
Towards Development of a Non-Toxigenic Clostridioides difficile Oral Spore Vaccine against Toxigenic C. difficile



Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. Preparation of Genomic DNA
2.3. Illumina Sequencing
2.4. Bioinformatics Analysis
2.5. NTCD Spore Preparation
2.6. Ex Vivo Spore Germination Assay
2.7. Cloning of Antigens in pMTL84123 for Expression in T7
2.8. Western Immunoblotting of Whole Cells and Supernatants to Confirm Antigen Expression and Localisation
2.9. Immunisations of Hamsters
2.10. Direct ELISA to Measure Total sIgA in Intestinal Lavages
2.11. Indirect ELISA to Measure IgG Titre in Sera
2.12. Adherence Blocking Assay
2.13. Toxin Neutralisation Assay
2.14. Ethics Statement
2.15. Statistical Analysis
3. Results
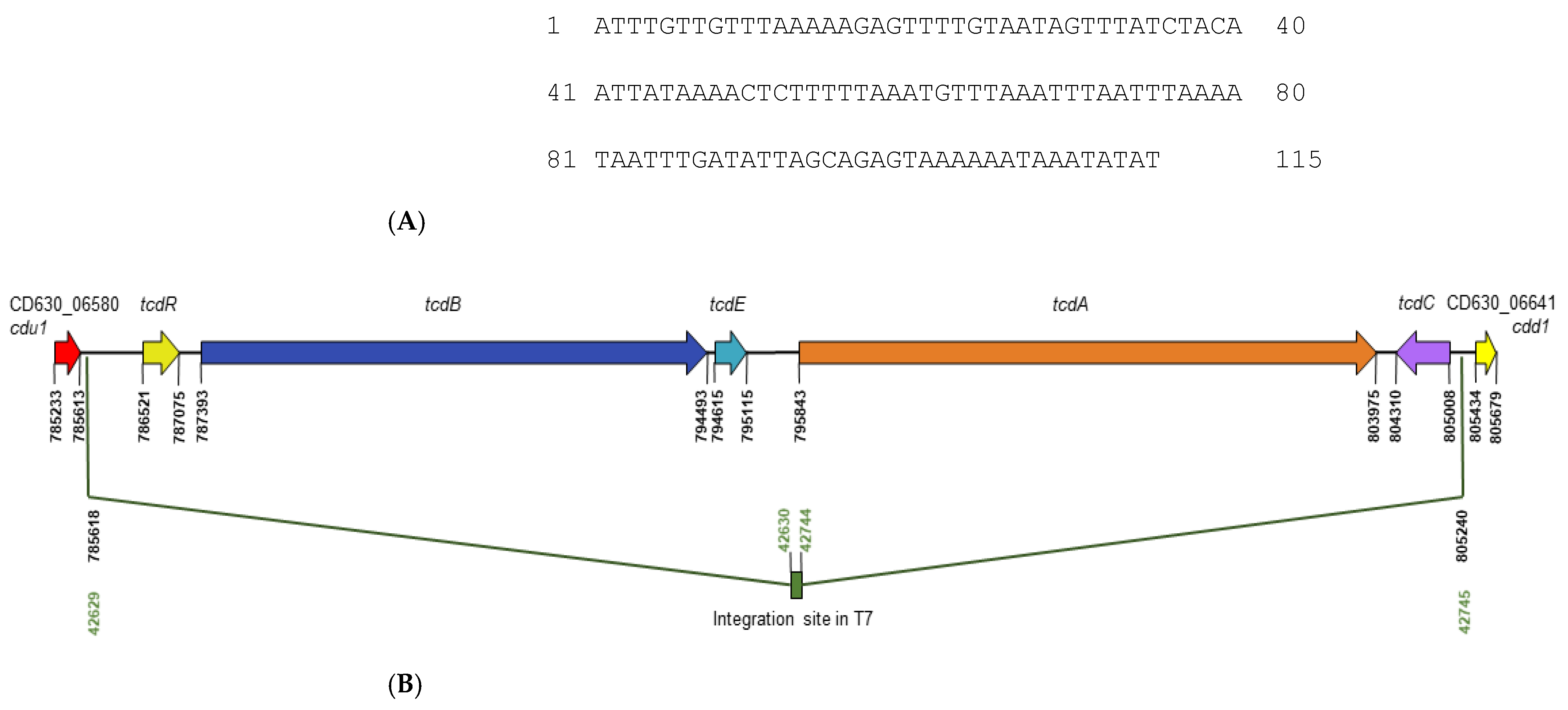
3.1. Characterisation of NTCD Chassis Strain, T7
3.1.1. NTCD T7 Lacks paLoc
3.1.2. NTCD T7 Encodes Non-Toxin Antigens
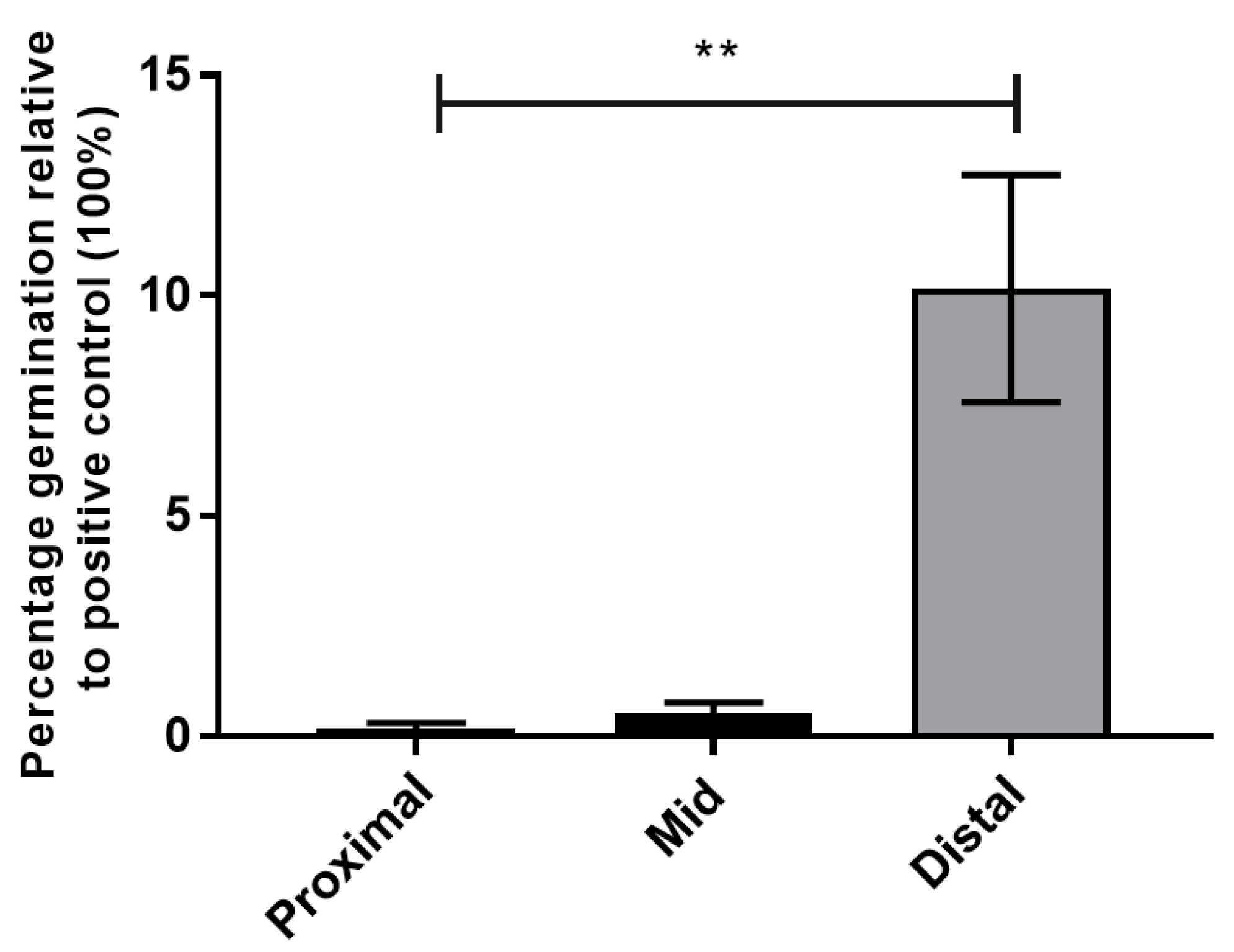
3.1.3. Spores of NTCD T7 Germinate in the Distal Small Intestine
3.2. Recombinant Overexpression of Antigens, CD0873 and TcdB-RBD, in NTCD T7
3.2.1. Cloning of Antigens in pMTL84123 for Expression in NTCD T7
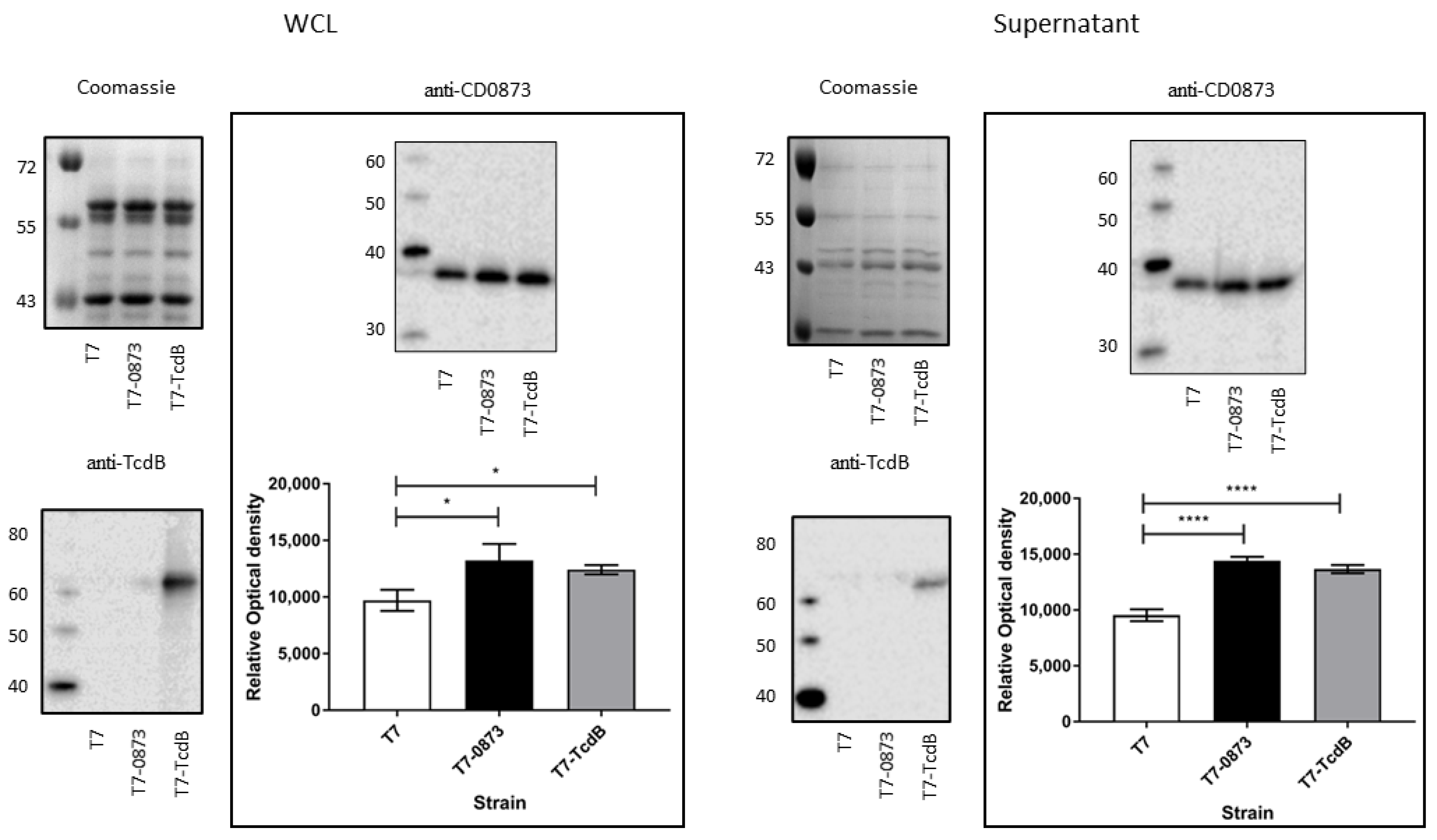
3.2.2. Confirmation of Expression of Antigens in Strains T7-0873 and T7-TcdB
3.3. Immunisation Regimen and In Vitro Cell Models to Assess Antibody Functionalities
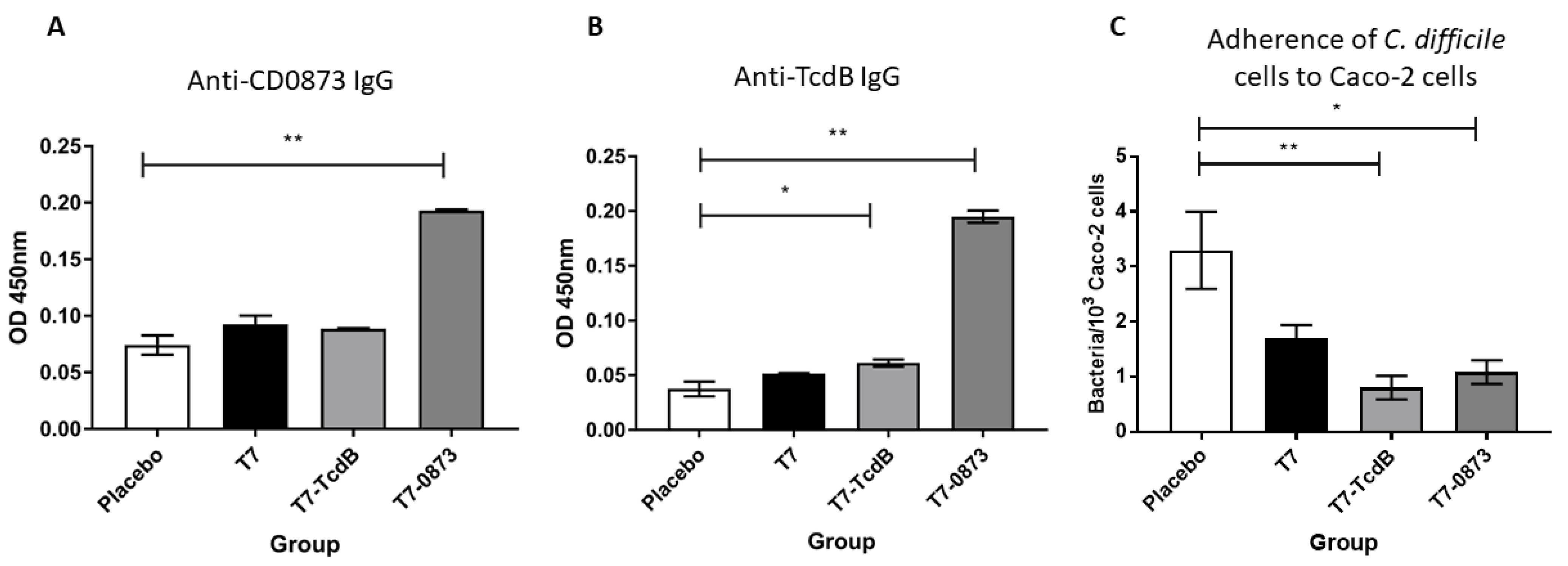
3.3.1. Intestinal Immune Responses in Vaccinated Hamsters
3.3.2. Systemic Immune Responses in Vaccinated Hamsters
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fekety, R.; Shah, A.B. Diagnosis and treatment of Clostridium difficile colitis. JAMA 1993, 269, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Mikawa, M.; Nakashio, S.; Takabatake, M.; Okado, I.; Yamakawa, K.; Serikawa, T.; Okumura, S.; Nishida, S. Isolation of Clostridium difficile from the feces and the antibody in sera of young and elderly adults. Microbiol. Immunol. 