
Crystallization of Form II Paracetamol with the Assistance of Carboxylic Acids toward Batch and Continuous Processes


Abstract
:
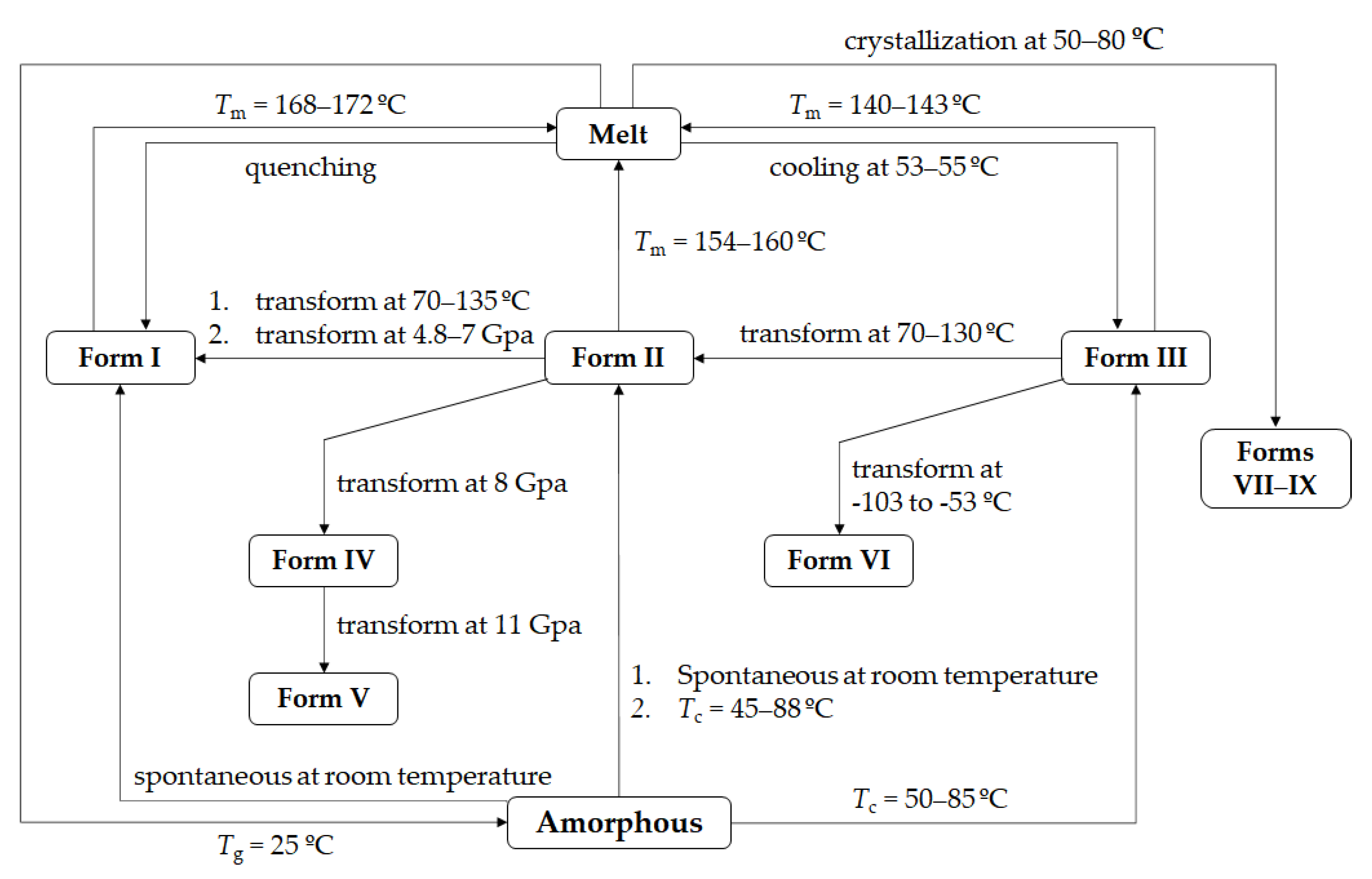
1. Introduction
2. Experimental Section
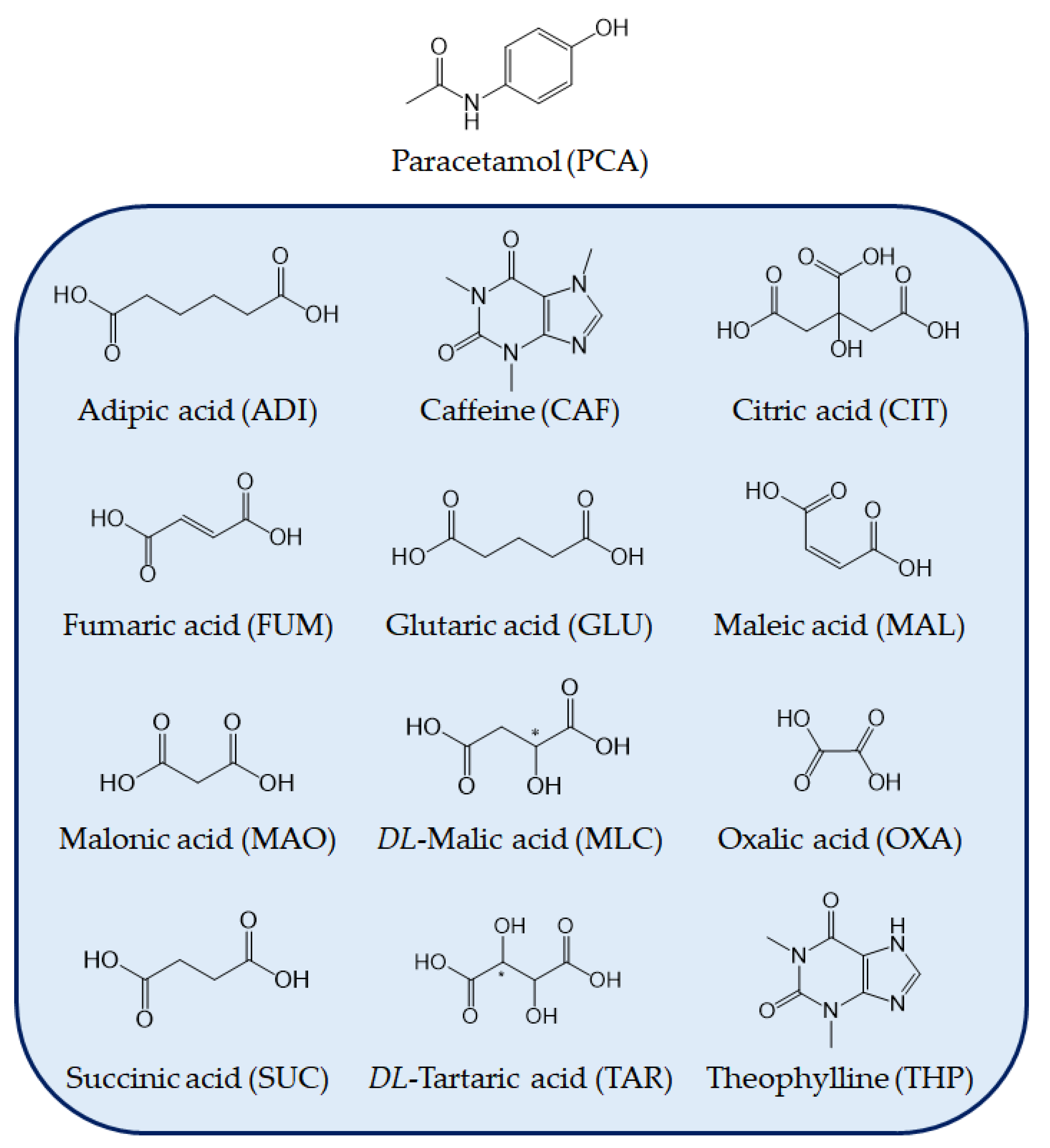
2.1. Materials
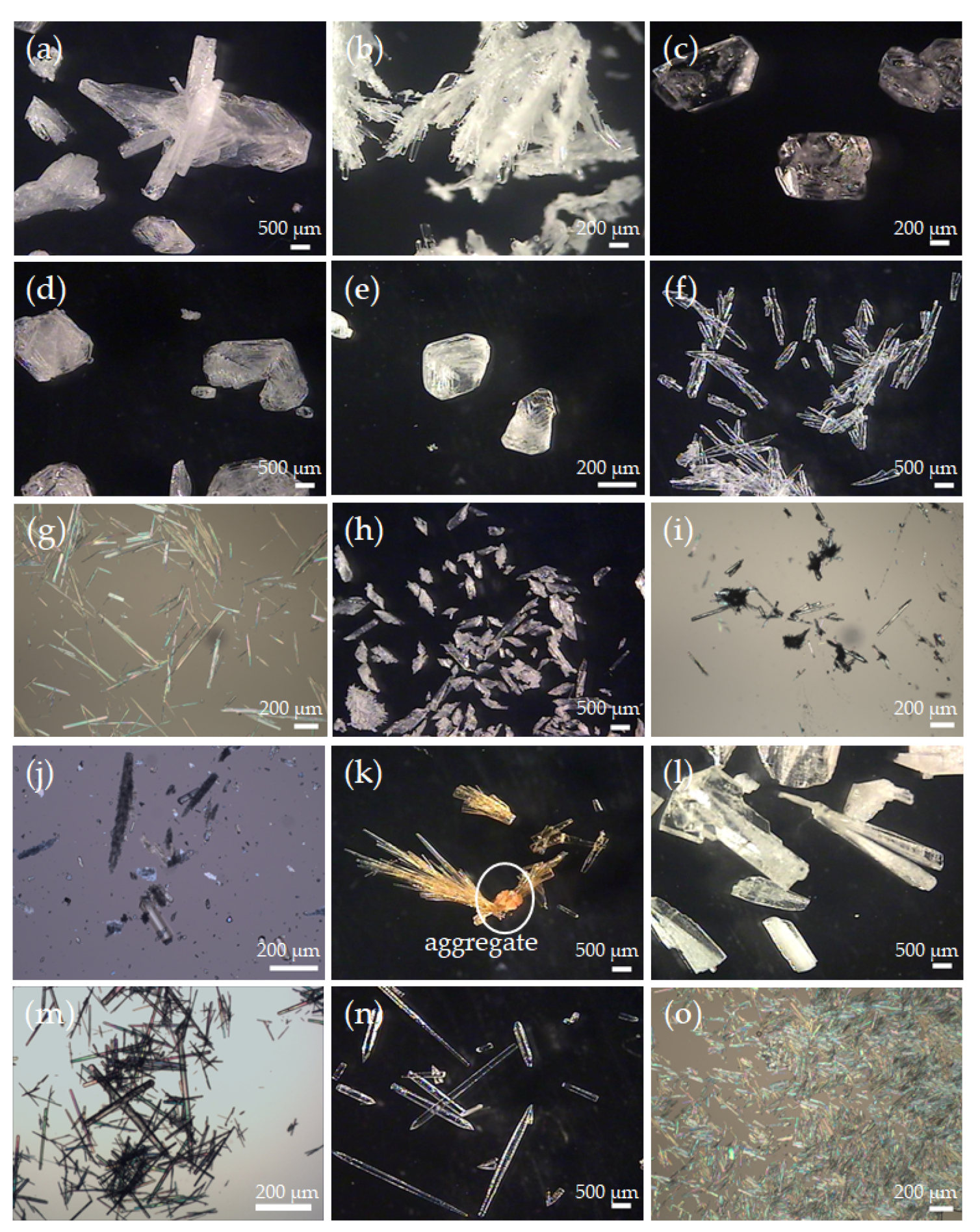
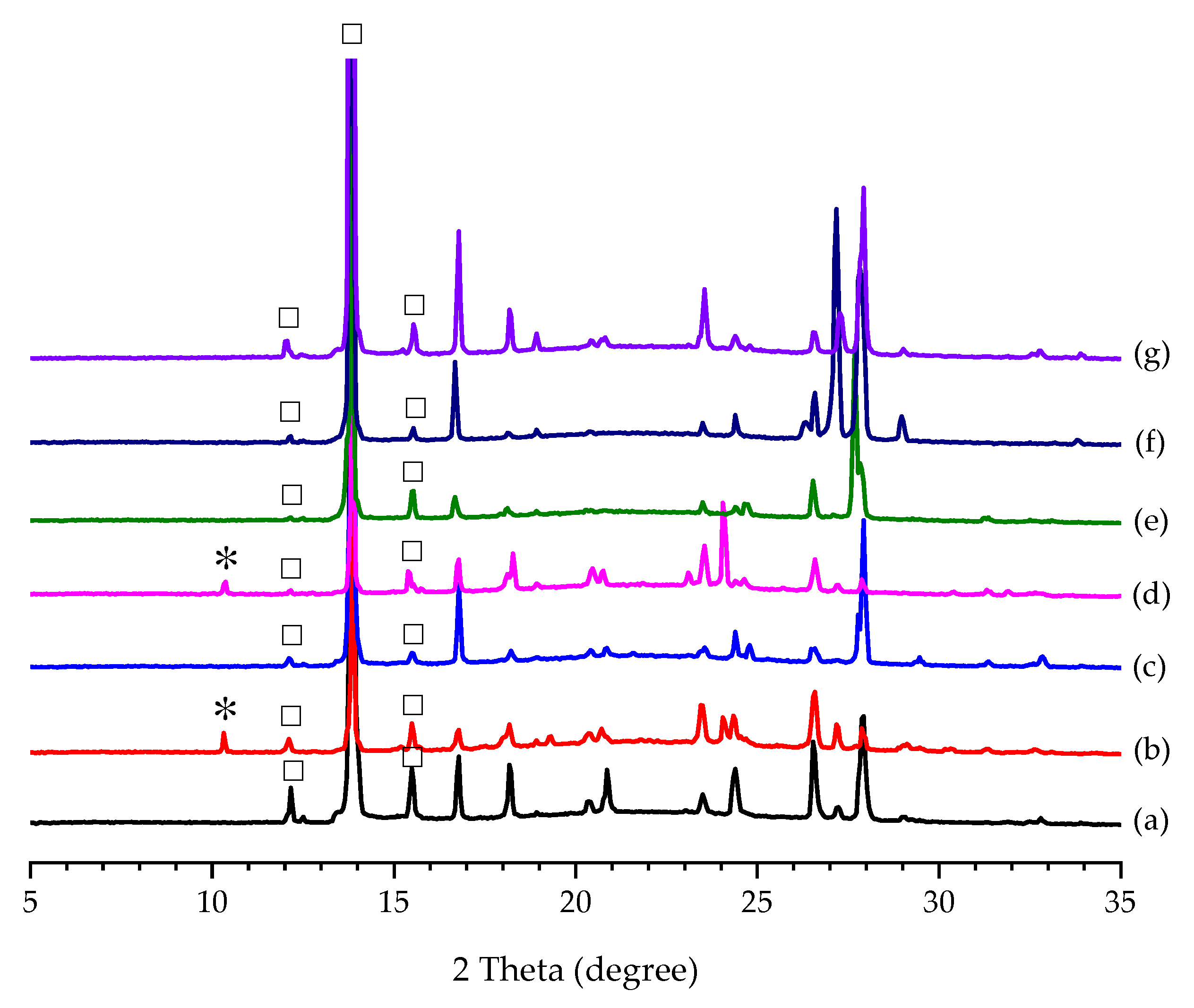
2.2. Additive Screening for the Preparation of Form II PCA
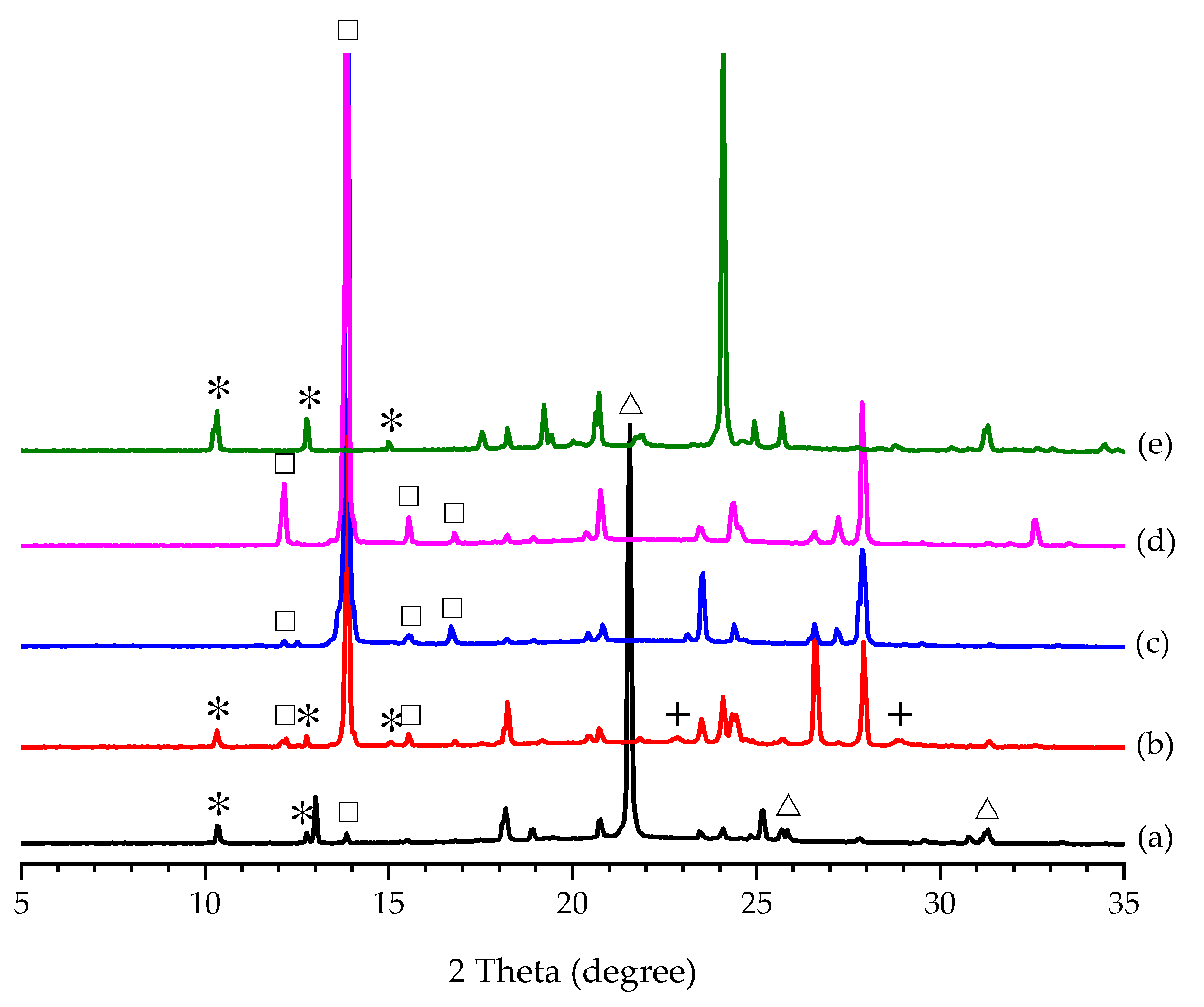
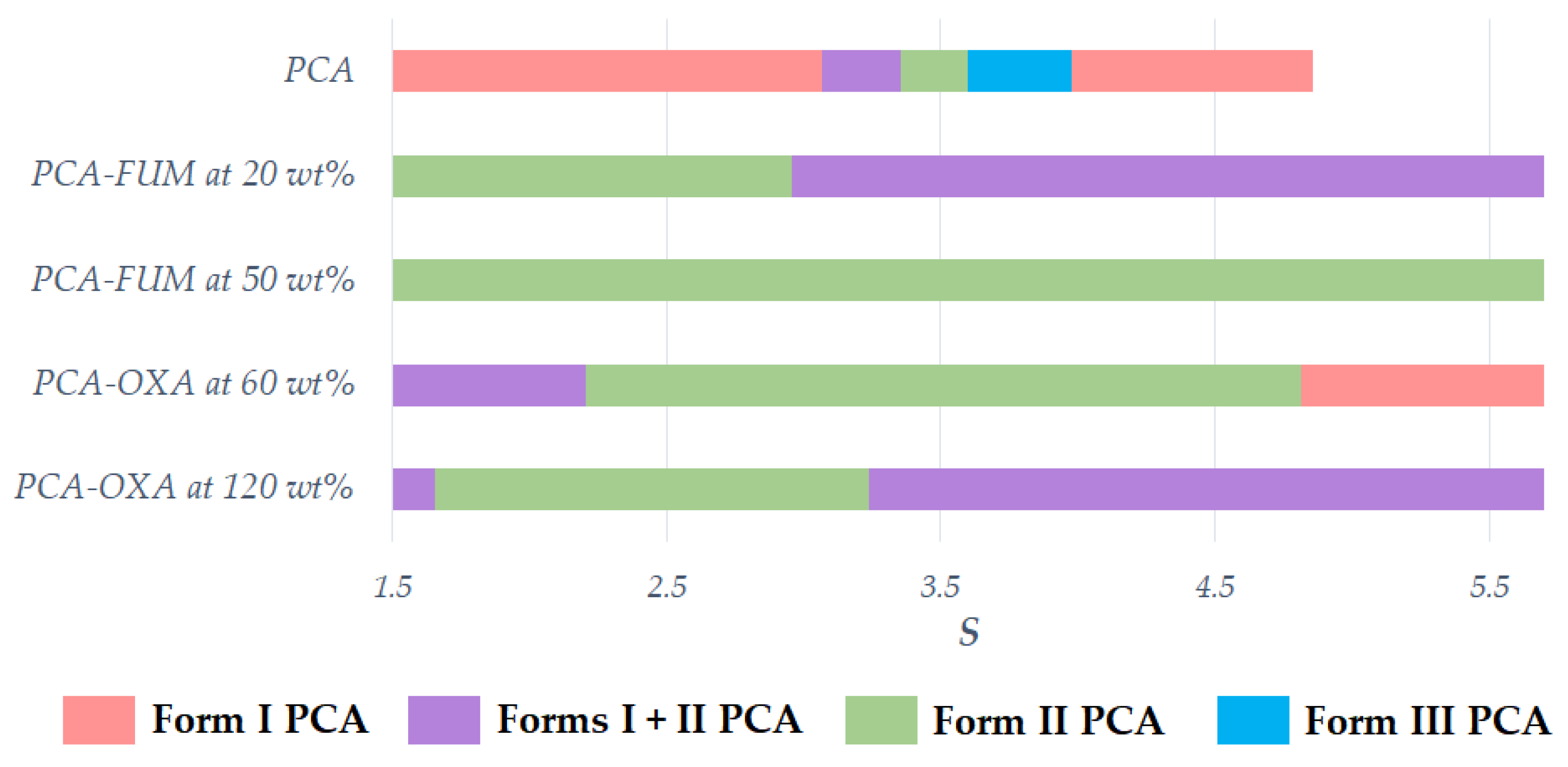
2.3. Effect of Additive Amount on the Polymorphic Formation of PCA
2.4. Effect of Seeding on the Polymorphic Formation of PCA
2.5. Liquid-Assisted Grinding
2.6. Effect of the Degree of Supersaturation on the Polymorphic Formation of PCA with the Assistance of FUM and OXA
2.7. Solubility Measurement in the Aqueous Solutions of FUM and OXA
2.8. Cake Washing for the Removal of FUM
2.9. Batch Additive-Assisted Cooling Crystallization of PCA in a Stirred Vessel
2.10. Continuous Additive-Assisted Cooling Crystallization of PCA in a Tubular Crystallizer
2.11. Instrumental Analysis
3. Results and Discussion
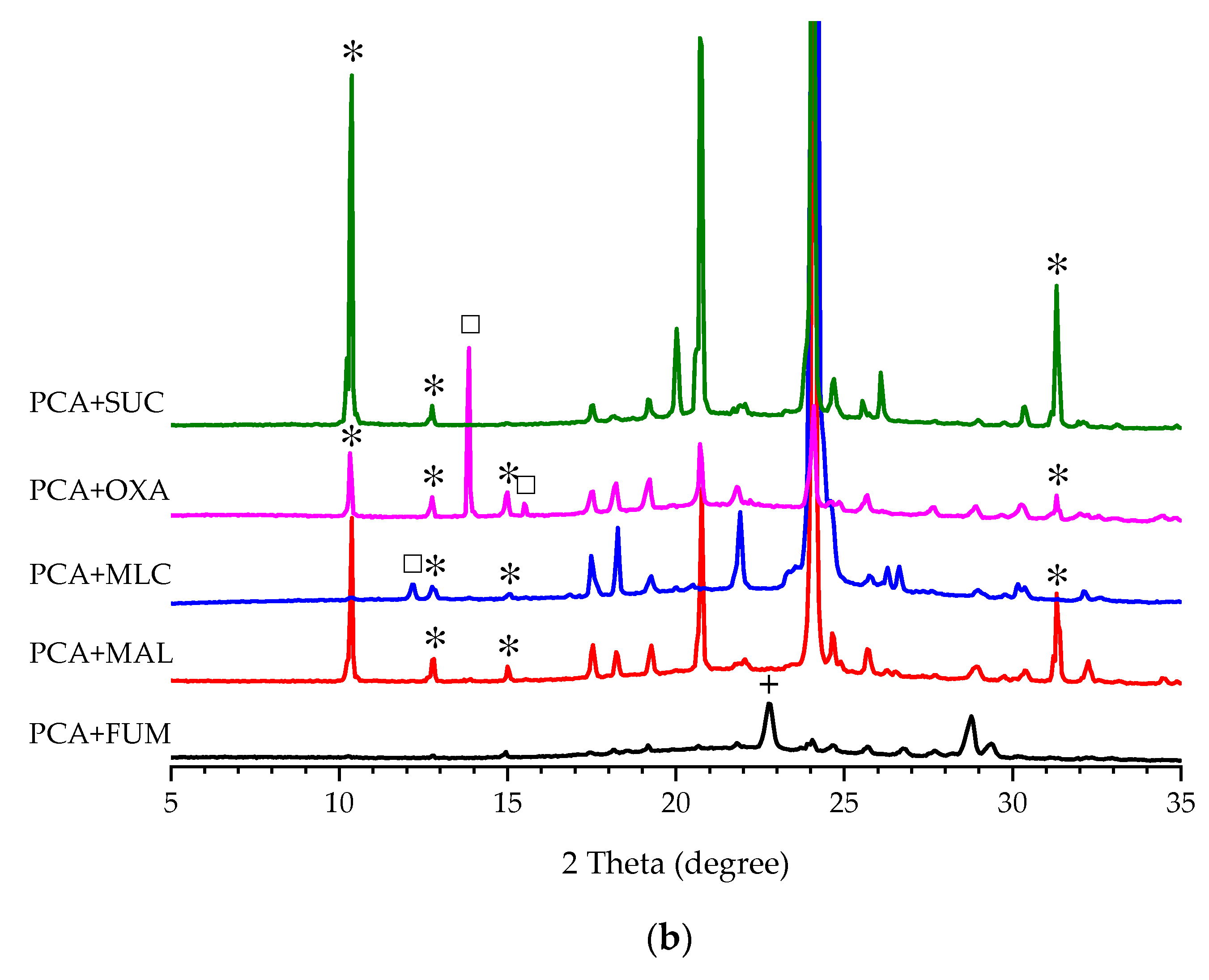
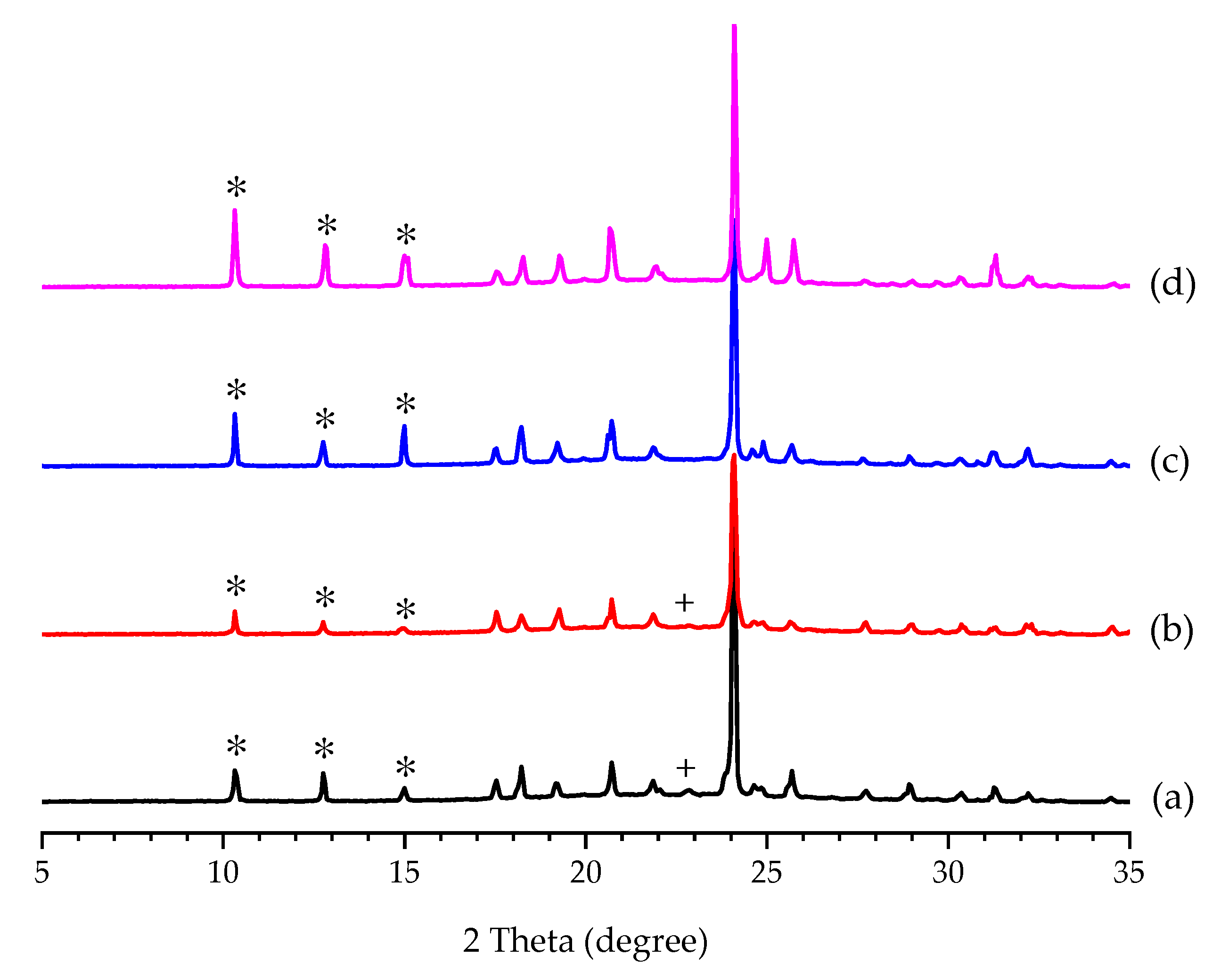
3.1. Additive Screening for the Preparation of Form II PCA
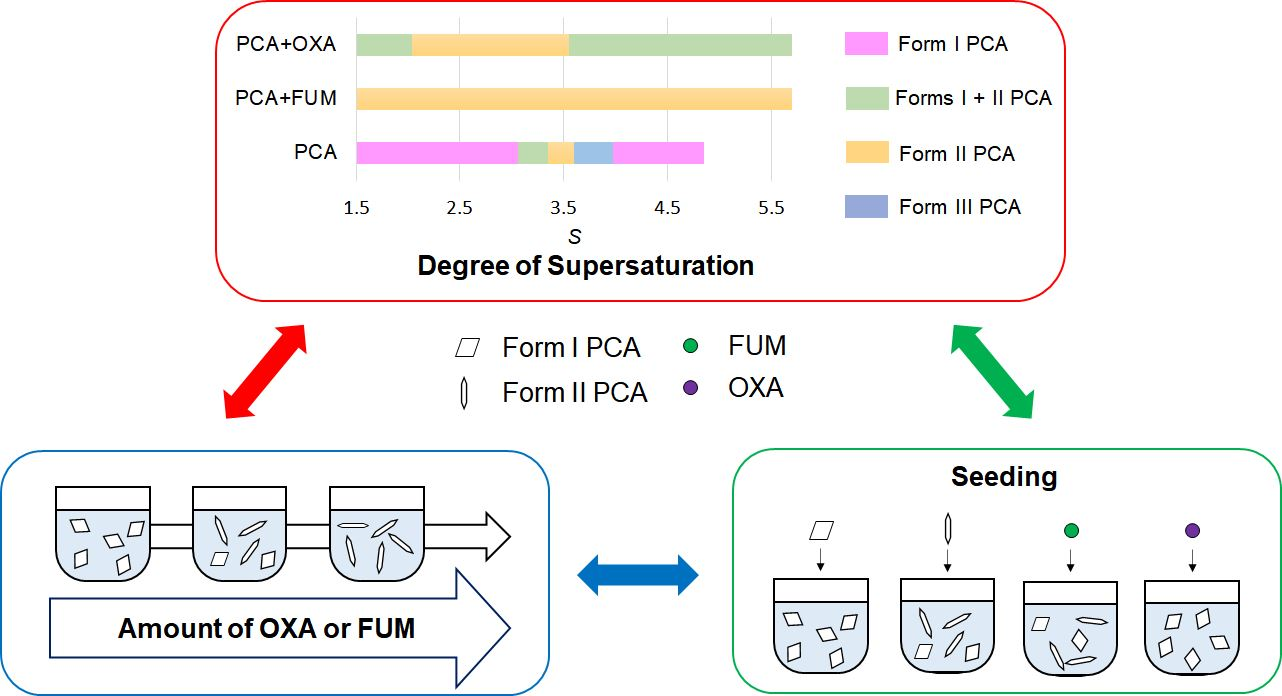
3.2. Effect of Additive Amount on the Polymorphic Formation of PCA
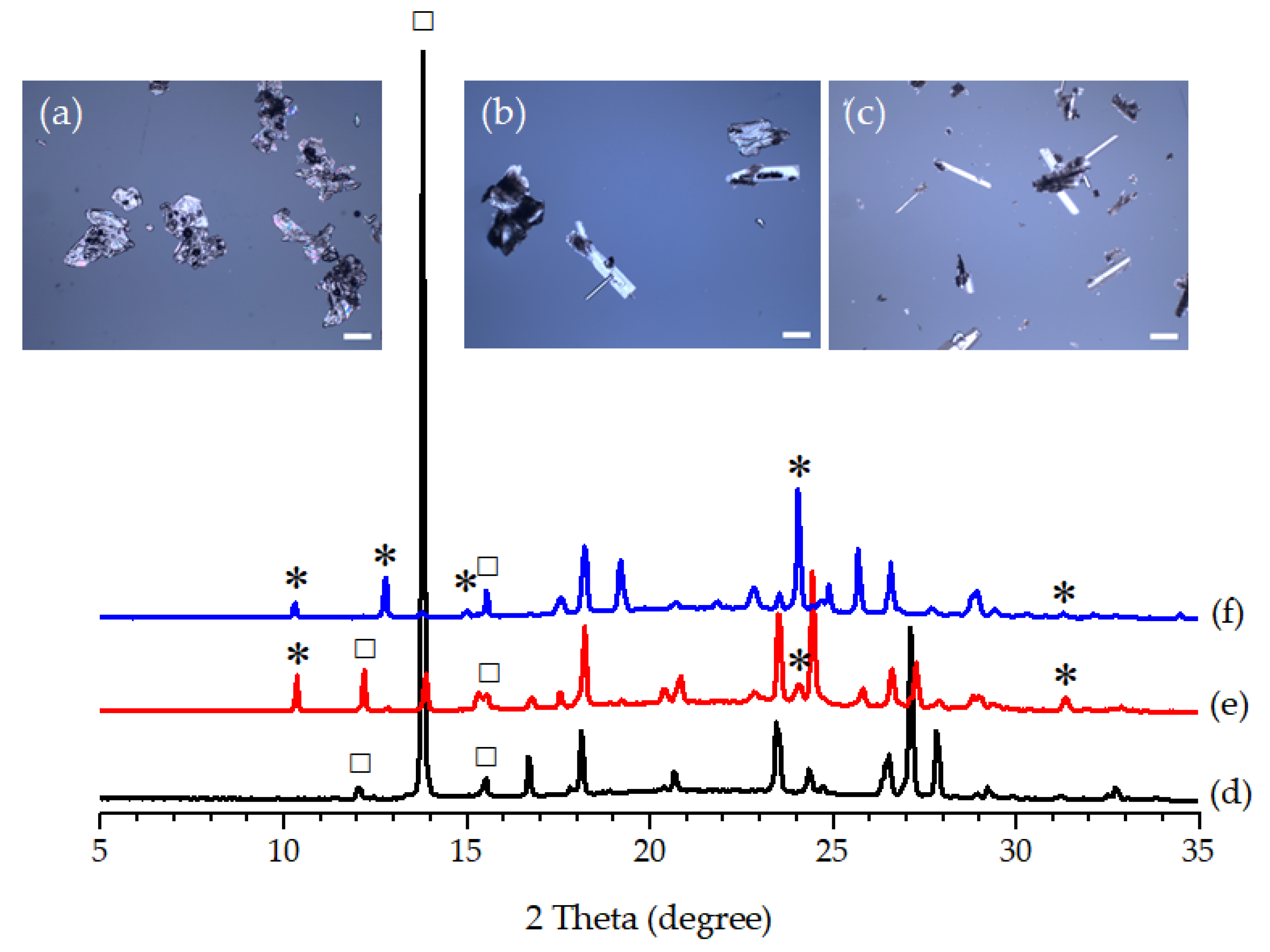
3.3. Effect of Seeding on the Polymorphic Formation of PCA
3.4. Effect of the Degree of Supersaturation on the Polymorphic Formation of PCA with the Assistance of FUM and OXA
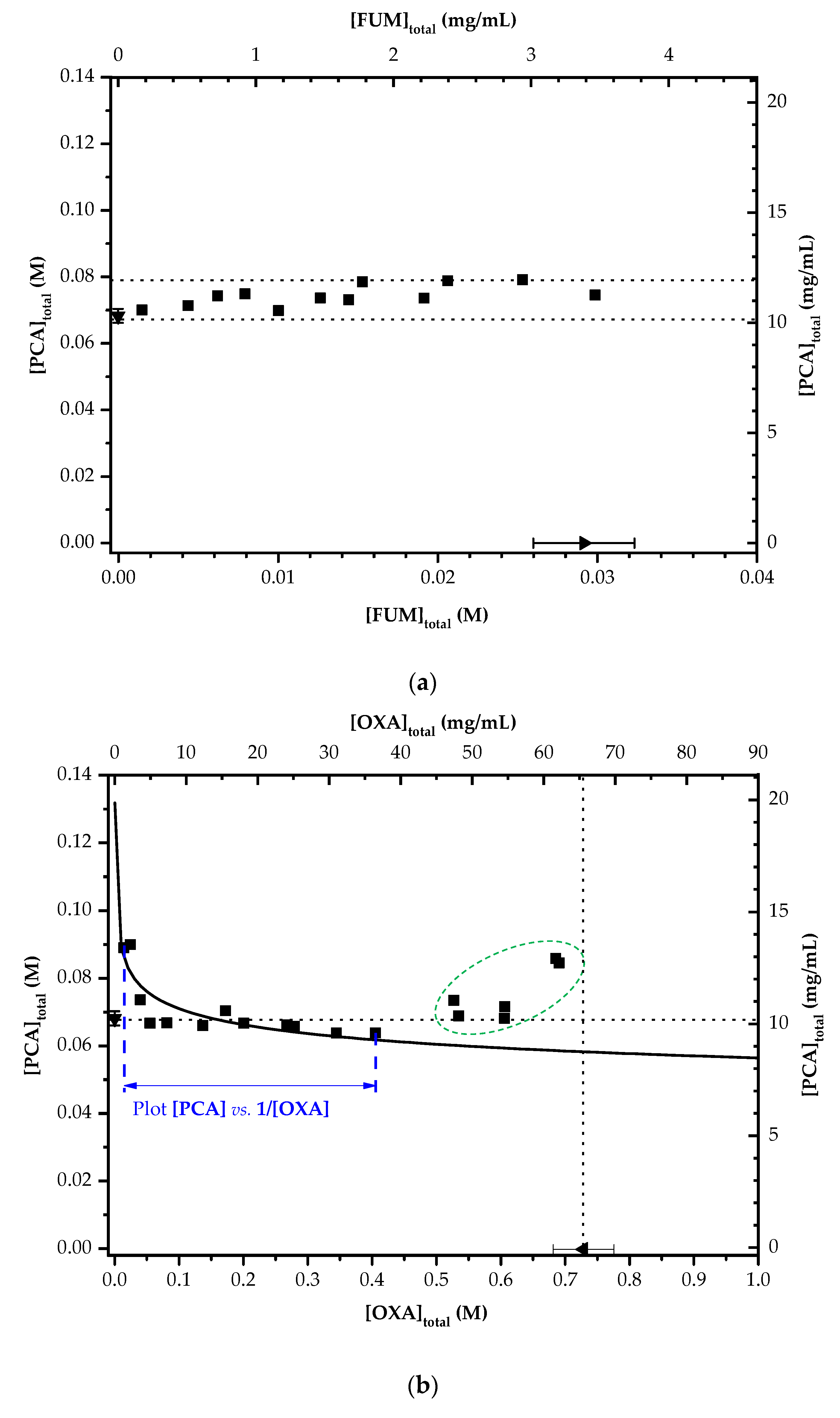
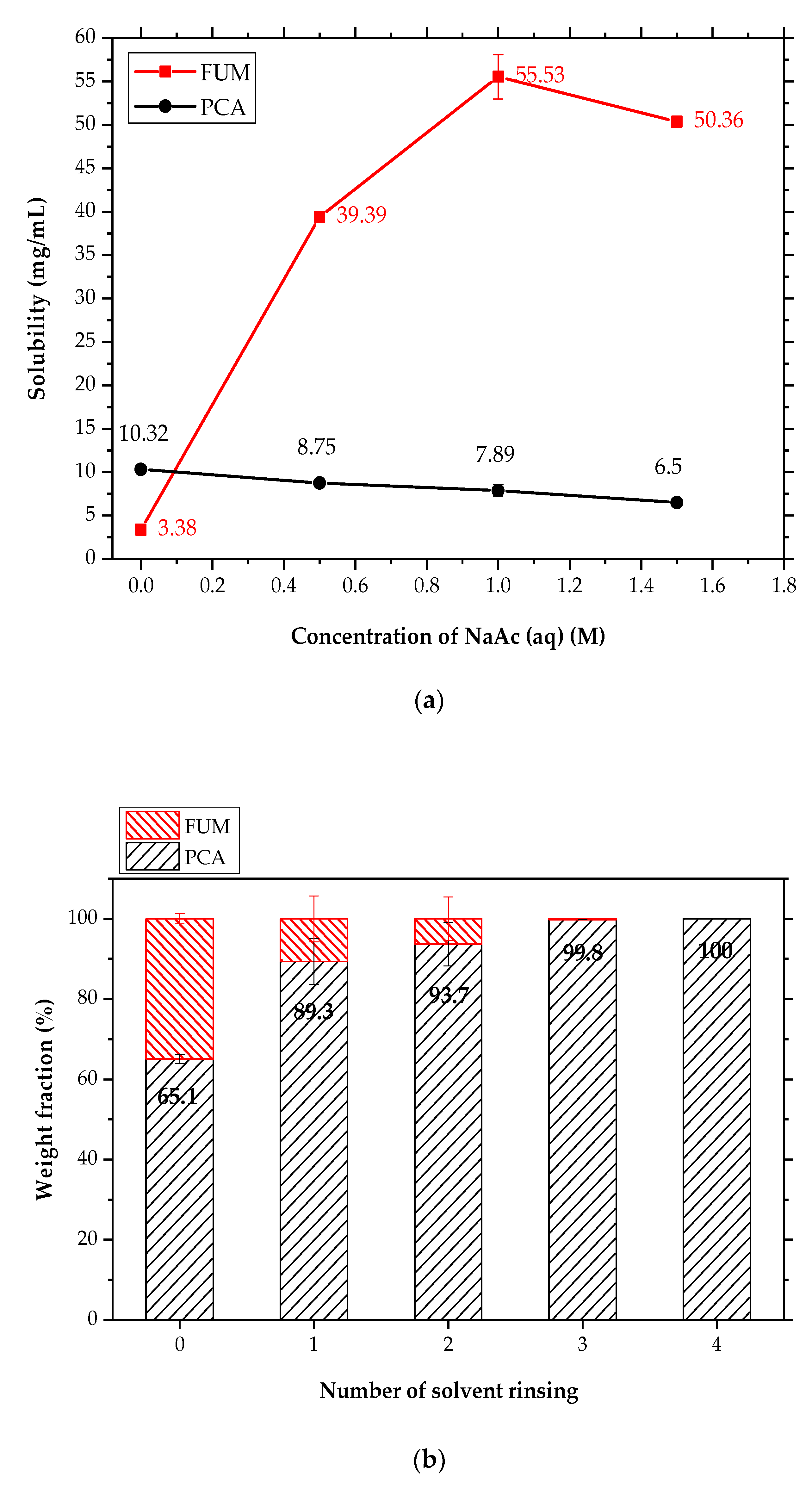
3.5. Solubility Diagrams of the PCA–FUM and PCA-OXA Aqueous Solutions
3.6. Removal of FUM Crystals from the Mixture of Form II PCA and FUM by Solvent Rinsing
3.7. Batch Additive-Assisted Cooling Crystallization of PCA in a Stirred Vessel
3.8. Continuous Additive-Assisted Cooling Crystallization of PCA in a Tubular Crystallizer
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brittain, H.G. Polymorphism and solvatomorphism 2010. J. Pharm. Sci. 2012, 101, 464–484. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.H.; Young, V., Jr.; Grant, D.J. Polymorph screening: Influence of solvents on the rate of solvent-mediated polymorphic transformation. J. Pharm. Sci. 2001, 90, 1878–1890. [Google Scholar] [CrossRef] [PubMed]
- Munroe, A.; Rasmuson, Å.C.; Hodnett, B.K.; Croker, D.M. Relative stabilities of the five polymorphs of sulfathiazole. Cryst. Growth Des. 2012, 12, 2825–2835. [Google Scholar] [CrossRef]
- Lee, H.L.; Cheng, Y.S.; Yeh, K.L.; Lee, T. A novel hydrate form of sodium dodecyl sulfate and its crystallization process. ACS Omega 2021, 6, 15770–15781. [Google Scholar] [CrossRef]
