
Leveraging Affinity Interactions to Prolong Drug Delivery of Protein Therapeutics


Abstract
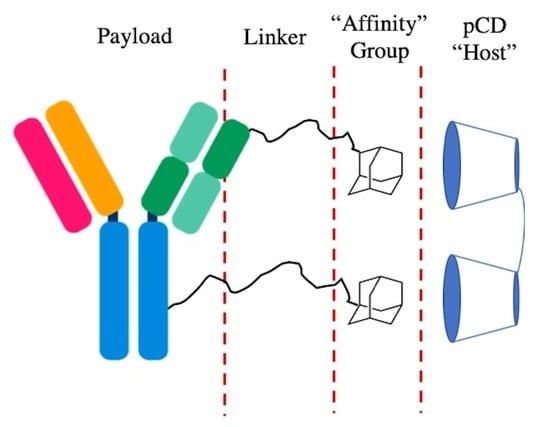
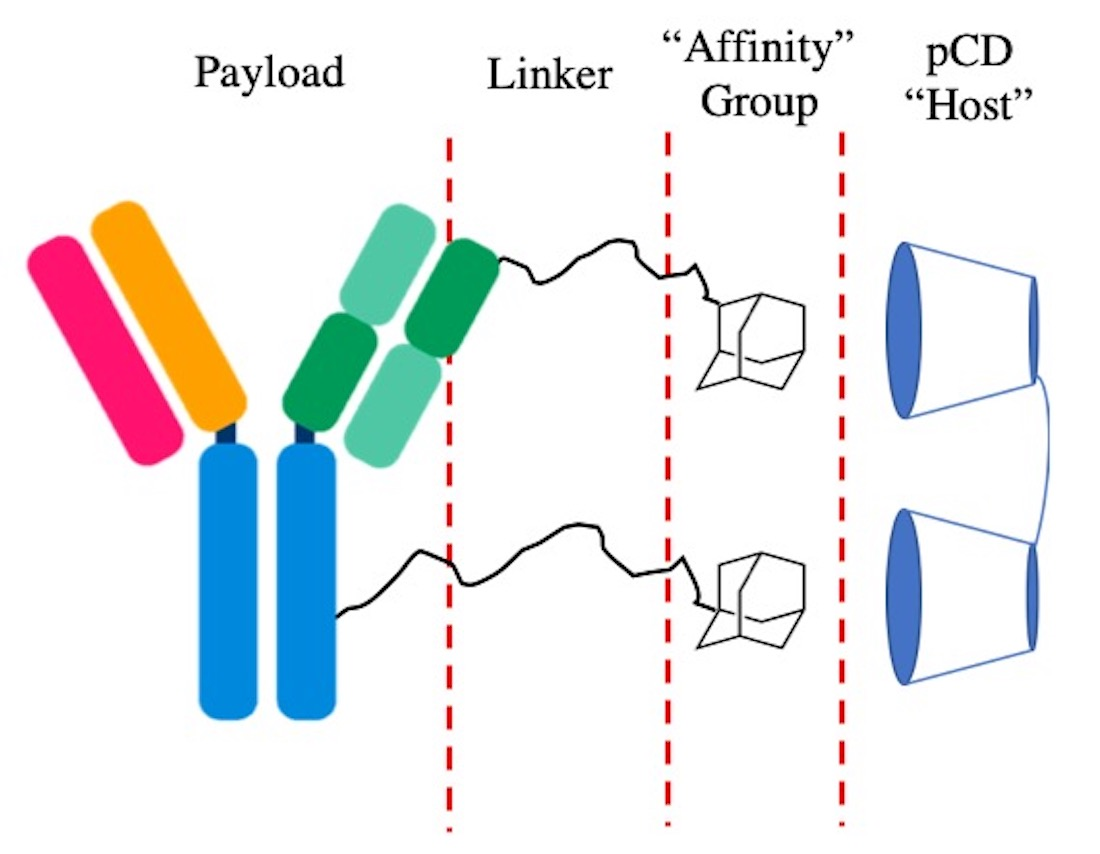
:
1. Introduction
2. Materials and Methods
2.1. Materials
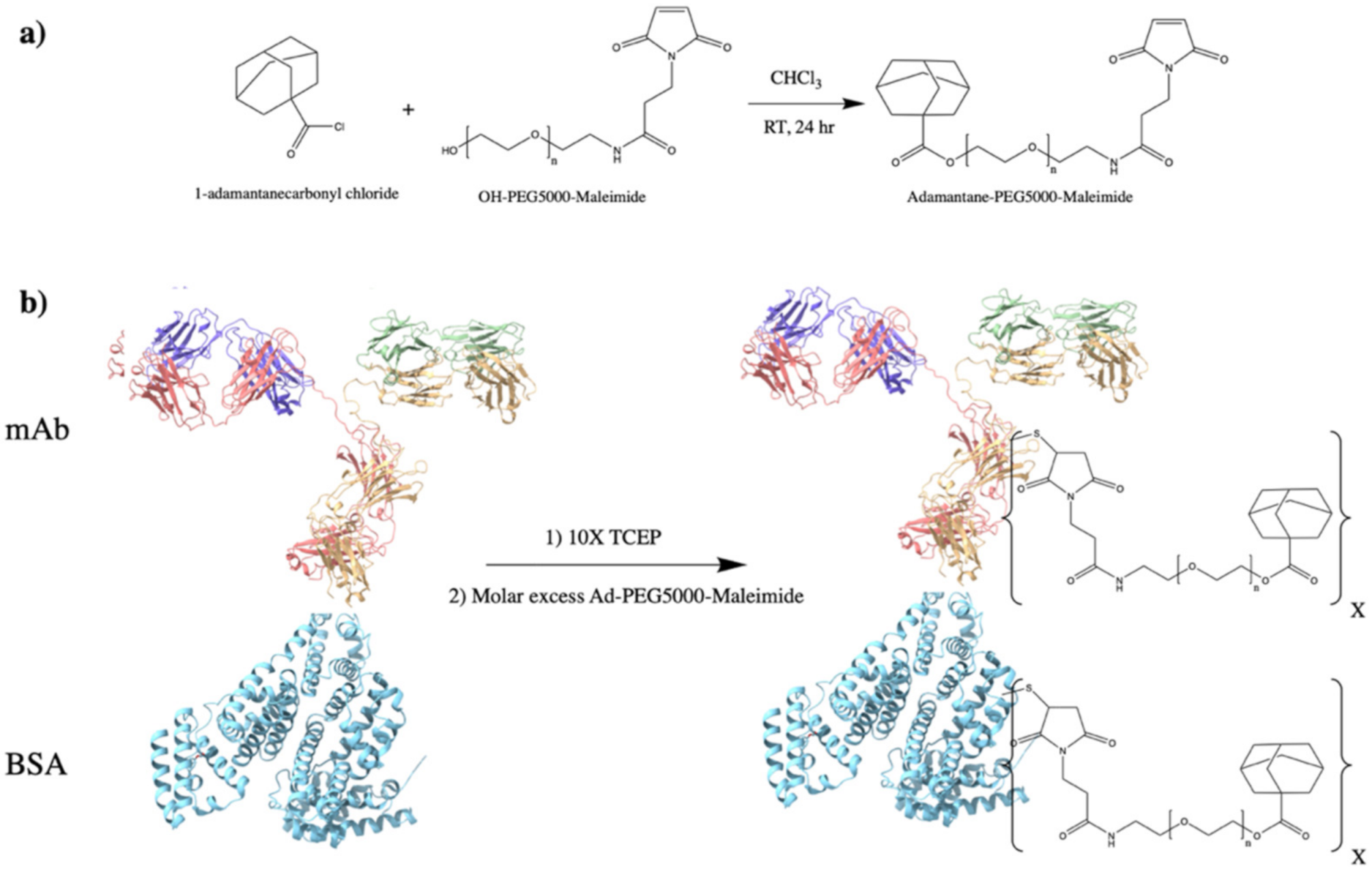
2.2. Protein–PEG–Adamantane Conjugation Synthesis
2.3. Nuclear Magnetic Resonance
2.4. Rate of Hydrolysis for (Ad–PEG5000–Mal) Species
2.5. pCD Polymer Synthesis (MPs and SRPL)
2.6. Affinity Testing—Surface Plasmon Resonance
2.7. Drug Loading and Release
2.8. Modified ELISA
2.9. Statistical Analysis
3. Results
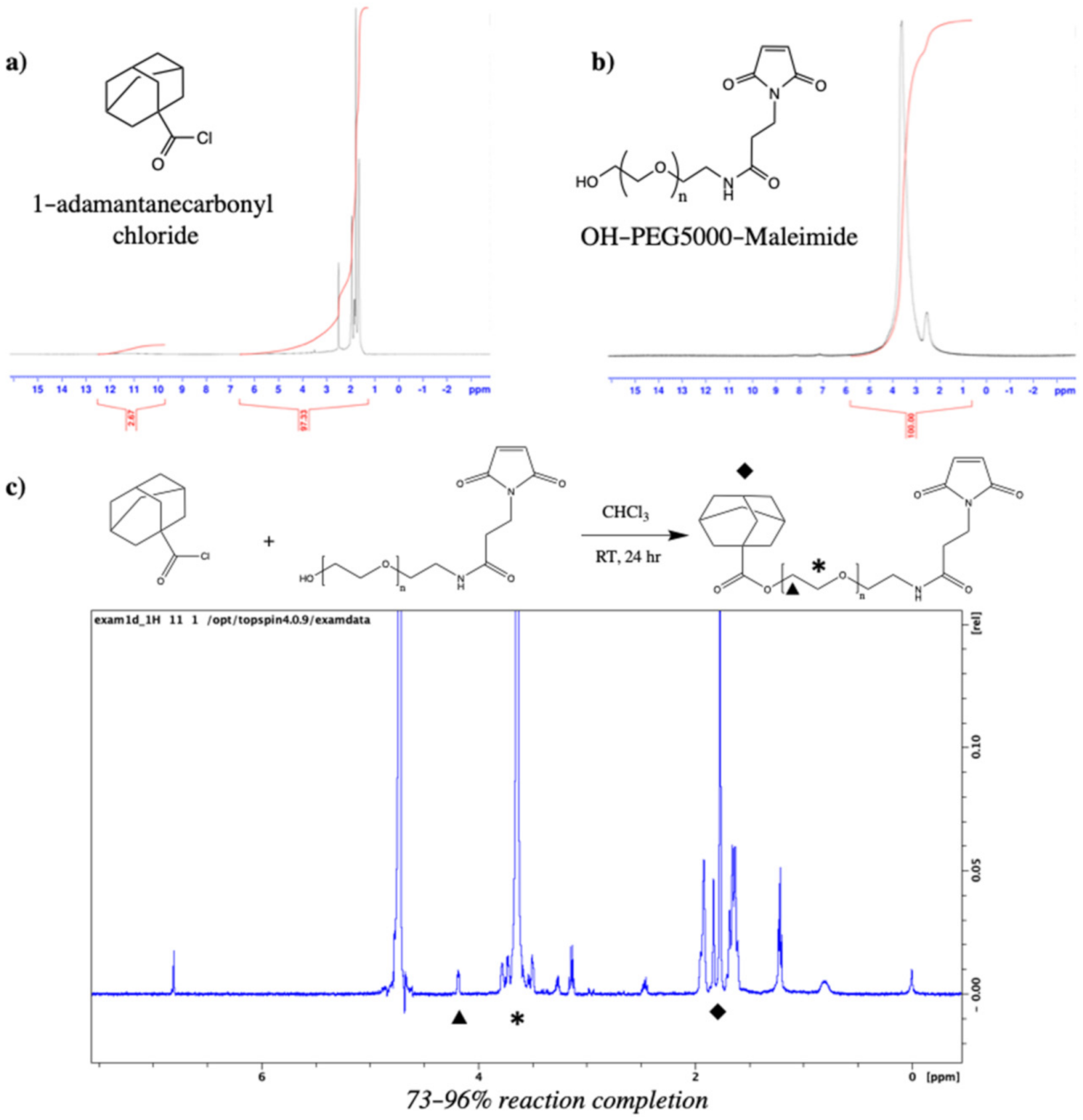
3.1. PEG–Adamantane (PEG–Ad) “Tether” Synthesis
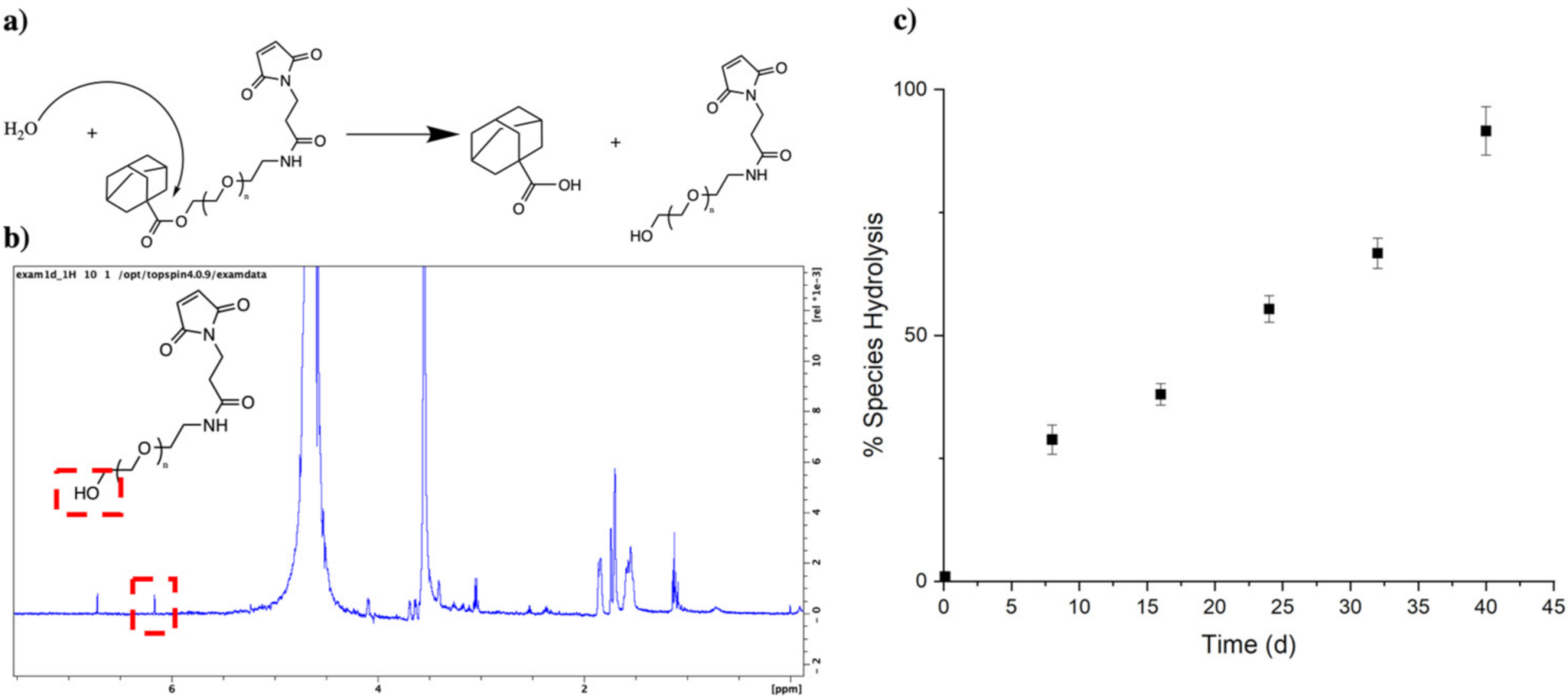
3.2. PEG–Ad “Tethers” Remain Stable for up to 40 Days
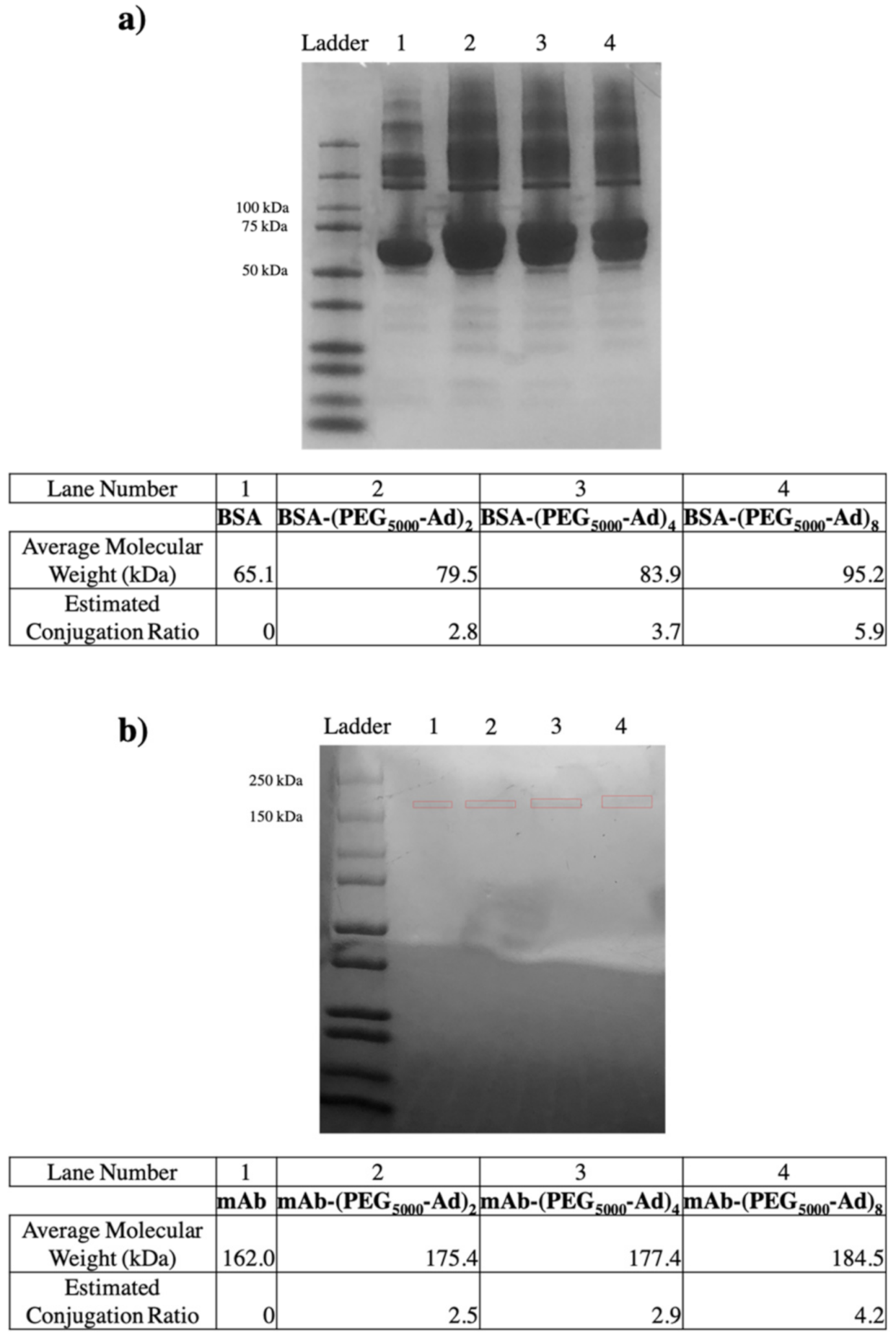
3.3. PEG–Ad “Tethers” Successfully Conjugated to Reduced Proteins
3.4. PEG–Ad “Tethers” Greatly Increased Protein–Polymer Conjugate Affinity for β-CD
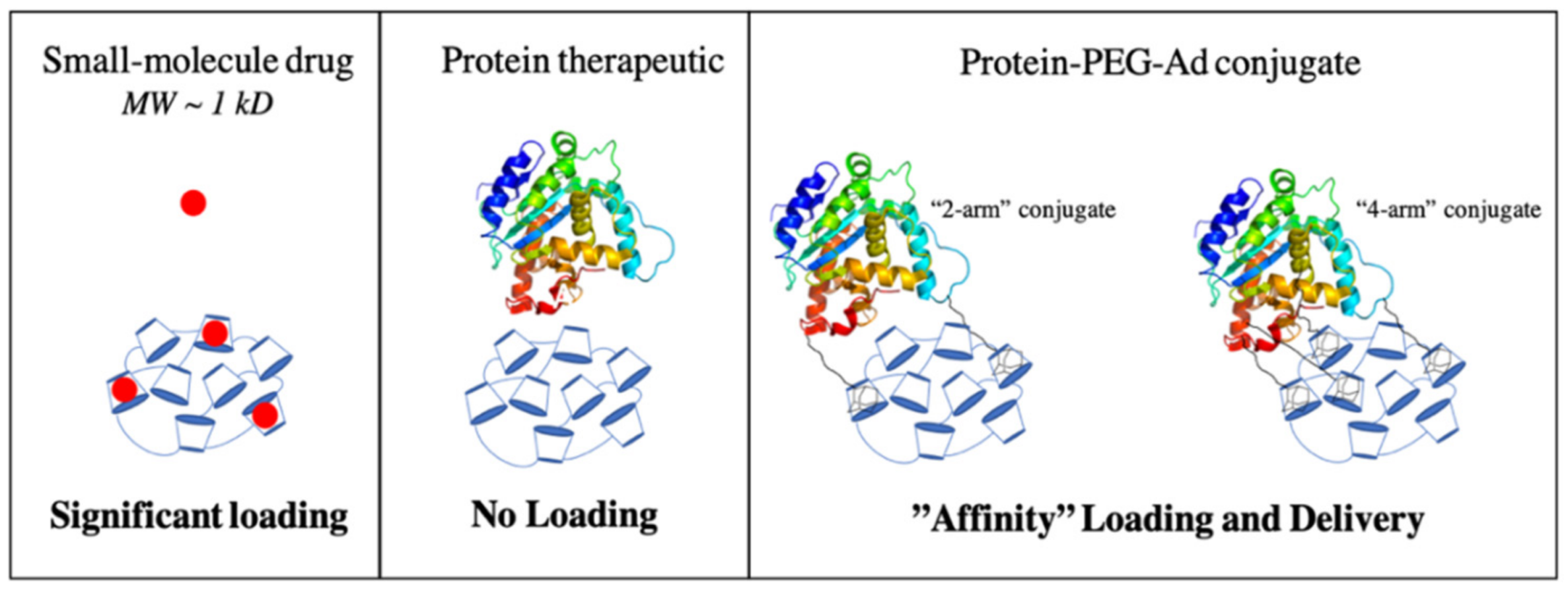
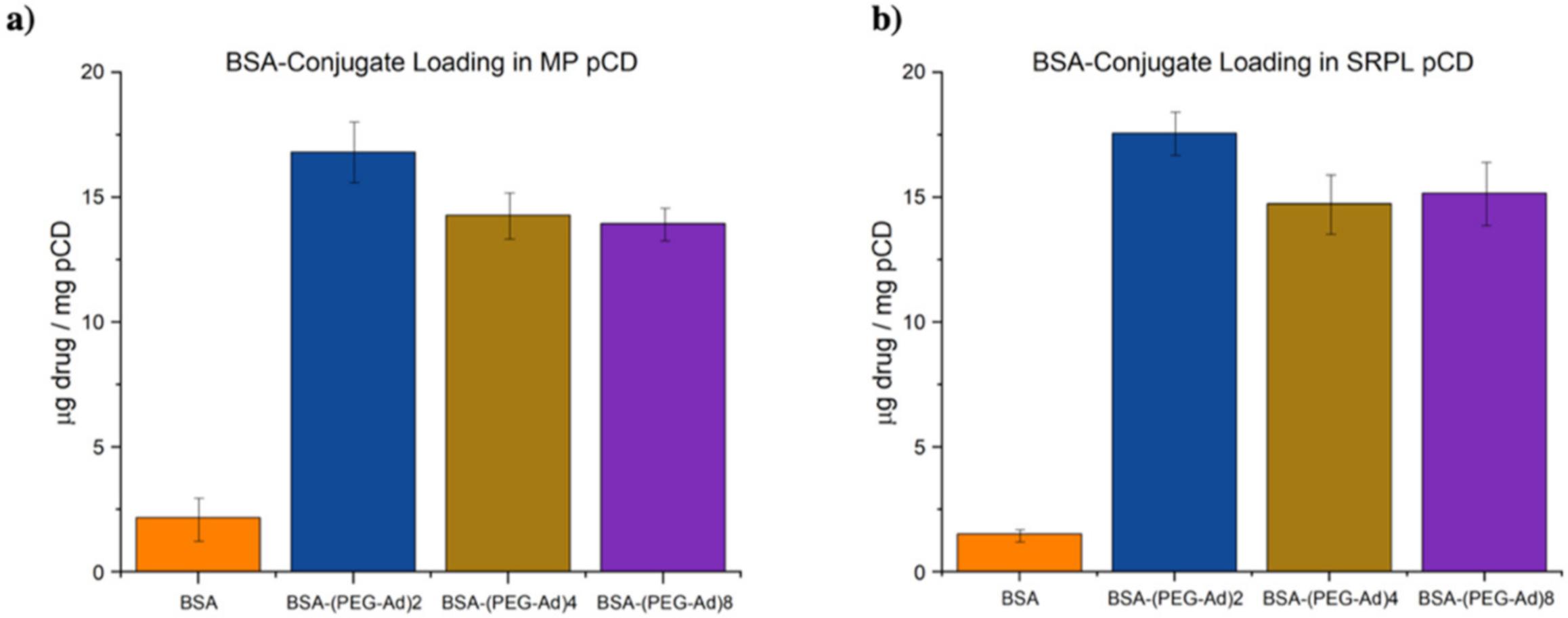
3.5. PEG–Ad Conjugation to BSA Greatly Increases Loading Capacity into pCD, Regardless of Form
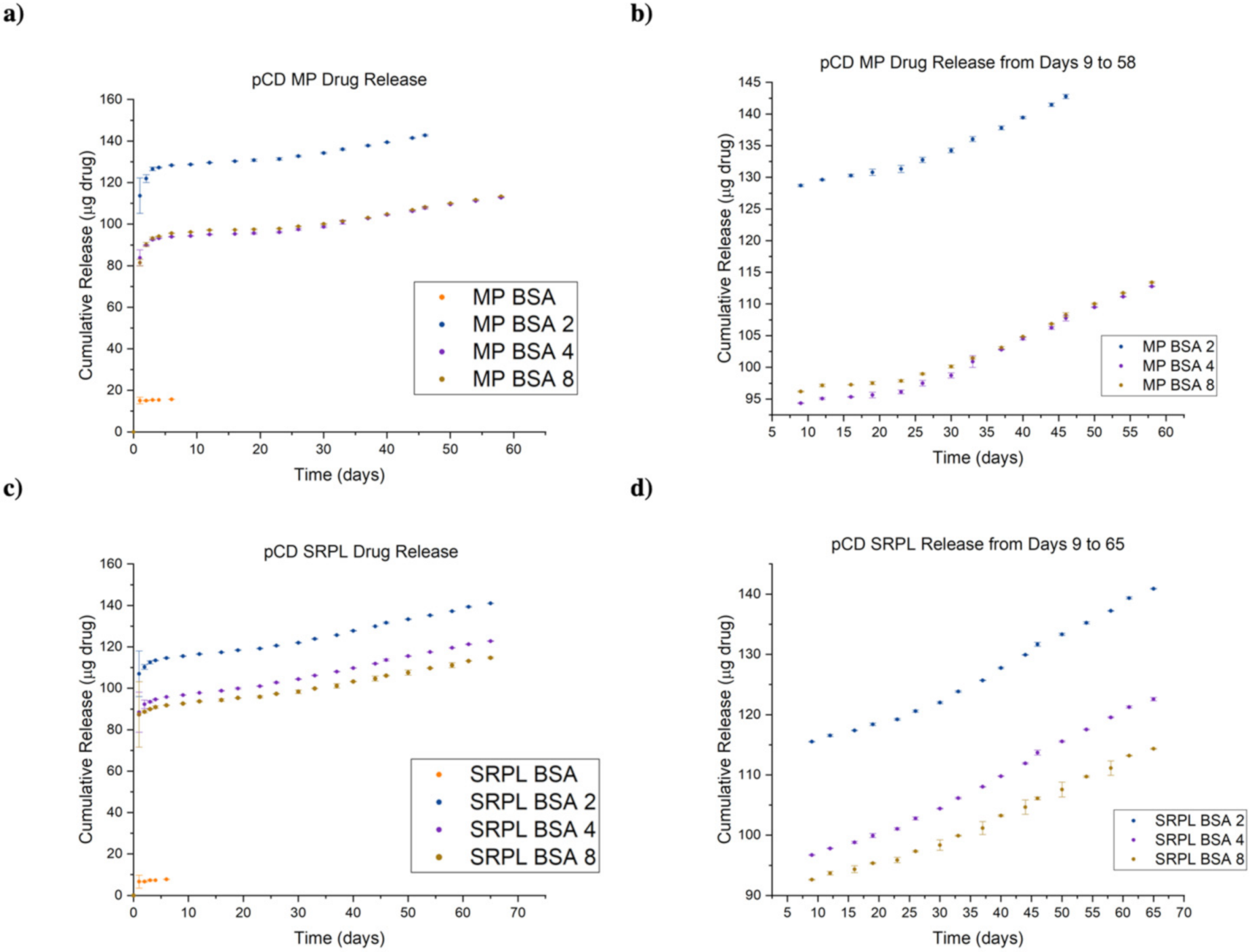
3.6. Protein–Polymer Conjugates Can Be Delivered for up to 65 Days in pCD Polymers
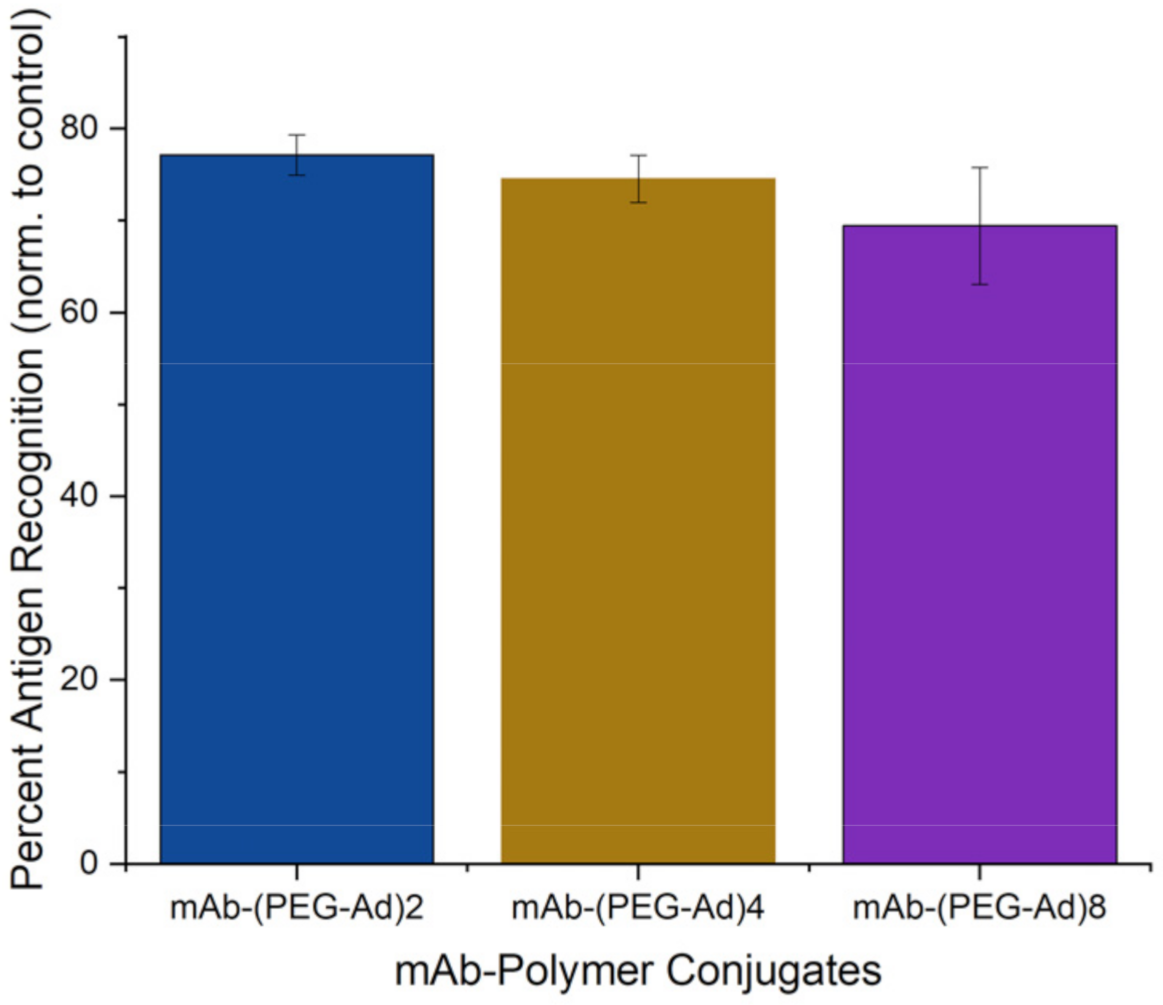
3.7. All mAb–Polymer Conjugates Maintained at Least 70% of Antigen-Recognition Ability
