
An Overview of Cell Membrane Perforation and Resealing Mechanisms for Localized Drug Delivery


Abstract
:1. Introduction
2. Overview of Physical Plasma Membrane Permeation Techniques
2.1. Microinjection
2.2. Sonoporation
2.3. Electroporation
2.4. Photoporation
3. Plasma Membrane Repair Mechanisms
3.1. Physical Intuition
3.2. Repair Triggers
3.3. Plasma Membrane Repair Hypotheses
4. Cytoskeletal Remodeling during Perforation
4.1. Initial Reaction: Deconstruction of Actin Network
4.2. Resealing and Remodeling: Actomyosin Contractile Ring
4.3. Resealing and Remodeling: S100A11-A2
4.4. Resealing and Remodeling: Repair Cap
4.5. Resealing and Remodeling: Exo/Endocytosis Events
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef] [Green Version]
- Manzari, M.T.; Shamay, Y.; Kiguchi, H.; Rosen, N.; Scaltriti, M.; Heller, D.A. Targeted drug delivery strategies for precision medicines. Nat. Rev. Mater. 2021, 6, 351–370. [Google Scholar] [CrossRef]
- Stewart, M.P.; Langer, R.; Jensen, K.F. Intracellular delivery by membrane disruption: Mechanisms, strategies, and concepts. Chem. Rev. 2018, 118, 7409–7531. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; FaroKhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Langer, R. New Methods of Drug Delivery. Science 1990, 249, 1527–1533. [Google Scholar] [CrossRef]
- Dong, P.; Rakesh, K.P.; Manukumar, H.M.; Mohammed, Y.H.E.; Karthik, C.S.; Sumathi, S.; Mallu, P.; Qin, H.L. Innovative nano-carriers in anticancer drug delivery-a comprehensive review. Bioorg. Chem. 2019, 85, 325–336. [Google Scholar] [CrossRef]
- Andrews, N.W.; Corrotte, M. Plasma membrane repair. Curr. Biol. 2018, 28, R392–R397. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, A.J.; Perez, F. Plasma membrane repair: The adaptable cell life-insurance. Curr. Opin. Cell Biol. 2017, 47, 99–107. [Google Scholar] [CrossRef]
- McNeil, P.L.; Kirchhausen, T. An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol. 2005, 6, 499–505. [Google Scholar] [CrossRef]
- Horn, A.; Jaiswal, J.K. Cellular mechanisms and signals that coordinate plasma membrane repair. Cell. Mol. Life Sci. 2018, 75, 3751–3770. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, L.C. Single-cell microinjection technology in cell biology. BioEssays 2008, 30, 606–610. [Google Scholar] [CrossRef]
- Gordon, J.W.; Scangos, G.A.; Plotkin, D.J.; Barbosa, J.A.; Ruddle, F.H. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc. Natl. Acad. Sci. USA. 1980, 77, 7380–7384. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, A.; Zakhartchenko, V.; Weppert, M.; Sebald, H.; Wenigerkind, H.; Brem, G.; Wolf, E.; Pfeifer, A. Generation of transgenic cattle by lentiviral gene transfer into oocytes. Biol. Reprod. 2004, 71, 405–409. [Google Scholar] [CrossRef]
- Auerbach, A.B. Production of functional transgenic mice by DNA pronuclear microinjection. Acta Biochim. Pol. 2004, 51, 9–31. [Google Scholar] [CrossRef] [Green Version]
- Bongso, T.A.; Sathananthan, A.H.; Wong, P.C.; Ratnam, S.S.; Ng, S.C.; Anandakumar, C.; Ganatra, S. Human fertilization by micro-injection of immotile spermatozoa. Hum. Reprod. 1989, 4, 175–179. [Google Scholar] [CrossRef]
- Szabo, T.L. Diagnostic Ultrasound Imaging: Inside and Out, 1st ed.; Elsevier: London, UK, 2004. [Google Scholar]
- Medwin, H. Counting bubbles acoustically: A review. Ultrasonics 1977, 15, 7–13. [Google Scholar] [CrossRef]
- Averkiou, M.A.; Bruce, M.F.; Powers, J.E.; Sheeran, P.S.; Burns, P.N. Imaging Methods for Ultrasound Contrast Agents. Ultrasound Med. Biol. 2020, 46, 498–517. [Google Scholar] [CrossRef] [Green Version]
- Helfield, B. A review of phospholipid encapsulated ultrasound contrast agent microbubble physics. Ultrasound Med. Biol. 2019, 45, 282–300. [Google Scholar] [CrossRef]