- Ferrari, E.S.; Davey, R.J.; Cross, W.I.; Gillon, A.L.; Towler, C.S. Crystallization in polymorphic systems: The solution-mediated transformation of β to α glycine. Cryst. Growth Des. 2003, 3, 53–60. [Google Scholar] [CrossRef]
- Nogueira, B.A.; Castiglioni, C.; Fausto, R. Color polymorphism in organic crystals. Commun. Chem. 2020, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, T.J. State-of-the-art chocolate manufacture: A review. Compr. Rev. Food Sci. Food Saf. 2017, 16, 1313–1344. [Google Scholar] [CrossRef] [Green Version]
- Wille, R.L.; Lutton, E.S. Polymorphism of cocoa butter. J. Am. Oil Chem. Soc. 1966, 43, 491–496. [Google Scholar] [CrossRef]
- Barišić, V.; Kopjar, M.; Jozinović, A.; Flanjak, I.; Ačkar, Đ.; Miličević, B.; Šubarić, D.; Babić, J. The chemistry behind chocolate production. Molecules 2019, 24, 3163. [Google Scholar] [CrossRef] [Green Version]
- Gupta, H.; Kumar, S.; Roy, S.K.; Gaud, R.S. Patent protection strategies. J. Pharm. BioAllied Sci. 2010, 2, 2–7. [Google Scholar] [CrossRef]
- Karpinski, P.H. Polymorphism of active pharmaceutical ingredients. Chem. Eng. Technol. 2006, 29, 233–237. [Google Scholar] [CrossRef]
- Miller, J.M.; Collman, B.M.; Greene, L.R.; Grant, D.J.; Blackburn, A.C. Identifying the stable polymorph early in the drug discovery-development process. Pharm. Dev. Tech. 2005, 10, 291–297. [Google Scholar]
- Haїsa, M.; Kashino, S.; Kawai, R.; Maeda, H. The monoclinic form of p-hydroxyacetanilide. Acta Crystallogr. B Struct. Cryst. Cryst. Chem. 1976, 32, 1283–1285. [Google Scholar] [CrossRef] [Green Version]
- Haїsa, M.; Kashino, S.; Maeda, H. The orthorhombic form of p-hydroxyacetanilide. Acta Crystallogr. B Struct. Cryst. Cryst. Chem. 1974, 30, 2510–2512. [Google Scholar] [CrossRef]
- Perrin, M.-A.; Neumann, M.A.; Elmaleh, H.; Zaske, L. Crystal structure determination of the elusive paracetamol Form III. Chem. Commun. 2009, 22, 3181–3183. [Google Scholar] [CrossRef]
- Smith, S.J.; Bishop, M.M.; Montgomery, J.M.; Hamilton, T.P.; Vohra, Y.K. Polymorphism in paracetamol: Evidence of additional forms IV and V at high pressure. J. Phys. Chem. A 2014, 118, 6068–6077. [Google Scholar] [CrossRef]
- Reiss, C.A.; van Mechelen, J.B.; Goubitzc, K.; Peschar, R. Reassessment of paracetamol orthorhombic Form III and determination of a novel low temperature monoclinic Form III-m from powder diffraction data. Acta Crystallogr. C Struct. Chem. 2018, 74, 392–399. [Google Scholar] [CrossRef] [Green Version]
- Shtukenberg, A.G.; Tan, M.; Vogt-Maranto, L.; Chan, E.J.; Xu, W.; Yang, J.; Tuckerman, M.E.; Hu, C.T.; Kahr, B. Melt crystallization for paracetamol polymorphism. Cryst. Growth Des. 2019, 19, 4070–4080. [Google Scholar] [CrossRef]
- Parkin, A.; Parsons, S.; Pulham, C.R. Paracetamol monohydrate at 150 K. Acta Crystallogr. E 2002, 58, o1345–o1347. [Google Scholar] [CrossRef]