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Urquhart, L. Top companies and drugs by sales in 2019. Nat. Rev. Drug Discov. 2020, 19, 228. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Shepard, H.M.; Phillips, G.L.; Thanos, C.D.; Feldmann, M. Developments in therapy with monoclonal antibodies and related proteins. Clin. Med. 2017, 17, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Baum, A.; Pascal, K.E.; Russo, V.; Giordano, S.; Wloga, E.; Fulton, B.O.; Yan, Y.; Koon, K.; Patel, K.; et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020, 369, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Marovich, M.; Mascola, J.R.; Cohen, M.S. Monoclonal antibodies for prevention and treatment of COVID-19. JAMA 2020, 324, 131. [Google Scholar] [CrossRef]
- Patel, A.; Patel, M.; Yang, X.; Mitra, A. Recent advances in protein and peptide drug delivery: A special emphasis on polymeric nanoparticles. Protein Pept. Lett. 2014, 21, 1102–1120. [Google Scholar] [CrossRef] [Green Version]
- Piepenhagen, P.A.; Vanpatten, S.; Hughes, H.; Waire, J.; Murray, J.; Andrews, L.; Edmunds, T.; O’Callaghan, M.; Thurberg, B.L. Use of direct fluorescence labeling and confocal microscopy to determine the biodistribution of two protein therapeutics, Cerezyme® and Ceredase®. Microsc. Res. Tech. 2010, 73, 694–703. [Google Scholar]
- Schaefer, E.; Abbaraju, S.; Walsh, M.; Newman, D.; Salmon, J.; Amin, R.; Weiss, S.; Grau, U.; Velagaleti, P.; Gilger, B. Sustained release of protein therapeutics from subcutaneous thermosensitive biocompatible and biodegradable pentablock copolymers (PTS gels). J. Drug Deliv. 2016, 2016, 2407459. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.B.; Solorio, L.; Wu, H.; Krupka, T.; Exner, A.A. Effect of injection site on in situ implant formation and drug release in vivo. J. Control. Release 2010, 147, 350–358. [Google Scholar] [CrossRef] [Green Version]