1981, 25, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Terveer, E.M.; Crobach, M.J.; Sanders, I.M.; Vos, M.C.; Verduin, C.M.; Kuijper, E.J. Detection of Clostridium difficile in Feces of Asymptomatic Patients Admitted to the Hospital. J. Clin. Microbiol. 2017, 55, 403–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rea, M.C.; Dobson, A.; O’Sullivan, O.; Crispie, F.; Fouhy, F.; Cotter, P.D.; Shanahan, F.; Kiely, B.; Hill, C.; Ross, R.P. Effect of broad- and narrow-spectrum antimicrobials on Clostridium difficile and microbial diversity in a model of the distal colon. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4639–4644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trzasko, A.; Leeds, J.A.; Praestgaard, J.; Lamarche, M.J.; McKenney, D. Efficacy of LFF571 in a hamster model of Clostridium difficile infection. Antimicrob. Agents Chemother. 2012, 56, 4459–4462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buffie, C.G.; Jarchum, I.; Equinda, M.; Lipuma, L.; Gobourne, A.; Viale, A.; Ubeda, C.; Xavier, J.; Pamer, E.G. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect. Immun. 2012, 80, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Leuzzi, R.; Adamo, R.; Scarselli, M. Vaccines against Clostridium difficile. Hum. Vaccines Immunother. 2014, 10, 1466–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, W.K.; Lyras, D.; Lacy, D.B.; Wilcox, M.H.; Kuijper, E.J. Clostridium difficile infection. Nat. Rev. Dis. Primers 2016, 2, 16020. [Google Scholar] [CrossRef] [Green Version]
- Khanna, S.; Gerding, D.N. Current and future trends in clostridioides (clostridium) difficile infection management. Anaerobe 2019, 58, 95–102. [Google Scholar] [CrossRef]
- Balsells, E.; Shi, T.; Leese, C.; Lyell, I.; Burrows, J.; Wiuff, C.; Campbell, H.; Kyaw, M.H.; Nair, H. Global burden of Clostridium difficile infections: A systematic review and meta-analysis. J. Glob. Health 2019, 9, 010407. [Google Scholar] [CrossRef]