- Fabbiani, F.P.; Allan, D.R.; David, W.I.; Moggach, S.A.; Parsons, S.; Pulham, C.R. High-pressure recrystallisation—A route to new polymorphs and solvates. CrystEngComm 2004, 6, 504–511. [Google Scholar] [CrossRef]
- McGregor, P.A.; Allan, D.R.; Parsons, S.; Pulham, C.R. Preparation and crystal structure of a trihydrate of paracetamol. J. Pharm. Sci. 2002, 91, 1308–1311. [Google Scholar] [CrossRef] [PubMed]
- Oswald, I.D.; Allan, D.R.; McGregor, P.A.; Motherwell, W.S.; Parsons, S.; Pulham, C.R. The formation of paracetamol (acetaminophen) adducts with hydrogen-bond acceptors. Acta Crystallogr. B 2002, 58, 1057–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oswald, I.D.; Motherwell, W.S.; Parsons, S.; Pulham, C.R. A paracetamol-morpholine adduct. Acta Crystallogr. E 2002, 58, o1290–o1292. [Google Scholar] [CrossRef]
- Fabbiani, F.P.; Allan, D.R.; Dawson, A.; David, W.I.; McGregor, P.A.; Oswald, I.D.; Parsons, S.; Pulham, C.R. Pressure-induced formation of a solvate of paracetamol. Chem. Comm. 2003, 24, 3004–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Martino, P.; Guyot-Hermann, A.M.; Conflant, P.; Drache, M.; Guyot, J.C. A new pure paracetamol for direct compression: The orthorhombic form. Int. J. Pharm. 1996, 128, 1–8. [Google Scholar] [CrossRef]
- Kachrimanis, K.; Fucke, K.; Noisternig, M.; Siebenhaar, B.; Griesser, U.J. Effects of moisture and residual solvent on the phase stability of orthorhombic paracetamol. Pharm. Res. 2008, 25, 1440–1449. [Google Scholar] [CrossRef]
- Telford, R.; Seaton, C.C.; Clout, A.; Buanz, A.; Gaisford, S.; Williams, G.R.; Prior, T.J.; Okoye, C.H.; Munshi, T.; Scowen, I.J. Stabilisation of metastable polymorphs: The case of paracetamol form III. Chem. Commun. 2016, 52, 12028–12031. [Google Scholar] [CrossRef] [Green Version]
- Sudha, C.; Srinivasan, K. Supersaturation dependent nucleation control and separation of mono, ortho and unstable polymorphs of paracetamol by swift cooling crystallization technique. CrystEngComm 2013, 15, 1914–1921. [Google Scholar] [CrossRef]
- Sudha, C.; Srinivasan, K. Nucleation control and separation of paracetamol polymorphs through swift cooling crystallization process. J. Cryst. Growth 2014, 401, 248–251. [Google Scholar] [CrossRef]
- Méndez del Río, J.R.; Rousseau, R.W. Batch and tubular-batch crystallization of paracetamol: Crystal size distribution and polymorph formation. Cryst. Growth Des. 2006, 6, 1407–1414. [Google Scholar] [CrossRef]
- Mikhailenko, M.A. Growth of large single crystals of the orthorhombic paracetamol. J. Cryst. Growth 2004, 265, 616–618. [Google Scholar] [CrossRef]
- Nichols, G.; Frampton, C.S. Physicochemical characterization of the orthorhombic polymorph of paracetamol crystallized from solution. J. Pharm. Sci. 1998, 87, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Al-Zoubi, N.; Kachrimanis, K.; Malamataris, S. Effects of harvesting and cooling on crystallization and transformation of orthorhombic paracetamol in ethanolic solution. Eur. J. Pharm. Sci. 2002, 17, 13–21. [Google Scholar] [CrossRef]