- Chen, H.; Kreider, W.; Brayman, A.A.; Bailey, M.R.; Matula, T.J. Blood vessel deformations on microsecond time scales by ultrasonic cavitation. Phys. Rev. Lett. 2011, 106, 034301. [Google Scholar] [CrossRef] [Green Version]
- Cho, E.E.; Drazic, J.; Ganguly, M.; Stefanovic, B.; Hynynen, K. Two-photon fluorescence microscopy study of cerebrovascular dynamics in ultrasound-induced blood-brain barrier opening. J. Cereb. Blood Flow Metab. 2011, 31, 1852–1862. [Google Scholar] [CrossRef] [Green Version]
- Helfield, B.L.; Chen, X.; Qin, B.; Watkins, S.C.; Villanueva, F.S. Mechanistic Insight into Sonoporation with Ultrasound-Stimulated Polymer Microbubbles. Ultrasound Med. Biol. 2017, 43, 2678–2689. [Google Scholar] [CrossRef]
- Van Wamel, A.; Bouakaz, A.; Versluis, M.; de Jong, N. Micromanipulation of endothelial cells: Ultrasound-microbubble-cell interaction. Ultrasound Med. Biol. 2004, 30, 1255–1258. [Google Scholar] [CrossRef]
- Hu, Y.; Wan, J.M.F.; Yu, A.C.H. Membrane perforation and recovery dynamics in microbubble-mediated sonoporation. Ultrasound Med. Biol. 2013, 39, 2393–2405. [Google Scholar] [CrossRef]
- Helfield, B.; Chen, X.; Watkins, S.C.; Villanueva, F.S. Biophysical insight into mechanisms of sonoporation. Proc. Natl. Acad. Sci. USA 2016, 113, 9983–9988. [Google Scholar] [CrossRef] [Green Version]
- Qin, P.; Xu, L.; Han, T.; Du, L.; Yu, A.C.H. Effect of non-acoustic parameters on heterogeneous sonoporation mediated by single-pulse ultrasound and microbubbles. Ultrason. Sonochem. 2016, 31, 107–115. [Google Scholar] [CrossRef]
- Kooiman, K.; Foppen-Harteveld, M.; van der Steen, A.F.W.; de Jong, N. Sonoporation of endothelial cells by vibrating targeted microbubbles. J. Control. Release 2011, 154, 35–41. [Google Scholar] [CrossRef]
- Zeghimi, A.; Uzbekov, R.; Arbeille, B.; Escoffre, J.M.; Bouakaz, A. Ultrastructural modifications of cell membranes and organelles induced by sonoporation. In Proceedings of the IEEE International Ultrasonics Symposium, Dresden, Germany, 7–10 October 2012; pp. 2045–2048. [Google Scholar] [CrossRef]
- Mehier-Humbert, S.; Bettinger, T.; Yan, F.; Guy, R.H. Plasma membrane poration induced by ultrasound exposure: Implication for drug delivery. J. Control. Release 2005, 104, 213–222. [Google Scholar] [CrossRef]
- Schlicher, R.K.; Radhakrishna, H.; Tolentino, T.P.; Apkarian, R.P.; Zarnitsyn, V.; Prausnitz, M.R. Mechanism of intracellular delivery by acoustic cavitation. Ultrasound Med. Biol. 2006, 32, 915–924. [Google Scholar] [CrossRef]
- Prentice, P.; Cuschieri, A.; Dholakia, K.; Prausnitz, M.; Campbell, P. Membrane disruption by optically controlled microbubble cavitation. Nat. Phys. 2005, 1, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Liu, H.; Mayer, M.; Deng, C.X. Spatiotemporally controlled single cell sonoporation. Proc. Natl. Acad. Sci. USA 2012, 109, 16486–16491. [Google Scholar] [CrossRef] [Green Version]
- Helfield, B.; Black, J.J.; Qin, B.; Pacella, J.; Chen, X.; Villanueva, F.S. Fluid Viscosity Affects the Fragmentation and Inertial Cavitation Threshold of Lipid-Encapsulated Microbubbles. Ultrasound Med. Biol. 2016, 42, 782–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Thrall, B.D.; Miller, D.L. Transfection of a reporter plasmid into cultured cells by sonoporation in vitro. Ultrasound Med. Biol. 1997, 23, 953–959. [Google Scholar] [CrossRef]
- Chen, H.H.; Matkar, P.N.; Afrasiabi, K.; Kuliszewski, M.A.; Leong-Poi, H. Prospect of ultrasound-mediated gene delivery in cardiovascular applications. Expert Opin. Biol. Ther. 2017, 16, 815–826. [Google Scholar] [CrossRef]
- Hynynen, K. Ultrasound for drug and gene delivery to the brain. Adv. Drug Deliv. Rev. 2008, 60, 1209–1217. [Google Scholar] [CrossRef] [Green Version]
- Escoffre, J.M.; Piron, J.; Novell, A.; Bouakaz, A. Doxorubicin delivery into tumor cells with ultrasound and microbubbles. Mol. Pharm. 2011, 8, 799–806. [Google Scholar] [CrossRef]