- Martins, S.; Sarmento, B.; Ferreira, D.C.; Souto, E.B. Lipid-based colloidal carriers for peptide and protein delivery—Liposomes versus lipid nanoparticles. Int. J. Nanomed. 2007, 2, 595–607. [Google Scholar]
- Villegas, M.R.; Baeza, A.; Vallet-Regí, M. Nanotechnological strategies for protein delivery. Molecules 2018, 23, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, J.C. Nanoparticle-based systems for delivery of protein therapeutics to the spinal cord. Front. Neurosci. 2018, 12, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Rao, L.; Yu, G.; Cook, T.R.; Chen, X.; Huang, F. Supramolecular cancer nanotheranostics. Chem. Soc. Rev. 2021, 50, 2839–2891. [Google Scholar] [CrossRef]
- Ding, Y.; Tong, Z.; Jin, L.; Ye, B.; Zhou, J.; Sun, Z.; Yang, H.; Hong, L.; Huang, F.; Wang, W.; et al. An NIR discrete metallacycle constructed from perylene bisimide and tetraphenylethylene fluorophores for imaging-guided cancer radio-chemotherapy. Adv. Mater. 2022, 34, 2106388. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, M.; Schneider, J.P. Peptide hydrogels for affinity-controlled release of therapeutic cargo: Current and potential strategies. J. Pept. Sci. 2022, 28, e3377. [Google Scholar] [CrossRef] [PubMed]
- Shuvaev, V.V.; Dziubla, T.; Wiewrodt, R.; Muzykantov, V.R. Streptavidin–Biotin Crosslinking of Therapeutic Enzymes with Carrier Antibodies: Nanoconjugates for Protection against Endothelial Oxidative Stress. In Bioconjugation Protocols 003–020; Humana Press: Totowa, NJ, USA, 2004. [Google Scholar] [CrossRef]