- Mori, Y.; Maruyama, M.; Takahashi, Y.; Yoshikawa, H.Y.; Okada, S.; Adachi, H.; Sugiyama, S.; Takano, K.; Murakami, S.; Matsumura, H.; et al. Metastable crystal growth of acetaminophen using solution-mediated phase transformation. Appl. Phys. Express 2017, 10, 015501. [Google Scholar] [CrossRef]
- Nicoud, L.; Licordari, F.; Myerson, A.S. Polymorph control in batch seeded crystallizers. A case study with paracetamol. CrystEngComm 2019, 21, 2105–2118. [Google Scholar] [CrossRef] [Green Version]
- Capes, J.S.; Cameron, R.E. Contact line crystallization to obtain metastable polymorphs. Cryst. Growth Des. 2007, 7, 108–112. [Google Scholar] [CrossRef]
- Chadwick, K.; Myerson, A.; Trout, B. Polymorphic control by heterogeneous nucleation-A new method for selecting crystalline substrates. CrystEngComm 2011, 13, 6625–6627. [Google Scholar] [CrossRef]
- Ehmann, H.M.; Werzer, O. Surface mediated structures: Stabilization of metastable polymorphs on the example of paracetamol. Cryst. Growth Des. 2014, 14, 3680–3684. [Google Scholar] [CrossRef]
- Bolla, G.; Myerson, A.S. SURMOF induced polymorphism and crystal morphological engineering of acetaminophen polymorphs: Advantage of heterogeneous nucleation. CrystEngComm 2018, 20, 2084–2088. [Google Scholar] [CrossRef] [Green Version]
- Lang, M.; Grzesiak, A.L.; Matzger, A.J. The use of polymer heteronuclei for crystalline polymorph selection. J. Am. Chem. Soc. 2002, 124, 14834–14835. [Google Scholar] [CrossRef]
- Mori, Y.; Maruyama, M.; Takahashi, Y.; Ikeda, K.; Fukukita, S.; Yoshikawa, H.Y.; Okada, S.; Adachi, H.; Sugiyama, S.; Takano, K.; et al. Selective crystallization of metastable phase of acetaminophen by ultrasonic irradiation. Appl. Phys. Express 2015, 8, 065501. [Google Scholar] [CrossRef]
- Kaur Bhangu, S.; Ashokkumar, M.; Lee, J. Ultrasound assisted crystallization of paracetamol: Crystal size distribution and polymorph control. Cryst. Growth Des. 2016, 16, 1934–1941. [Google Scholar] [CrossRef]
- Lee, H.L.; Lin, H.Y.; Lee, T. Large-scale crystallization of a pure metastable polymorph by reaction coupling. Org. Process Res. Dev. 2014, 18, 539–545. [Google Scholar] [CrossRef]
- Thomas, L.H.; Wales, C.; Zhao, L.; Wilson, C.C. Paracetamol form II: An elusive polymorph through facile multicomponent crystallization routes. Cryst. Growth Des. 2011, 11, 1450–1452. [Google Scholar] [CrossRef]