- Bulner, S.; Prodeus, A.; Gariepy, J.; Hynynen, K.; Goertz, D.E. Enhancing Checkpoint Inhibitor Therapy with Ultrasound Stimulated Microbubbles. Ultrasound Med. Biol. 2019, 45, 500–512. [Google Scholar] [CrossRef]
- Meng, Y.; Reilly, R.M.; Pezo, R.C.; Trudeau, M.; Sahgal, A.; Singnurkar, A.; Perry, J.; Myrehaug, S.; Pople, C.B.; Davidson, B.; et al. MR-guided focused ultrasound enhances delivery of trastuzumab to Her2-positive brain metastases. Sci. Transl. Med. 2021, 13, eabj4011. [Google Scholar] [CrossRef]
- Kopechek, J.A.; Carson, A.R.; McTiernan, C.F.; Chen, X.; Hasjim, B.; Lavery, L.; Sen, M.; Grandis, J.R.; Villanueva, F.S. Ultrasound targeted microbubble destruction-mediated delivery of a transcription factor decoy inhibits STAT3 signaling and tumor growth. Theranostics 2015, 5, 1378–1387. [Google Scholar] [CrossRef]
- Lentacker, I.; De Smedt, S.C.; Sanders, N.N. Drug loaded microbubble design for ultrasound triggered delivery. Soft Matter 2009, 5, 2161–2170. [Google Scholar] [CrossRef]
- Carson, A.R.; McTiernan, C.F.; Lavery, L.; Grata, M.; Leng, X.; Wang, J.; Chen, X.; Villanueva, F.S. Ultrasound-targeted microbubble destruction to deliver siRNA cancer therapy. Cancer Res. 2012, 72, 6191–6199. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.M.; McMahon, D.; Hynynen, K. Ultrafast three-dimensional microbubble imaging in vivo predicts tissue damage volume distributions during nonthermal brain ablation. Theranostics 2020, 10, 7211–7230. [Google Scholar] [CrossRef] [PubMed]
- Aycock, K.N.; Davalos, R.V. Irreversible Electroporation: Background, Theory, and Review of Recent Developments in Clinical Oncology. Bioelectricity 2019, 1, 214–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwan, H.P. Electrical properties of tissue and cell suspensions. Adv. Biol. Med. Phys. 1957, 5, 147–209. [Google Scholar] [CrossRef] [PubMed]
- Gehl, J. Electroporation: Theory and methods, perspectives for drug delivery, gene therapy and research. Acta Physiol. Scand. 2003, 177, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Teissié, J.; Rols, M.P. An experimental evaluation of the critical potential difference inducing cell membrane electropermeabilization. Biophys. J. 1993, 65, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Shil, P.; Bidaye, S.; Vidyasagar, P.B. Analysing the effects of surface distribution of pores in cell electroporation for a cell membrane containing cholesterol. J. Phys. D Appl. Phys. 2008, 41, 055502. [Google Scholar] [CrossRef]
- Krassowska, W.; Filev, P.D. Modeling electroporation in a single cell. Biophys. J. 2007, 92, 404–417. [Google Scholar] [CrossRef] [Green Version]
- Kotnik, T.; Rems, L.; Tarek, M.; Miklavcic, D. Membrane Electroporation and Electropermeabilization: Mechanisms and Models. Annu. Rev. Biophys. 2019, 48, 63–91. [Google Scholar] [CrossRef]
- Frandsen, S.K.; Vissing, M.; Gehl, J. A comprehensive review of calcium electroporation —A novel cancer treatment modality. Cancers 2020, 12, 290. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, M.; Py, B.F.; Bosc, C.; Laubreton, D.; Moutin, M.J.; Marvel, J.; Flamant, F.; Markossian, S. Electroporation of mice zygotes with dual guide RNA/Cas9 complexes for simple and efficient cloning-free genome editing. Sci. Rep. 2018, 8, 474. [Google Scholar] [CrossRef] [Green Version]
- Aslan, H.; Zilberman, Y.; Arbeli, V.; Sheyn, D.; Matan, Y.; Liebergall, M.; Li, J.Z.; Helm, G.A.; Gazit, D.; Gazit, Z. Nucleofection-based Ex vivo nonviral gene delivery to human stem cells as a platform for tissue regeneration. Tissue Eng. 2006, 12, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Chicaybam, L.; Abdo, L.; Viegas, M.; Marques, L.V.C.; de Sousa, P.; Batista-Silva, L.R.; Alves-Monteiro, V.; Bonecker, S.; Monte-Mór, B.; Bonamino, M.H. Transposon-mediated generation of CAR-T cells shows efficient anti B-cell leukemia response after ex vivo expansion. Gene Ther. 2020, 27, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Lizano, C.; Sanz, S.; Luque, J.; Pinilla, M. In vitro study of alcohol dehydrogenase and acetaldehyde dehydrogenase encapsulated into human erythrocytes by an electroporation procedure. Biochim. Biophys. Acta Gen. Subj. 1998, 1425, 328–336. [Google Scholar] [CrossRef]