- Huynh, V.; Wylie, R.G. Competitive affinity release for long-term delivery of antibodies from hydrogels. Angew. Chem. Int. Ed. 2018, 57, 3406–3410. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Cantor, C.R. Cooperative biotin binding by streptavidin. J. Biol. Chem. 1990, 265, 3369–3373. [Google Scholar] [CrossRef]
- Utzeri, G.; Matias, P.M.C.; Murtinho, D.; Valente, A.J.M. Cyclodextrin-based nanosponges: Overview and opportunities. Front. Chem. 2022, 10, 859406. [Google Scholar]
- Shoukat, H.; Pervaiz, F.; Khan, M.; Rehman, S.; Akram, F.; Abid, U.; Noreen, S.; Nadeem, M.; Qaiser, R.; Ahmad, R.; et al. Development of β-cyclodextrin/polyvinypyrrolidone-co-poly (2-acrylamide-2-methylpropane sulphonic acid) hybrid nanogels as nano-drug delivery carriers to enhance the solubility of Rosuvastatin: An in vitro and in vivo evaluation. PLoS ONE 2022, 17, e0263026. [Google Scholar] [CrossRef]
- Xiong, R.; Xu, R.X.; Huang, C.; De Smedt, S.; Braeckmans, K. Stimuli-responsive nanobubbles for biomedical applications. Chem. Soc. Rev. 2021, 50, 5746–5776. [Google Scholar] [CrossRef]
- Cromwell, W.C.; Byström, K.; Eftink, M.R. Cyclodextrin-adamantanecarboxylate inclusion complexes: Studies of the variation in cavity size. J. Phys. Chem. 1985, 89, 326–332. [Google Scholar] [CrossRef]