- Agnew, L.R.; Cruickshank, D.L.; McGlone, T.; Wilson, C.C. Controlled production of the elusive metastable form II of acetaminophen (paracetamol): A fully scalable templating approach in a cooling environment. Chem. Commun. 2016, 52, 7368–7371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnew, L.R.; McGlone, T.; Wheatcroft, H.P.; Robertson, A.; Parsons, A.R.; Wilson, C.C. Continuous crystallization of paracetamol (acetaminophen) form II: Selective access to a metastable solid form. Cryst. Growth Des. 2017, 17, 2418–2427. [Google Scholar] [CrossRef]
- Nicoud, L.; Licordari, F.; Myerson, A.S. Polymorph Control in MSMPR Crystallizers. A Case Study with Paracetamol. Org. Proc. Res. Dev. 2019, 23, 794–806. [Google Scholar] [CrossRef]
- Cruz, P.C.; Rocha, F.A.; Ferreira, A.M. Application of selective crystallization methods to isolate the metastable polymorphs of paracetamol: A review. Org. Proc. Res. Dev. 2019, 23, 2592–2607. [Google Scholar] [CrossRef]
- Mohammed, M.; Syed, M.F.; Bhatt, M.J.; Hoffman, E.J.; Aslan, K. Rapid and selective crystallization of acetaminophen using metal-assisted and microwave-accelerated evaporative crystallization. Nano Biomed. Eng. 2012, 4, 35. [Google Scholar] [CrossRef] [Green Version]
- Kollamaram, G.; Hopkins, S.C.; Glowacki, B.A.; Croker, D.M.; Walker, G.M. Inkjet printing of paracetamol and indomethacin using electromagnetic technology: Rheological compatibility and polymorphic selectivity. Eur. J. Pharm. Sci. 2018, 115, 248–257. [Google Scholar] [CrossRef]
- Niinomi, H.; Sugiyama, T.; Uda, S.; Tagawa, M.; Ujihara, T.; Miyamoto, K.; Omatsu, T. Plasmonic trapping-induced crystallization of acetaminophen. Cryst. Growth Des. 2019, 19, 529–537. [Google Scholar] [CrossRef]
- Al-Ani, A.J.; Herdes, C.; Wilson, C.C.; Castro-Dominguez, B. Engineering a new access route to metastable polymorphs with electrical confinement. Cryst. Growth Des. 2020, 20, 1451–1457. [Google Scholar] [CrossRef]
- Wang, S.; Wang, S.; Jiang, L.; Wang, M.; Wei, Y.; Sun, J.; Qu, L. Polymorph-controlled crystallization of acetaminophen through femtosecond laser irradiation. Cryst. Growth Des. 2019, 19, 3265–3271. [Google Scholar] [CrossRef]
- Liu, Y.; Gabriele, B.; Davey, R.J.; Cruz-Cabeza, A.J. Concerning elusive crystal forms: The case of paracetamol. J. Am. Chem. Soc. 2020, 142, 6682–6689. [Google Scholar] [CrossRef] [PubMed]
- Urwin, S.J.; Yerdelen, S.; Houson, I.; ter Horst, J.H. Impact of impurities on crystallization and product quality: A case study with paracetamol. Crystals 2021, 11, 1344. [Google Scholar] [CrossRef]
- Yeh, K.L.; Lee, T. Selective formation of form II paracetamol through the assistance of paracetamol co-crystal as templates in a solution. CrystEngComm 2021, 23, 3940–3945. [Google Scholar] [CrossRef]
- Chen, J.; Sarma, B.; Evans, J.M.; Myerson, A.S. Pharmaceutical crystallization. Cryst. Growth Des. 2011, 11, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Zhu, Z.; Jimenez, E.; Papageorgiou, C.D.; Waetzig, J.; Hardy, A.; Langston, M.; Braatz, R.D. Continuous-flow tubular crystallization in slugs spontaneously induced by hydrodynamics. Cryst. Growth Des. 2014, 14, 851–860. [Google Scholar] [CrossRef]