- Heller, R.; Jaroszeski, M.; Atkin, A.; Moradpour, D.; Gilbert, R.; Wands, J.; Nicolau, C. In vivo gene electroinjection and expression in rat liver. FEBS Lett. 1996, 389, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.; Chen, L.; Moser, M.A.J.; Zhang, W.; Qin, Z.; Zhang, B. Electroporation-Based Therapy for Brain Tumors: A Review. J. Biomech. Eng. 2021, 143, 100802. [Google Scholar] [CrossRef]
- Maor, E.; Ivorra, A.; Leor, J.; Rubinsky, B. The effect of irreversible electroporation on blood vessels. Technol. Cancer Res. Treat. 2007, 6, 307–312. [Google Scholar] [CrossRef]
- Lee, J.M.; Choi, H.S.; Chun, H.J.; Kim, E.S.; Keum, B.; Seo, Y.S.; Jeen, Y.T.; Lee, H.S.; Um, S.H.; Kim, C.D.; et al. EUS-guided irreversible electroporation using endoscopic needle-electrode in porcine pancreas. Surg. Endosc. 2019, 33, 658–662. [Google Scholar] [CrossRef]
- Rubinsky, J.; Onik, G.; Mikus, P.; Rubinsky, B. Optimal Parameters for the Destruction of Prostate Cancer Using Irreversible Electroporation. J. Urol. 2008, 180, 2668–2674. [Google Scholar] [CrossRef]
- Stevenson, D.J.; Gunn-Moore, F.J.; Campbell, P.; Dholakia, K. Single cell optical transfection. J. R. Soc. Interface 2010, 7, 863–871. [Google Scholar] [CrossRef]
- Schneckenburger, H. Laser-assisted optoporation of cells and tissues—A mini-review. Biomed. Opt. Express 2019, 10, 2883. [Google Scholar] [CrossRef]
- Peng, Q.; Juzeniene, A.; Chen, J.; Svaasand, L.O.; Warloe, T.; Giercksky, K.E.; Moan, J. Lasers in medicine. Rep. Prog. Phys. 2008, 71, 056701. [Google Scholar] [CrossRef]
- Vogel, A.; Noack, J.; Hüttman, G.; Paltauf, G. Mechanisms of femtosecond laser nanosurgery of cells and tissues. Appl. Phys. B Lasers Opt. 2005, 81, 1015–1047. [Google Scholar] [CrossRef]
- Soughayer, J.S.; Krasieva, T.; Jacobson, S.C.; Ramsey, J.M.; Tromberg, B.J.; Allbritton, N.L. Characterization of cellular optoporation with distance. Anal. Chem. 2000, 72, 1342–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Q.; Hu, W.; Ohta, A.T. Efficient single-cell poration by microsecond laser pulses. Lab Chip 2015, 15, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukakoshi, M.; Kurata, S.; Nomiya, Y.; Ikawa, Y.; Kasuya, T. A novel method of DNA transfection by laser microbeam cell surgery. Appl. Phys. B Photophysics Laser Chem. 1984, 35, 135–140. [Google Scholar] [CrossRef]
- Lei, M.; Xu, H.; Yang, H.; Yao, B. Femtosecond laser-assisted microinjection into living neurons. J. Neurosci. Methods 2008, 174, 215–218. [Google Scholar] [CrossRef]
- Uchugonova, A.; König, K.; Bueckle, R.; Isemann, A.; Tempea, G. Targeted transfection of stem cells with sub-20 femtosecond laser pulses. Opt. Express 2008, 16, 9357. [Google Scholar] [CrossRef]
- Bergeron, E.; Boutopoulos, C.; Martel, R.; Torres, A.; Rodriguez, C.; Niskanen, J.; Lebrun, J.J.; Winnik, F.M.; Sapieha, P.; Meunier, M. Cell-specific optoporation with near-infrared ultrafast laser and functionalized gold nanoparticles. Nanoscale 2015, 7, 17836–17847. [Google Scholar] [CrossRef]
- Jaiswal, J.K.; Lauritzen, S.P.; Scheffer, L.; Sakaguchi, M.; Bunkenborg, J.; Simon, S.M.; Kallunki, T.; Jäättelä, M.; Nylandsted, J. S100A11 is required for efficient plasma membrane repair and survival of invasive cancer cells. Nat. Commun. 2014, 5, 3795. [Google Scholar] [CrossRef] [Green Version]
- Croissant, C.; Gounou, C.; Bouvet, F.; Tan, S.; Bouter, A. Annexin-A6 in Membrane Repair of Human Skeletal Muscle Cell: A Role in the Cap Subdomain. Cells 2020, 9, 1742. [Google Scholar] [CrossRef]