- Czepiel, J.; Dróżdż, M.; Pituch, H.; Kuijper, E.J.; Perucki, W.; Mielimonka, A.; Goldman, S.; Wultańska, D.; Garlicki, A.; Biesiada, G. Clostridium difficile infection: Review. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2019, 38, 1211–1221. [Google Scholar] [CrossRef] [Green Version]
- Sidler, J.A.; Battegay, M.; Tschudin-Sutter, S.; Widmer, A.F.; Weisser, M. Enterococci, Clostridium difficile and ESBL-producing bacteria: Epidemiology, clinical impact and prevention in ICU patients. Swiss Med. Wkly. 2014, 144, w14009. [Google Scholar] [CrossRef]
- Cohen, S.H.; Gerding, D.N.; Johnson, S.; Kelly, C.P.; Loo, V.G.; McDonald, L.C.; Pepin, J.; Wilcox, M.H. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA). Infect. Control. Hosp. Epidemiol. 2010, 31, 431–455. [Google Scholar] [CrossRef]
- Kuijper, E.J.; Coignard, B.; Tüll, P. Emergence of Clostridium difficile-associated disease in North America and Europe. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2006, 12 (Suppl. 6), 2–18. [Google Scholar] [CrossRef] [Green Version]
- Leffler, D.A.; Lamont, J.T. Clostridium difficile Infection. N. Engl. J. Med. 2015, 373, 287–288. [Google Scholar] [CrossRef] [Green Version]
- Kochan, T.J.; Shoshiev, M.S.; Hastie, J.L.; Somers, M.J.; Plotnick, Y.M.; Gutierrez-Munoz, D.F.; Foss, E.D.; Schubert, A.M.; Smith, A.D.; Zimmerman, S.K.; et al. Germinant Synergy Facilitates Clostridium difficile Spore Germination under Physiological Conditions. mSphere 2018, 3, e00335-18. [Google Scholar] [CrossRef] [Green Version]
- Koenigsknecht, M.J.; Theriot, C.M.; Bergin, I.L.; Schumacher, C.A.; Schloss, P.D.; Young, V.B. Dynamics and establishment of Clostridium difficile infection in the murine gastrointestinal tract. Infect. Immun. 2015, 83, 934–941. [Google Scholar] [CrossRef] [Green Version]