- Zhang, H.; Quon, J.; Alvarez, A.J.; Evans, J.; Myerson, A.S.; Trout, B. Development of continuous anti-solvent/cooling crystallization process using cascaded mixed suspension, mixed product removal crystallizers. Org. Proc. Res. Dev. 2012, 16, 915–924. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, S.; Du, S.; Wang, J.; Gong, J. Progress of pharmaceutical continuous crystallization. Engineering 2017, 3, 354–364. [Google Scholar] [CrossRef]
- Lai, T.T.C.; Ferguson, S.; Palmer, L.; Trout, B.L.; Myerson, A.S. Continuous crystallization and polymorph dynamics in the L-glutamic acid system. Org. Proc. Res. Dev. 2014, 18, 1382–1390. [Google Scholar] [CrossRef]
- Elbagerma, M.A.; Edwards, H.G.M.; Munshi, T.; Scowen, I.J. Identification of a new cocrystal of citric acid and paracetamol of pharmaceutical relevance. CrystEngComm 2011, 13, 1877–1884. [Google Scholar] [CrossRef] [Green Version]
- Karki, S.; Friščić, T.; Fabian, L.; Laity, P.R.; Day, G.M.; Jones, W. Improving mechanical properties of crystalline solids by cocrystal formation: New compressible forms of paracetamol. Adv. Meter. 2009, 21, 3905–3909. [Google Scholar] [CrossRef]
- Childs, S.L.; Stahly, G.P.; Park, A. The salt−cocrystal continuum: The influence of crystal structure on ionization state. Mol. Pharm. 2007, 4, 323–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latif., S.; Abbas, N.; Hussain, A.; Arshad, M.S.; Bukhari, N.I.; Afzal, H.; Riffat, S.; Ahmad, Z. Development of paracetamol-caffeine co-crystals to improve compressional, formulation and in vivo performance. Drug Dev. Ind. Pharm. 2018, 44, 1099–1108. [Google Scholar] [CrossRef]
- Suzuki, N.; Kawahata, M.; Yamaguchi, K.; Suzuki, T.; Tomono, K.; Fukami, T. Comparison of the relative stability of pharmaceutical cocrystals consisting of paracetamol and dicarboxylic acids. Drug Dev. Ind. Pharm. 2018, 44, 582–589. [Google Scholar] [CrossRef]
- Surena, S.; Sunsandeea, N.; Stolcova, M.; Hronecm, M.; Leepipatpiboonc, N.; Pancharoena, U.; Kheawhoma, S. Measurement on the solubility of adipic acid in various solvents at high temperature and its thermodynamics parameters. Fluid Phase Equilib. 2013, 360, 332–337. [Google Scholar] [CrossRef]
- Dang, L.; Du, W.; Black, S.; Wei, H. Solubility of fumaric acid in propan-2-ol, ethanol, acetone, propan-1-ol, and water. J. Chem. Eng. Data 2009, 54, 3112–3113. [Google Scholar] [CrossRef]
- Lee, H.L.; Lee, T. Direct co-crystal assembly from synthesis to cocrystallization. CrystEngComm 2015, 17, 9002–9006. [Google Scholar] [CrossRef]
- Nehm, S.J.; Rodríguez-Spong, B.; Rodríguez-Hornedo, N. Phase solubility diagrams of cocrystals are explained by solubility product and solution complexation. Cryst. Growth Des. 2006, 6, 592–600. [Google Scholar] [CrossRef]