- Boye, T.L.; Maeda, K.; Pezeshkian, W.; Sønder, S.L.; Haeger, S.C.; Gerke, V.; Simonsen, A.C.; Nylandsted, J. Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nat. Commun. 2017, 8, 1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häger, S.C.; Nylandsted, J. Annexins: Players of single cell wound healing and regeneration. Commun. Integr. Biol. 2019, 12, 162–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, G.Q.; Alderton, J.M.; Steinhardt, R.A. Calcium-regulated exocytosis is required for cell membrane resealing. J. Cell Biol. 1995, 131, 1747–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.S.; Tucker, W.C.; Chapman, E.R.; Steinhardt, R.A. Molecular regulation of membrane resealing in 3T3 fibroblasts. J. Biol. Chem. 2005, 280, 1652–1660. [Google Scholar] [CrossRef] [Green Version]
- Koerdt, S.N.; Gerke, V. Annexin A2 is involved in Ca2+-dependent plasma membrane repair in primary human endothelial cells. BBA Mol. Cell Res. 2017, 1864, 1046–1053. [Google Scholar] [CrossRef]
- Boucher, E.; Mandato, C.A. Plasma membrane and cytoskeleton dynamics during single-cell wound healing. Biochim. Biophys. Acta 2015, 1853, 2649–2661. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Weisleder, N.; Ko, J.K.; Komazaki, S.; Sunada, Y.; Nishi, M.; Takeshima, H.; Ma, J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 2009, 284, 15894–15902. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009, 11, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Dias, C.; Nylandsted, J. Plasma membrane integrity in health and disease: Significance and therapeutic potential. Cell Discov. 2021, 7, 4. [Google Scholar] [CrossRef]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT machinery is required for plasma membrane repair. Science 2014, 343, 1247136. [Google Scholar] [CrossRef]
- Scheffer, L.L.; Sreetama, S.C.; Sharma, N.; Medikayala, S.; Brown, K.J.; Defour, A.; Jaiswal, J.K. Mechanism of Ca2+-triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun. 2014, 5, 5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Gomes, T.; Corrotte, M.; Tam, C.; Andrews, N.W. Plasma membrane repair is regulated extracellularly by proteases released from lysosomes. PLoS ONE 2016, 11, e0152583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brochard-Wyart, F.; De Gennes, P.G.; Sandre, O. Transient pores in stretched vesicles: Role of leak-out. Phys. A Stat. Mech. Appl. 2000, 278, 32–51. [Google Scholar] [CrossRef] [Green Version]
- Terasaki, M.; Miyake, K.; McNeil, P.L. Large plasma membrane disruptions are rapidly resealed by Ca2+- dependent vesicle-vesicle fusion events. J. Cell Biol. 1997, 139, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Helfield, B.; Chen, X.; Watkins, S.C.; Villanueva, F.S. Transendothelial Perforations and the Sphere of Influence of Single-Site Sonoporation. Ultrasound Med. Biol. 2020, 46, 1686–1697. [Google Scholar] [CrossRef]
- Togo, T. Disruption of the plasma membrane stimulates rearrangement of microtubules and lipid traffic toward. J. Cell Sci. 2006, 1, 2780–2786. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-janvore, J.; Perez, F.; Piel, M. Laser Induced Wounding of the Plasma Membrane and Methods to Study the Repair Process; Elsevier Ltd.: Amsterdam, The Netherlands, 2015; Volume 125. [Google Scholar]
- Bushinsky, D.A.; Monk, R.D. Calcium. Lancet 1998, 352, 306–311. [Google Scholar] [CrossRef]
- Deng, C.X.; Sieling, F.; Pan, H.; Cui, J. Ultrasound-induced cell membrane porosity. Ultrasound Med. Biol. 2004, 30, 519–526. [Google Scholar] [CrossRef]
- McNeil, P.L.; Vogel, S.S.; Miyake, K.; Terasaki, M. Patching plasma membrane disruptions with cytoplasmic membrane. J. Cell Sci. 2000, 113, 1891–1902. [Google Scholar] [CrossRef]