- Wilson, K.H. Efficiency of various bile salt preparations for stimulation of Clostridium difficile spore germination. J. Clin. Microbiol. 1983, 18, 1017–1019. [Google Scholar] [CrossRef] [Green Version]
- Lawler, A.J.; Lambert, P.A.; Worthington, T. A Revised Understanding of Clostridioides difficile Spore Germination. Trends Microbiol. 2020, 28, 744–752. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef] [Green Version]
- Theriot, C.M.; Bowman, A.A.; Young, V.B. Antibiotic-Induced Alterations of the Gut Microbiota Alter Secondary Bile Acid Production and Allow for Clostridium difficile Spore Germination and Outgrowth in the Large Intestine. mSphere 2016, 1, e00045-15. [Google Scholar] [CrossRef] [Green Version]
- Dang, T.H.; de la Riva, L.; Fagan, R.P.; Storck, E.M.; Heal, W.P.; Janoir, C.; Fairweather, N.F.; Tate, E.W. Chemical probes of surface layer biogenesis in Clostridium difficile. ACS Chem. Biol. 2010, 5, 279–285. [Google Scholar] [CrossRef] [Green Version]
- Kirby, J.M.; Ahern, H.; Roberts, A.K.; Kumar, V.; Freeman, Z.; Acharya, K.R.; Shone, C.C. Cwp84, a surface-associated cysteine protease, plays a role in the maturation of the surface layer of Clostridium difficile. J. Biol. Chem. 2009, 284, 34666–34673. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, W.J.; Bruxelle, J.F.; Kovacs-Simon, A.; Harmer, N.J.; Janoir, C.; Péchiné, S.; Acharya, K.R.; Michell, S.L. Molecular features of lipoprotein CD0873: A potential vaccine against the human pathogen Clostridioides difficile. J. Biol. Chem. 2019, 294, 15850–15861. [Google Scholar] [CrossRef] [Green Version]
- Karyal, C.; Hughes, J.; Kelly, M.L.; Luckett, J.C.; Kaye, P.V.; Cockayne, A.; Minton, N.P.; Griffin, R. Colonisation Factor CD0873, an Attractive Oral Vaccine Candidate against Clostridioides difficile. Microorganisms 2021, 9, 306. [Google Scholar] [CrossRef]