- Jayasankar, A.; Reddy, L.S.; Bethune, S.J.; Rodríguez-Hornedo, N. Role of cocrystal and solution chemistry on the formation and stability of cocrystals with different stoichiometry. Cryst. Growth Des. 2009, 9, 889–897. [Google Scholar] [CrossRef]
- Saleemi, A.; Onyemelukwe, I.I.; Nagy, Z. Effects of a structurally related substance on the crystallization of paracetamol. Front. Chem. Sci. Eng. 2013, 7, 79–87. [Google Scholar] [CrossRef]
- Alhalaweh, A.; Sokolowski, A.; Rodriguez-Hornedo, N.; Velaga, S.P. Solubility behavior and solution chemistry of indomethacin cocrystals in organic solvents. Cryst. Growth Des. 2011, 11, 3923–3929. [Google Scholar] [CrossRef]
- Yu, Z.Q.; Chow, P.S.; Tan, R.B.H. Operating regions in cooling cocrystallization of caffeine and glutaric acid in acetonitrile. Cryst. Growth Des. 2010, 10, 2382–2387. [Google Scholar] [CrossRef]
- Shahid, M.; Sanxaridou, G.; Ottoboni, S.; Lue, L.; Price, C. Exploring the role of anti-solvent effects during washing on active pharmaceutical ingredient purity. Org. Proc. Res. Dev. 2021, 25, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Florence, A.J. Relating induction time and metastable zone width. CrystEngComm 2017, 19, 3966–3978. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Additive | Screening Method 1 | Screening Method 2 |
|---|---|---|
| ADI | Form II PCA + ADI | Form I PCA + ADI |
| CAF | No crystal | PCA-CAF |
| CIT | Form I PCA | Form I PCA |
| FUM | Form II PCA + FUM | Form II PCA + FUM |
| GLU | Form I PCA | Form I PCA |
| MAL | Form I PCA | Form II PCA |
| MAO | Form I PCA | Form I PCA |
| MLC | Form I PCA | Form II PCA |
| OXA | Form I PCA | Form II PCA |
| SUC | Form II PCA | Form II PCA |
| THP | PCA-THP + Form II THP | PCA-THP + Form II THP |
| TAR | Form I PCA | Form I PCA |
| Additive | Weight Ratio of PCA to Additive | Weight Percentage of Additive (wt%) | Composition |
|---|---|---|---|
| ADI | 1:0.25 | 25 | Form I PCA |
| 1:0.5 | 50 | Form II PCA + ADI | |
| 1:0.75 | 75 | Forms I + II PCA + ADI | |
| 1:1 | 100 | Forms I + II PCA + ADI | |
| FUM | 1:0.1 | 10 | Form I PCA |
| 1:0.2 | 20 | Form II PCA + FUM | |
| 1:0.3 | 30 | Form II PCA + FUM | |
| 1:0.5 | 50 | Form II PCA + FUM | |
| MLC | 1:0.25 | 25 | Form I PCA |
| 1:0.5 | 50 | Form I PCA | |
| 1:0.75 | 75 | Form I PCA | |
| 1:1 | 100 | Form I PCA | |
| OXA | 1:0.3 | 30 | Form I PCA |
| 1:0.6 | 60 | Form I PCA | |
| 1:0.9 | 90 | Forms I + II PCA | |
| 1:1.2 | 120 | Form II PCA | |
| SUC | 1:0.25 | 25 | Form I PCA |
| 1:0.5 | 50 | Form I PCA | |
| 1:0.75 | 75 | Forms I + II PCA | |
| 1:1 | 100 | Form II PCA |
| Expt. | Additive | Weight Percent (%) | Agitation Speed (rpm) | Induction Temperature (°C) | PCA Yield (%) | Composition |
|---|---|---|---|---|---|---|