- Potez, S.; Luginbühl, M.; Monastyrskaya, K.; Hostettler, A.; Draeger, A.; Babiychuk, E.B. Tailored protection against plasmalemmal injury by annexins with different Ca2+ sensitivities. J. Biol. Chem. 2011, 286, 17982–17991. [Google Scholar] [CrossRef] [Green Version]
- Steinhardt, R.A.; Bi, G.; Alderton, J.M. Cell Membrane Resealing by a Vesicular Mechanism Similar to Neurotransmitter Release. Science 1994, 263, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, F.G.; Handy, D.E.; Loscalzo, J. Redox regulation in the extracellular environment. Circ. J. 2008, 72, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Pan, Z.; Nishi, M.; Komazaki, S.; Takeshima, H.; Ma, J. MG53 regulates membrane budding and exocytosis in muscle cells. J. Biol. Chem. 2009, 284, 3314–3322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, M.S.F.; Caldwell, R.W.; Chiao, H.; Miyake, K.; McNeil, P.L. Contraction-induced cell wounding and release of fibroblast growth factor in heart. Circ. Res. 1995, 76, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Karatekin, E.; Sandre, O.; Guitouni, H.; Borghi, N.; Puech, P.-H.; Brochard-Wyart, F. Cascades of transient pores in giant vesicles: Line tension and transport. Biophys. J. 2003, 84, 1734–1749. [Google Scholar] [CrossRef] [Green Version]
- Draeger, A.; Monastyrskaya, K.; Babiychuk, E.B. Plasma membrane repair and cellular damage control: The annexin survival kit. Biochem. Pharmacol. 2011, 81, 703–712. [Google Scholar] [CrossRef]
- Bouter, A.; Gounou, C.; Bérat, R.; Tan, S.; Gallois, B.; Granier, T.; D’Estaintot, B.L.; Pöschl, E.; Brachvogel, B.; Brisson, A.R. Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat. Commun. 2011, 2, 270. [Google Scholar] [CrossRef] [Green Version]
- Martinez, I.; Chakrabarti, S.; Hellevik, T.; Morehead, J.; Fowler, K.; Andrews, N.W. Synaptotagmin vii regulates Ca2+-dependent exocytosis of lysosomes in fibroblasts. J. Cell Biol. 2000, 148, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.K.; Huynh, C.; Proux-Gillardeaux, V.; Galli, T.; Andrews, N.W. Identification of SNAREs Involved in Synaptotagmin VII-regulated Lysosomal Exocytosis. J. Biol. Chem. 2004, 279, 20471–20479. [Google Scholar] [CrossRef] [Green Version]
- Mercier, V.; Larios, J.; Molinard, G.; Goujon, A.; Matile, S.; Gruenberg, J.; Roux, A. Endosomal membrane tension regulates ESCRT-III-dependent intra-lumenal vesicle formation. Nat. Cell Biol. 2020, 22, 947–959. [Google Scholar] [CrossRef]
- Castro-Gomes, T.; Koushik, A.B.; Andrews, N.W. ESCRT: Nipping the wound in the bud? Trends Biochem. Sci. 2014, 39, 307–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of injured plasma membrane by rapid Ca2+ dependent endocytosis. J. Cell Biol. 2008, 180, 905–914. [Google Scholar] [CrossRef] [Green Version]
- Tam, C.; Idone, V.; Devlin, C.; Fernandes, M.C.; Flannery, A.; He, X.; Schuchman, E.; Tabas, I.; Andrews, N.W. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell Biol. 2010, 189, 1027–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassmé, H.; Riethmüller, J.; Gulbins, E. Biological aspects of ceramide-enriched membrane domains. Prog. Lipid Res. 2007, 46, 161–170. [Google Scholar] [CrossRef]
- Sonnemann, K.J.; Bement, W.M. Wound repair: Toward understanding and integration of single-cell and multicellular wound responses. Annu. Rev. Cell Dev. Biol. 2011, 27, 237–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandato, C.A.; Bement, W.M. Actomyosin Transports Microtubules and Microtubules Control Actomyosin Recruitment during Xenopus Oocyte Wound Healing. Curr. Biol. 2003, 13, 1096–1105. [Google Scholar] [CrossRef] [Green Version]
- Abreu-Blanco, M.T.; Watts, J.J.; Verboon, J.M.; Parkhurst, S.M. Cytoskeleton responses in wound repair. Cell. Mol. Life Sci. 2012, 69, 2469–2483. [Google Scholar] [CrossRef] [Green Version]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef]