- Hennequin, C.; Porcheray, F.; Waligora-Dupriet, A.; Collignon, A.; Barc, M.; Bourlioux, P.; Karjalainen, T. GroEL (Hsp60) of Clostridium difficile is involved in cell adherence. Microbiology 2001, 147, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Janoir, C. Virulence factors of Clostridium difficile and their role during infection. Anaerobe 2016, 37, 13–24. [Google Scholar] [CrossRef]
- Tasteyre, A.; Barc, M.C.; Collignon, A.; Boureau, H.; Karjalainen, T. Role of FliC and FliD flagellar proteins of Clostridium difficile in adherence and gut colonization. Infect. Immun. 2001, 69, 7937–7940. [Google Scholar] [CrossRef] [Green Version]
- Voth, D.E.; Ballard, J.D. Clostridium difficile toxins: Mechanism of action and role in disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar] [CrossRef] [Green Version]
- Barbut, F.; Richard, A.; Hamadi, K.; Chomette, V.; Burghoffer, B.; Petit, J.C. Epidemiology of recurrences or reinfections of Clostridium difficile-associated diarrhea. J. Clin. Microbiol. 2000, 38, 2386–2388. [Google Scholar] [CrossRef]
- Gerding, D.N.; Lessa, F.C. The epidemiology of Clostridium difficile infection inside and outside health care institutions. Infect. Dis. Clin. N. Am. 2015, 29, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Tonna, I.; Welsby, P.D. Pathogenesis and treatment of Clostridium difficile infection. Postgrad. Med. J. 2005, 81, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Katchar, K.; Taylor, C.P.; Tummala, S.; Chen, X.; Sheikh, J.; Kelly, C.P. Association between IgG2 and IgG3 subclass responses to toxin A and recurrent Clostridium difficile-associated disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2007, 5, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Kyne, L.; Warny, M.; Qamar, A.; Kelly, C.P. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet 2001, 357, 189–193. [Google Scholar] [CrossRef]
- Medicine NUSNLo. Study of a Candidate Clostridium difficile Toxoid Vaccine in Subjects at Risk for C. difficile Infection. Available online: https://clinicaltrials.gov/ct2/show/NCT01887912 (accessed on 16 April 2022).