- Wang, N.X.; von Recum, H.A. Affinity-Based Drug Delivery. Macromol. Biosci. 2011, 11, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Halpern, J.M.; Gormley, C.A.; Keech, M.A.; von Recum, H.A. Thermomechanical properties, antibiotic release, and bioactivity of a sterilized cyclodextrin drug delivery system. J. Mater. Chem. B 2014, 2, 2764–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haley, R.M.; von Recum, H.A. Localized and targeted delivery of NSAIDs for treatment of inflammation: A review. Exp. Biol. Med. 2019, 244, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Cyphert, E.L.; von Recum, H.A. Emerging technologies for long-term antimicrobial device coatings: Advantages and limitations. Exp. Biol. Med. 2017, 242, 788–798. [Google Scholar] [CrossRef]
- Rivera-Delgado, E.; von Recum, H.A. Using affinity to provide long-term delivery of antiangiogenic drugs in cancer therapy. Mol. Pharm. 2017, 14, 899–907. [Google Scholar] [CrossRef]
- Singh, R.P.; Hidalgo, T.; Cazade, P.-A.; Darcy, R.; Cronin, M.F.; Dorin, I.; O’Driscoll, C.M.; Thompson, D. Self-assembled cationic β-cyclodextrin nanostructures for siRNA delivery. Mol. Pharm. 2019, 16, 1358–1366. [Google Scholar] [CrossRef] [Green Version]
- He, D.; Deng, P.; Yang, L.; Tan, Q.; Liu, J.; Yang, M.; Zhang, J. Molecular encapsulation of rifampicin as an inclusion complex of hydroxypropyl-β-cyclodextrin: Design; characterization and in vitro dissolution. Colloids Surfaces B Biointerfaces 2013, 103, 580–585. [Google Scholar] [CrossRef]
- Mealy, J.E.; Rodell, C.B.; Burdick, J.A. Sustained small molecule delivery from injectable hyaluronic acid hydrogels through host-guest mediated retention. J. Mater. Chem. B 2015, 3, 8010–8019. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, R.; Tang, L.; Yang, L. Nonviral delivery systems of mRNA vaccines for cancer gene therapy. Pharmaceutics 2022, 14, 512. [Google Scholar] [CrossRef]