- Porat-Shliom, N.; Milberg, O.; Masedunskas, A.; Weigert, R. Multiple roles for the actin cytoskeleton during regulated exocytosis. Cell. Mol. Life Sci. 2013, 70, 2099–2121. [Google Scholar] [CrossRef] [Green Version]
- Mellgren, R.L.; Zhang, W.; Miyake, K.; Mcneil, P.L. Calpain Is Required for the Rapid, Calcium-dependent Repair of Wounded Plasma Membrane. J. Biol. Chem. 2007, 282, 2567–2575. [Google Scholar] [CrossRef] [Green Version]
- Togo, T.; Krasieva, T.B.; Steinhardt, R.A. A Decrease in Membrane Tension Precedes Successful Cell-Membrane Repair. Mol. Biol. Cell 2000, 11, 4339–4346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godin, L.M.; Vergen, J.; Prakash, Y.S.; Pagano, R.E.; Hubmayr, R.D. Spatiotemporal dynamics of actin remodeling and endomembrane trafficking in alveolar epithelial type I cell wound healing. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L615–L623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, E.T.; Salmon, E.D.; Erickson, H.P. How Calcium Causes Microtubule Depolymerization. Cell Motil. Cytoskeleton. 1997, 135, 125–135. [Google Scholar] [CrossRef]
- Bi, G.; Morris, R.L.; Liao, G.; Alderton, J.M.; Scholey, J.M.; Steinhardt, R.A. Kinesin- and Myosin-driven Steps of Vesicle Recruitment for Ca 2. J. Cell Biol. 1997, 138, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Mandato, C.A.; Bement, W.M. Contraction and polymerization cooperate to assemble and close actomyosin rings around Xenopus oocyte wounds. J. Cell Biol. 2000, 154, 785–797. [Google Scholar] [CrossRef]
- Piekny, A.; Werner, M.; Glotzer, M. Cytokinesis: Welcome to the Rho zone. Trends Cell Biol. 2005, 15, 651–658. [Google Scholar] [CrossRef]
- Benink, H.A.; Bement, W.M. Concentric zones of active RhoA and Cdc42 around single cell wounds. J. Cell Biol. 2005, 168, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Prehoda, K.E.; Scott, J.A.; Mullins, R.D.; Lim, W.A. Integration of Multiple Signals Through Cooperative Regulation of the N-WASP—Arp2/3 Complex. Science 2000, 290, 801–807. [Google Scholar] [CrossRef] [Green Version]
- Kobb, A.B.; Rothenberg, K.E.; Fernandez-Gonzalez, R. Actin and myosin dynamics are independent during Drosophila embryonic wound repair. Mol. Biol. Cell 2019, 30, 2901–2912. [Google Scholar] [CrossRef]
- Abreu-Blanco, M.T.; Verboon, J.M.; Parkhurst, S.M. Coordination of Rho family GTPase activities to orchestrate cytoskeleton responses during cell wound repair. Curr. Biol. 2014, 24, 144–155. [Google Scholar] [CrossRef] [Green Version]
- Russo, J.M.; Florian, P.; Shen, L.E.; Graham, W.V.; Maria, S.; Gitter, A.H.; Mrsny, R.J.; Turner, J.R. Distinct Temporal-Spatial Roles for Rho Kinase and Myosin Light Chain Kinase in Epithelial Purse-String Wound Closure. Gastroenterology. 2006, 128, 987–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danjo, Y.; Gipson, I.K. Actin ‘purse string’ filaments are anchored by E-cadherin-mediated adherens junctions at the leading edge of the epithelial wound, providing coordinated cell movement. J. Cell Biol. 1998, 3331, 3323–3331. [Google Scholar] [CrossRef] [PubMed]
- Tamada, M.; Perez, T.D.; Nelson, W.J.; Sheetz, M.P. Two distinct modes of myosin assembly and dynamics during epithelial wound closure. J. Cell Biol. 2007, 176, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Dekraker, C.; Boucher, E.; Mandato, C.A. Regulation and Assembly of Actomyosin Contractile Rings in Cytokinesis and Cell Repair. Anat. Rec. 2018, 2066, 2051–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Chen, B.P.C.; Azuma, N.; Hu, Y.; Wu, S.Z.; Sumpio, B.E.; Shyy, J.Y.; Chien, S. Distinct roles for the small GTPases Cdc42 and Rho in endothelial responses to shear stress. J. Clin. Investig. 1999, 103, 1141–1150. [Google Scholar] [CrossRef] [Green Version]
- Horn, A.; Vandermeulen, J.H.; Defour, A.; Hogarth, M.; Reed, A.; Scheffer, L.; Chandel, N.S.; Jaiswal, J.K. Mitochondrial redox signaling enables repair of injured skeletal muscle cells. Sci. Signal. 2018, 10, eaaj1978. [Google Scholar] [CrossRef] [Green Version]