- Medicine NUSNLo. Clostridium difficile Vaccine Efficacy Trial (Clover). Available online: https://clinicaltrials.gov/ct2/show/NCT03090191 (accessed on 16 April 2022).
- de Bruyn, G.; Gordon, D.L.; Steiner, T.; Tambyah, P.; Cosgrove, C.; Martens, M.; Bassily, E.; Chan, E.S.; Patel, D.; Chen, J.; et al. Safety, immunogenicity, and efficacy of a Clostridioides difficile toxoid vaccine candidate: A phase 3 multicentre, observer-blind, randomised, controlled trial. Lancet Infect. Dis. 2021, 21, 252–262. [Google Scholar] [CrossRef]
- Pfizer.com. Phase 3 CLOVER Trial for Pfizer’s Investigational Clostridioides difficile Vaccine Indicates Strong Potential Effect in Reducing Duration and Severity of Disease Based on Secondary Endpoints. Available online: https://www.pfizer.com/news/press-release/press-release-detail/phase-3-clover-trial-pfizers-investigational-clostridioides (accessed on 16 April 2022).
- Corthésy, B.; Kaufmann, M.; Phalipon, A.; Peitsch, M.; Neutra, M.R.; Kraehenbuhl, J.P. A pathogen-specific epitope inserted into recombinant secretory immunoglobulin A is immunogenic by the oral route. J. Biol. Chem. 1996, 271, 33670–33677. [Google Scholar] [CrossRef] [Green Version]
- Corthésy, B.; Benureau, Y.; Perrier, C.; Fourgeux, C.; Parez, N.; Greenberg, H.; Schwartz-Cornil, I. Rotavirus anti-VP6 secretory immunoglobulin A contributes to protection via intracellular neutralization but not via immune exclusion. J. Virol. 2006, 80, 10692–10699. [Google Scholar] [CrossRef] [Green Version]
- Johal, S.S.; Lambert, C.P.; Hammond, J.; James, P.D.; Borriello, S.P.; Mahida, Y.R. Colonic IgA producing cells and macrophages are reduced in recurrent and non-recurrent Clostridium difficile associated diarrhoea. J. Clin. Pathol. 2004, 57, 973–979. [Google Scholar] [CrossRef]
- Warny, M.; Vaerman, J.P.; Avesani, V.; Delmée, M. Human antibody response to Clostridium difficile toxin A in relation to clinical course of infection. Infect. Immun. 1994, 62, 384–389. [Google Scholar] [CrossRef] [Green Version]
- Gerding, D.N.; Sambol, S.P.; Johnson, S. Non-toxigenic Clostridioides (Formerly Clostridium) difficile for Prevention of C. difficile Infection: From Bench to Bedside Back to Bench and Back to Bedside. Front. Microbiol. 2018, 9, 1700. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, S.; Bouillaut, L.; Li, C.; Duan, Z.; Zhang, K.; Ju, X.; Tzipori, S.; Sonenshein, A.L.; Sun, X. Oral Immunization with Nontoxigenic Clostridium difficile Strains Expressing Chimeric Fragments of TcdA and TcdB Elicits Protective Immunity against C. difficile Infection in Both Mice and Hamsters. Infect. Immun. 2018, 86, e00489-18. [Google Scholar] [CrossRef] [Green Version]
- Nagaro, K.J.; Phillips, S.T.; Cheknis, A.K.; Sambol, S.P.; Zukowski, W.E.; Johnson, S.; Gerding, D.N. Nontoxigenic Clostridium difficile protects hamsters against challenge with historic and epidemic strains of toxigenic BI/NAP1/027 C. difficile. Antimicrob. Agents Chemother. 2013, 57, 5266–5270. [Google Scholar] [CrossRef] [Green Version]
- Sambol, S.P.; Merrigan, M.M.; Tang, J.K.; Johnson, S.; Gerding, D.N. Colonization for the prevention of Clostridium difficile disease in hamsters. J. Infect. Dis. 2002, 186, 1781–1789. [Google Scholar] [CrossRef] [Green Version]
- Karyal, C.; Palazi, P.; Hughes, J.; Griffiths, R.C.; Persaud, R.R.; Tighe, P.J.; Mitchell, N.J.; Griffin, R. Mimicking Native Display of CD0873 on Liposomes Augments Its Potency as an Oral Vaccine against Clostridioides difficile. Vaccines 2021, 9, 1453. [Google Scholar] [CrossRef]
- Liu, Y.W.; Chen, Y.H.; Chen, J.W.; Tsai, P.J.; Huang, I.H. Immunization with Recombinant TcdB-Encapsulated Nanocomplex Induces Protection against Clostridium difficile Challenge in a Mouse Model. Front. Microbiol. 2017, 8, 1411. [Google Scholar] [CrossRef]
- Woods, C.; Humphreys, C.M.; Rodrigues, R.M.; Ingle, P.; Rowe, P.; Henstra, A.M.; Köpke, M.; Simpson, S.D.; Winzer, K.; Minton, N.P. A novel conjugal donor strain for improved DNA transfer into Clostridium spp. Anaerobe 2019, 59, 184–191. [Google Scholar] [CrossRef]