- Thatiparti, T.R.; Averell, N.; Overstreet, D.; Von Recum, H.A. Multiplexing interactions to control antibiotic release from cyclodextrin hydrogels. Macromol. Biosci. 2011, 11, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Dogan, A.; von Recum, H. Engineering selective molecular tethers to enhance suboptimal drug properties. Acta Biomater. 2020, 115, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef] [Green Version]
- Rohner, N.A.; Dogan, A.B.; Robida, O.A.; von Recum, H.A. Serum biomolecules unable to compete with drug refilling into cyclodextrin polymers regardless of the form. J. Mater. Chem. B 2019, 7, 5320–5327. [Google Scholar] [CrossRef] [PubMed]
- Cyphert, E.L.; Learn, G.D.; Hurley, S.K.; Lu, C.; von Recum, H.A. An additive to pmma bone cement enables postimplantation drug refilling, broadens range of compatible antibiotics, and prolongs antimicrobial therapy. Adv. Healthc. Mater. 2018, 7, 1800812. [Google Scholar] [CrossRef]
- Chen, X.; Dong, C.; Wei, K.; Yao, Y.; Feng, Q.; Zhang, K.; Han, F.; Fuk-Tat Mak, F.; Li, B.; Bian, L. Supramolecular hydrogels cross-linked by preassembled host–guest PEG cross-linkers resist excessive, ultrafast, and non-resting cyclic compression. NPG Asia Mater. 2018, 10, 788–799. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Delgado, E.; Djuhadi, A.; Danda, C.; Kenyon, J.; Maia, J.; Caplan, A.I.; von Recum, H.A. Injectable liquid polymers extend the delivery of corticosteroids for the treatment of osteoarthritis. J. Control. Release 2018, 284, 112–121. [Google Scholar] [CrossRef]
