| PHOS | 8.17 | 7.7 | 7.97 | 9.6 | 7.7 | PHOS | 7.63 | 7.43 | 7.1 | 7.3 | 6.2 |
| CRE | 0.27 | 0.33 | 0.27 | 0.3 | 0.33 | CRE | 0.27 | 0.27 | 0.2 | 0.23 | 0.37 |
| GLU | 128.7 | 103.7 | 108.3 | 117.7 | 119.7 | GLU | 117.3 | 116.3 | 96.3 | 98.7 | 99 |
| NA+ | 154.7 | 154.3 | 153.7 | 159 | 154 | NA+ | 155.7 | 156 | 154.7 | 155.3 | 155.7 |
| K+ | 6.9 | 6.43 | 6.87 | 6.67 | 5.97 | K+ | 6.93 | 7 | 7.05 | 7.13 | 6.1 |
| TP | 6.03 | 5.8 | 6 | 6.07 | 5.9 | TP | 6.07 | 5.9 | 6.17 | 6.13 | 6.27 |
| GLOB | 1.53 | 1.43 | 1.43 | 1.5 | 1.53 | GLOB | 1.57 | 1.37 | 1.5 | 1.5 | 1.67 |
| C | Complete Blood Count-Acute | D | Complete Blood Count-Chronic | ||||||||
| Untreated | MSN | C-MSN | 2KPEG-MSN | 35KPEG-MSN | Untreated | MSN | C-MSN | 2KPEG-MSN | 35KPEG-MSN | ||
| RBC | 10.2 | 10.5 | 10.3 | 10.3 | 10.5 | RBC | 10.4 | 10.2 | 11.4 | 10.7 | 10.8 |
| HGB | 16.6 | 17.2 | 16.4 | 16.2 | 16.7 | HGB | 16.7 | 15.8 | 17.1 | 16.4 | 16.3 |
| HCT | 56.9 | 58.4 | 55.4 | 55.1 | 58.3 | HCT | 56.8 | 54.5 | 57.6 | 56.2 | 56.1 |
| MCV | 56 | 55.5 | 54 | 53.6 | 55.4 | MCV | 54.6 | 53.4 | 50.6 | 52.6 | 51.9 |
| MCH | 16.3 | 16.3 | 16 | 15.7 | 15.8 | MCH | 16 | 15.5 | 15 | 15.3 | 15.1 |
| MCHC | 29.1 | 29.4 | 29.5 | 29.3 | 28.6 | MCHC | 29.3 | 29 | 29.7 | 29.2 | 29 |
| RDW-SD | 28.9 | 29.7 | 28.8 | 29 | 29.5 | RDW-SD | 30.1 | 30.1 | 29.1 | 29.3 | 29.7 |
| RDW-CV | 20.7 | 21.7 | 21.6 | 21.8 | 21.6 | RDW-CV | 22.1 | 22.6 | 24.1 | 22.9 | 23.4 |
| RET | 409 | 396 | 440 | 367 | 541 | RET | 282 | 380 | 562 | 477 | 564 |
| RET% | 4.05 | 3.78 | 4.32 | 3.57 | 5.18 | RET% | 2.69 | 3.77 | 4.96 | 4.4 | 5.22 |
| PLT | 904 | 776 | 979 | 878 | 1086 | PLT | 1173 | 1113 | 1548 | 1338 | 1360 |
| PDW | 9.1 | 9.37 | 8.5 | 10.1 | 9.13 | PDW | 9.53 | 7.67 | 9.83 | 8.13 | 7.73 |
| MPV | 8.1 | 7.97 | 8.23 | 8.47 | 8.3 | MPV | 8.43 | 8 | 9.3 | 8.23 | 8.17 |
| P-LCR | 12 | 10.9 | 9.4 | 12.9 | 10.1 | P-LCR | 12.5 | 6.5 | 10.7 | 7.1 | 6.7 |
| PCT | 0.73 | 0.62 | 0.8 | 0.74 | 0.92 | PCT | 0.99 | 0.9 | 1.41 | 1.1 | 1.1 |
| WBC | 4.44 | 5.67 | 6.7 | 6.7 | 6.02 | WBC | 4.41 | 5.32 | 7.74 | 3.81 | 6.09 |
| NEUT | 0.52 | 0.78 | 1.46 | 0.8 | 0.67 | NEUT | 0.46 | 0.79 | 0.85 | 0.45 | 0.79 |
| LYMPH | 3.79 | 4.72 | 5.03 | 5.74 | 5.19 | LYMPH | 3.81 | 4.37 | 3.77 | 3.23 | 5.1 |
| MONO | 0.02 | 0.03 | 0.01 | 0.03 | 0.04 | MONO | 0.01 | 0.03 | 0.02 | 0.03 | 0.05 |
| EO | 0.09 | 0.14 | 0.19 | 0.13 | 0.11 | EO | 0.12 | 0.13 | 0.1 | 0.09 | 0.15 |
| BASO | 0.01 | 0 | 0.01 | 0.01 | 0.01 | BASO | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MacCuaig, W.M.; Samykutty, A.; Foote, J.; Luo, W.; Filatenkov, A.; Li, M.; Houchen, C.; Grizzle, W.E.; McNally, L.R. Toxicity Assessment of Mesoporous Silica Nanoparticles upon Intravenous Injection in Mice: Implications for Drug Delivery. Pharmaceutics 2022, 14, 969. https://doi.org/10.3390/pharmaceutics14050969
MacCuaig WM, Samykutty A, Foote J, Luo W, Filatenkov A, Li M, Houchen C, Grizzle WE, McNally LR. Toxicity Assessment of Mesoporous Silica Nanoparticles upon Intravenous Injection in Mice: Implications for Drug Delivery. Pharmaceutics. 2022; 14(5):969. https://doi.org/10.3390/pharmaceutics14050969
Chicago/Turabian StyleMacCuaig, William M., Abhilash Samykutty, Jeremy Foote, Wenyi Luo, Alexander Filatenkov, Min Li, Courtney Houchen, William E. Grizzle, and Lacey R. McNally. 2022. "Toxicity Assessment of Mesoporous Silica Nanoparticles upon Intravenous Injection in Mice: Implications for Drug Delivery" Pharmaceutics 14, no. 5: 969. https://doi.org/10.3390/pharmaceutics14050969