| 1 | - | - | 300 | 52 ± 2.7 | 81.17 ± 0.83 | Form I PCA |
| 2 | - | - | 200 | 38.3 ± 1.6 | 80.75 ± 0.74 | Form I PCA |
| 3 | - | - | 100 | 38.8 ± 1.4 | 77.94 ± 0.89 | Form I PCA |
| 4 | FUM | 20 | 300 | 40.9 ± 2.9 | 79.47 ± 5.33 | Form I PCA + FUM |
| 5 | FUM | 20 | 200 | 41.1 ± 1.8 | 82.97 ± 1.84 | Form I PCA + FUM |
| 6 | FUM | 20 | 100 | 33.2 ± 4.3 | 77.91 ± 3.01 | Form I PCA + FUM |
| 7 | FUM | 50 | 300 | 43.7 ± 4.5 | 79.06 ± 2.26 | Form I PCA + FUM |
| 8 | FUM | 50 | 200 | 38.8 ± 6.4 | 80.76 ± 1.71 | Form I PCA + FUM |
| 9 | FUM | 50 | 100 | 39.2 ± 4.3 | 78.47 ± 3.32 | Form I PCA + FUM |
| 10 | OXA | 60 | 300 | 34.9 ± 6.2 | 70.94 ± 2.85 | Form I PCA |
| 11 | OXA | 60 | 200 | 33.5 ± 6.8 | 71.94 ± 2.01 | Form I PCA |
| 12 | OXA | 60 | 100 | 27.8 ± 9.1 | 70.23 ± 3.73 | Form I PCA |
| 13 | OXA | 120 | 300 | 23.8 ± 11.3 | 60.71 ± 17.29 | Form I PCA |
| 14 | OXA | 120 | 200 | 17.8 ± 0.8 | 58.06 ± 5.39 | Form I PCA |
| 15 | OXA | 120 | 100 | 19.8 ± 3.5 | 57.48 ± 3.11 | Form I PCA |
| 16 | - | - | - | 33.9 ± 5.3 | 73.74 ± 4.51 | Form I PCA |
| 17 | FUM | 20 | - | 18.2 ± 5.7 | 66.64 ± 5.46 | Forms I + II PCA + FUM |
| 18 | FUM | 50 | - | 28.2 ± 2.8 | 72.26 ± 3.72 | Forms I + II PCA + FUM |
| 19 | OXA | 60 | - | 11.2 ± 1.1 | 21.21 ± 7.62 | Form I PCA |
| 20 | OXA | 120 | - | 11.2 ± 1.2 | 1.91 ± 1.40 | Form I PCA |
| Expt. | PCA (g) | FUM (wt%) | Flow Rate (mL/min) | Yield (%) | Product Composition | |
|---|---|---|---|---|---|---|
| Collected from Outlet | Remaining in the Crystallizer | |||||
| 21 | 20 | 20 | 75 | 1.4 | Form I PCA | Form I PCA + FUM |
| 22 | 20 | 20 | 150 | 0.15 | Forms I + II PCA | Form I + II PCA + FUM |
| 23 | 20 | 30 | ˙75 | 0.35 | Forms I + II PCA + FUM | Form I + II PCA + FUM |
| 24 | 20 | 30 | 150 | 0.4 | Forms I + II PCA + FUM | Form I + II PCA + FUM |
| 25 | 20 | 50 | 75 | - | Clogging | Form I PCA + FUM |
| 26 | 20 | 50 | 150 | 0.15 | Form II PCA + FUM | Form II PCA + FUM |
| 27 | 15 | 50 | 75 | 4.27 | Form I PCA + FUM | Forms I + II PCA + FUM |
| 28 | 15 | 50 | 150 | 3.13 | Form II PCA + FUM | Form II PCA + FUM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, K.-L.; Lee, H.-L.; Lee, T. Crystallization of Form II Paracetamol with the Assistance of Carboxylic Acids toward Batch and Continuous Processes. Pharmaceutics 2022, 14, 1099. https://doi.org/10.3390/pharmaceutics14051099
Yeh K-L, Lee H-L, Lee T. Crystallization of Form II Paracetamol with the Assistance of Carboxylic Acids toward Batch and Continuous Processes. Pharmaceutics. 2022; 14(5):1099. https://doi.org/10.3390/pharmaceutics14051099
Chicago/Turabian StyleYeh, Kuan-Lin, Hung-Lin Lee, and Tu Lee. 2022. "Crystallization of Form II Paracetamol with the Assistance of Carboxylic Acids toward Batch and Continuous Processes" Pharmaceutics 14, no. 5: 1099. https://doi.org/10.3390/pharmaceutics14051099