- Rohner, N.A.; Purdue, L.N.; von Recum, H.A. Affinity-Based Polymers Provide Long-Term Immunotherapeutic Drug Delivery Across Particle Size Ranges Optimal for Macrophage Targeting. J. Pharm. Sci. 2021, 110, 1693–1700. [Google Scholar] [CrossRef]
- Rohner, N.A.; Schomisch, S.J.; Marks, J.M.; von Recum, H.A. Cyclodextrin polymer preserves sirolimus activity and local persistence for antifibrotic delivery over the time course of wound healing. Mol. Pharm. 2019, 16, 1766–1774. [Google Scholar] [CrossRef]
- Thatiparti, T.R.; Shoffstall, A.J.; von Recum, H.A. Cyclodextrin-based device coatings for affinity-based release of antibiotics. Biomaterials 2010, 31, 2335–2347. [Google Scholar] [CrossRef]
- Nejadmoghaddam, M.-R.; Minai-Tehrani, A.; Ghahremanzadeh, R.; Mahmoudi, M.; Dinarvand, R.; Zarnani, A.H. Antibody-drug conjugates: Possibilities and challenges. Avicenna J. Med. Biotechnol. 2019, 11, 3–23. [Google Scholar] [PubMed]
- Larrañeta, E.; Martínez-Ohárriz, C.; Vélaz, I.; Zornoza, A.; Machín, R.; Isasi, J.R. In vitro release from reverse poloxamine/α-cyclodextrin matrices: Modelling and comparison of dissolution profiles. J. Pharm. Sci. 2014, 103, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Aria, F.; Pagano, B.; Giancola, C. Thermodynamic properties of hydroxypropyl-β-cyclodextrin/guest interaction: A survey of recent studies. J. Therm. Anal. Calorim. 2022, 147, 4889–4897. [Google Scholar] [CrossRef]