- da Silva, R.A.G.; Karlyshev, A.V.; Oldfield, N.J.; Wooldridge, K.G.; Bayliss, C.D.; Ryan, A.; Griffin, R. Variant Signal Peptides of Vaccine Antigen, FHbp, Impair Processing Affecting Surface Localization and Antibody-Mediated Killing in Most Meningococcal Isolates. Front. Microbiol. 2019, 10, 2847. [Google Scholar] [CrossRef] [Green Version]
- Donald, R.G.K.; Flint, M.; Kalyan, N.; Johnson, E.; Witko, S.E.; Kotash, C.; Zhao, P.; Megati, S.; Yurgelonis, I.; Lee, P.K.; et al. A novel approach to generate a recombinant toxoid vaccine against Clostridium difficile. Microbiology 2013, 159, 1254–1266. [Google Scholar] [CrossRef]
- Heap, J.T.; Pennington, O.J.; Cartman, S.T.; Minton, N.P. A modular system for Clostridium shuttle plasmids. J. Microbiol. Methods 2009, 78, 79–85. [Google Scholar] [CrossRef]
- Natarajan, M.; Walk, S.T.; Young, V.B.; Aronoff, D.M. A clinical and epidemiological review of non-toxigenic Clostridium difficile. Anaerobe 2013, 22, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Braun, V.; Hundsberger, T.; Leukel, P.; Sauerborn, M.; von Eichel-Streiber, C. Definition of the single integration site of the pathogenicity locus in Clostridium difficile. Gene 1996, 181, 29–38. [Google Scholar] [CrossRef]
- Oberli, M.A.; Hecht, M.L.; Bindschädler, P.; Adibekian, A.; Adam, T.; Seeberger, P.H. A possible oligosaccharide-conjugate vaccine candidate for Clostridium difficile is antigenic and immunogenic. Chem. Biol. 2011, 18, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; St Michael, F.; Aubry, A.; Cairns, C.M.; Strong, P.C.; Hayes, A.C.; Logan, S.M. Investigating the candidacy of a lipoteichoic acid-based glycoconjugate as a vaccine to combat Clostridium difficile infection. Glycoconj. J. 2013, 30, 843–855. [Google Scholar] [CrossRef]
- Cox, A.D.; St Michael, F.; Aubry, A.; Strong, P.C.R.; Hayes, A.C.; Logan, S.M. Comparison of polysaccharide glycoconjugates as candidate vaccines to combat Clostridiodes (Clostridium) difficile. Glycoconj. J. 2021, 38, 493–508. [Google Scholar] [CrossRef]
- Péchiné, S.; Denève, C.; Le Monnier, A.; Hoys, S.; Janoir, C.; Collignon, A. Immunization of hamsters against Clostridium difficile infection using the Cwp84 protease as an antigen. FEMS Immunol. Med. Microbiol. 2011, 63, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Sandolo, C.; Péchiné, S.; Le Monnier, A.; Hoys, S.; Janoir, C.; Coviello, T.; Alhaique, F.; Collignon, A.; Fattal, E.; Tsapis, N. Encapsulation of Cwp84 into pectin beads for oral vaccination against Clostridium difficile. Eur. J. Pharm. Biopharm. 2011, 79, 566–573. [Google Scholar] [CrossRef]
- Péchiné, S.; Hennequin, C.; Boursier, C.; Hoys, S.; Collignon, A. Immunization using GroEL decreases Clostridium difficile intestinal colonization. PLoS ONE 2013, 8, e81112. [Google Scholar] [CrossRef]
- Bruxelle, J.F.; Mizrahi, A.; Hoys, S.; Collignon, A.; Janoir, C.; Péchiné, S. Immunogenic properties of the surface layer precursor of Clostridium difficile and vaccination assays in animal models. Anaerobe 2016, 37, 78–84. [Google Scholar] [CrossRef]
- Ghose, C.; Eugenis, I.; Sun, X.; Edwards, A.N.; McBride, S.M.; Pride, D.T.; Kelly, C.P.; Ho, D.D. Immunogenicity and protective efficacy of recombinant Clostridium difficile flagellar protein FliC. Emerg. Microbes Infect. 2016, 5, e8. [Google Scholar] [CrossRef] [Green Version]
- Péchiné, S.; Janoir, C.; Boureau, H.; Gleizes, A.; Tsapis, N.; Hoys, S.; Fattal, E.; Collignon, A. Diminished intestinal colonization by Clostridium difficile and immune response in mice after mucosal immunization with surface proteins of Clostridium difficile. Vaccine 2007, 25, 3946–3954. [Google Scholar] [CrossRef]