- Dennler, P.; Fischer, E.; Schibli, R. Antibody conjugates: From heterogeneous populations to defined reagents. Antibodies 2015, 4, 197–224. [Google Scholar] [CrossRef]
- Rivera-Delgado, E.; Sadeghi, Z.; Wang, N.X.; Kenyon, J.; Satyanarayan, S.; Kavran, M.; Flask, C.; Hijaz, A.Z.; A Von Recum, H. Local release from affinity-based polymers increases urethral concentration of the stem cell chemokine CCL7 in rats. Biomed. Mater. 2016, 11, 25022. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interaction (Host/Ligand) | KD (M) | Binding Free Energy (KJ/mol) |
|---|---|---|
| βCD/BSA | 0.001293 1 | −16.5 |
| βCD/BSA–(PEG5000–Ad)2 | 7.966 × 10−5 1 | −23.4 |
| βCD/BSA–(PEG5000–Ad)4 | 9.090 × 10−9 | −45.9 |
| βCD/BSA–(PEG5000–Ad)8 | 4.099 × 10−10 | −53.6 |
| βCD/mAb | 0.0001443 1 | −21.9 |
| βCD/mAb–(PEG5000–Ad)2 | 8.755 × 10−10 1 | −52.9 |
| βCD/mAb–(PEG5000–Ad)4 | 3.012 × 10−11 | −54.1 |
| βCD/mAb–(PEG5000–Ad)8 | 4.515 × 10−13 | −70.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dogan, A.B.; Dabkowski, K.E.; von Recum, H.A. Leveraging Affinity Interactions to Prolong Drug Delivery of Protein Therapeutics. Pharmaceutics 2022, 14, 1088. https://doi.org/10.3390/pharmaceutics14051088
Dogan AB, Dabkowski KE, von Recum HA. Leveraging Affinity Interactions to Prolong Drug Delivery of Protein Therapeutics. Pharmaceutics. 2022; 14(5):1088. https://doi.org/10.3390/pharmaceutics14051088
Chicago/Turabian StyleDogan, Alan B., Katherine E. Dabkowski, and Horst A. von Recum. 2022. "Leveraging Affinity Interactions to Prolong Drug Delivery of Protein Therapeutics" Pharmaceutics 14, no. 5: 1088. https://doi.org/10.3390/pharmaceutics14051088