- Ghose, C.; Eugenis, I.; Edwards, A.N.; Sun, X.; McBride, S.M.; Ho, D.D. Immunogenicity and protective efficacy of Clostridium difficile spore proteins. Anaerobe 2016, 37, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giel, J.L.; Sorg, J.A.; Sonenshein, A.L.; Zhu, J. Metabolism of bile salts in mice influences spore germination in Clostridium difficile. PLoS ONE 2010, 5, e8740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.; Drudy, D.; Kyne, L.; Brown, K.; Fairweather, N.F. Immunoreactive cell wall proteins of Clostridium difficile identified by human sera. J. Med. Microbiol. 2008, 57, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Kovacs-Simon, A.; Leuzzi, R.; Kasendra, M.; Minton, N.; Titball, R.W.; Michell, S.L. Lipoprotein CD0873 is a novel adhesin of Clostridium difficile. J. Infect. Dis. 2014, 210, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Cafardi, V.; Biagini, M.; Martinelli, M.; Leuzzi, R.; Rubino, J.T.; Cantini, F.; Norais, N.; Scarselli, M.; Serruto, D.; Unnikrishnan, M. Identification of a novel zinc metalloprotease through a global analysis of Clostridium difficile extracellular proteins. PLoS ONE 2013, 8, e81306. [Google Scholar] [CrossRef]
- Griffin, R.; Minton, N.P. Exposing hidden proteins in Clostridium difficile. [Poster presentation]. In Proceedings of the ClostPath 10th International Conference on the Molecular Biology and Pathogenesis of the Clostridia, Ann Arbor, MI, USA, 10 August 2017; University of Michigan: Ann Arbor, MI, USA, 2017. [Google Scholar]
- Senoh, M.; Iwaki, M.; Yamamoto, A.; Kato, H.; Fukuda, T.; Shibayama, K. Development of vaccine for Clostridium difficile infection using membrane fraction of nontoxigenic Clostridium difficile. Microb. Pathog. 2018, 123, 42–46. [Google Scholar] [CrossRef]
- Brouwer, M.S.; Roberts, A.P.; Hussain, H.; Williams, R.J.; Allan, E.; Mullany, P. Horizontal gene transfer converts non-toxigenic Clostridium difficile strains into toxin producers. Nat. Commun. 2013, 4, 2601. [Google Scholar] [CrossRef]
- Wang, Y.K.; Yan, Y.X.; Kim, H.B.; Ju, X.; Zhao, S.; Zhang, K.; Tzipori, S.; Sun, X. A chimeric protein comprising the glucosyltransferase and cysteine proteinase domains of toxin B and the receptor binding domain of toxin A induces protective immunity against Clostridium difficile infection in mice and hamsters. Hum. Vaccines Immunother. 2015, 11, 2215–2222. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.A.; Hitri, K.; Hosseini, S.; Kotowicz, N.; Bryan, D.; Mawas, F.; Wilkinson, A.J.; van Broekhoven, A.; Kearsey, J.; Cutting, S.M. Mucosal Antibodies to the C Terminus of Toxin A Prevent Colonization of Clostridium difficile. Infect. Immun. 2017, 85, e01060-16. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Annotated Function | Nucleotide Identity | Amino Acid Identity | Source |
|---|---|---|---|---|
| Cwp84 | Cell wall-binding cysteine protease | 2396/2412 (99%) | 802/803 (99%) | [58,59] |
| GroEL | Heat shock protein | 1625/1629 (99%) | 541/542 (99%) | [60] |
| CD0873 | ABC transporter substrate-binding protein. Adhesin | 1022/1023 (99%) | 340/340 (100%) | [24,25] |
| SLpA | S-layer precursor protein | 825/1026 (80%) out of 2160 bp | 427/733 (58%) | [61] |
| FlicC | Flagellin | 873/873 (100%) | 290/290 (100%) | [62] |
| FliD | Flagellin cap protein (tested in combination with flagellar preparation) | 1524/1524 (100%) | 507/507 (100%) | [63] |
| CdeC | Spore protein. (Exosporium morphogenetic protein) | 1213/1218 (99%) | 403/405 (99%) | [64] |
| CdeM | Spore protein. (Exosporium morphogenetic protein) | 482/483 (99%) | 160/160 (100%) | [64] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hughes, J.; Aston, C.; Kelly, M.L.; Griffin, R. Towards Development of a Non-Toxigenic Clostridioides difficile Oral Spore Vaccine against Toxigenic C. difficile. Pharmaceutics 2022, 14, 1086. https://doi.org/10.3390/pharmaceutics14051086
Hughes J, Aston C, Kelly ML, Griffin R. Towards Development of a Non-Toxigenic Clostridioides difficile Oral Spore Vaccine against Toxigenic C. difficile. Pharmaceutics. 2022; 14(5):1086. https://doi.org/10.3390/pharmaceutics14051086
Chicago/Turabian StyleHughes, Jaime, Carl Aston, Michelle L. Kelly, and Ruth Griffin. 2022. "Towards Development of a Non-Toxigenic Clostridioides difficile Oral Spore Vaccine against Toxigenic C. difficile" Pharmaceutics 14, no. 5: 1086. https://doi.org/10.3390/pharmaceutics14051086