- Ashraf, A.P.K.; Gerke, V. Plasma membrane wound repair is characterized by extensive membrane lipid and protein rearrangements in vascular endothelial cells. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118991. [Google Scholar] [CrossRef]
- Ebstrup, M.L.; Dias, C.; Sofie, A.; Heitmann, B.; Sønder, S.L.; Nylandsted, J. Actin Cytoskeletal Dynamics in Single-Cell Wound Repair. Int. J. Mol. Sci. 2021, 22, 10886. [Google Scholar] [CrossRef]
- Nightingale, T.; Cutler, D. The secretion of von Willebrand factor from endothelial cells; an increasingly complicated story. J. Thromb. Haemost. 2013, 11, 192–201. [Google Scholar] [CrossRef]
- Desnos, C.; Huet, S.; Fanget, I.; Chapuis, C.; Böttiger, C.; Racine, V.; Sibarita, J.B.; Henry, J.P.; Darchen, F. Myosin Va mediates docking of secretory granules at the plasma membrane. J. Neurosci. 2007, 27, 10636–10645. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Yamakuchi, M.; Lowenstein, C.J. SNAP23 Regulates Endothelial Exocytosis of von Willebrand Factor. PLoS ONE 2015, 4, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, T.D.; Cutler, D.F.; Cramer, L.P. Actin coats and rings promote regulated exocytosis. Trends Cell Biol. 2012, 22, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Miklavc, P.; Hecht, E.; Hobi, N.; Wittekindt, O.H.; Dietl, P.; Kranz, C.; Frick, M. Actin coating and compression of fused secretory vesicles are essential for surfactant secretion—A role for Rho, formins and myosin II. J. Cell Sci. 2012, 125, 2765–2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Corrotte, M.; Almeida, P.E.; Tam, C.; Castro-gomes, T.; Fernandes, M.C.; Millis, B.A.; Cortez, M.; Miller, H.; Song, W.; Maugel, T.K.; et al. Caveolae internalization repairs wounded cells and muscle fibers. eLife 2013, 2, e00926. [Google Scholar] [CrossRef]
- Bullock, A.; Pickavance, P.; Haddow, D.; MacNeil, S. Development of a calcium-chelating hydrogel for treatment of superficial burns and scalds. Regen. Med. 2010, 5, 55–64. [Google Scholar] [CrossRef]
- Hutcheson, J.D.; Schlicher, R.K.; Hicks, H.K.; Prausnitz, M.R. Saving cells from ultrasound-induced apoptosis: Quantification of cell death and uptake following sonication and effects of targeted calcium chelation. Ultrasound Med. Biol. 2010, 36, 1008–1021. [Google Scholar] [CrossRef] [Green Version]
- Kondoh, M.; Yagi, K. Tight Junction Modulators: Promising Candidates for Drug Delivery. Curr. Med. Chem. 2007, 14, 2482–2488. [Google Scholar] [CrossRef]
- Brown, R.C.; Davis, T.P. Calcium modulation of adherens and tight junction function: A potential mechanism for blood-brain barrier disruption after stroke. Stroke 2002, 33, 1706–1711. [Google Scholar] [CrossRef]
- Kawai, K.; Larson, B.J.; Ishise, H.; Carre, A.L.; Nishimoto, S.; Longaker, M.; Lorenz, H.P. Calcium-based nanoparticles accelerate skin wound healing. PLoS ONE 2011, 6, e27106. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, D.; Walker, K.; Moise, S.; Telling, N.D.; Harper, A.G.S. Controlling human platelet activation with calcium-binding nanoparticles. Nano Res. 2020, 13, 2697–2705. [Google Scholar] [CrossRef]
- Zalba, S.; ten Hagen, T.L.M. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat. Rev. 2017, 52, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrich, A.; Michalak, K. Lipids as a Target for Drugs Modulating Multidrug Resistance of Cancer Cells. Curr. Drug Targets 2005, 4, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Mehier-Humbert, S.; Bettinger, T.; Yan, F.; Guy, R. Influence of polymer adjuvants on the ultrasound-mediated transfection of cells in culture. Eur. J. Pharm. Biopharm. 2009, 72, 567–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, R.; Borgens, R.B. Anatomical repair of nerve membranes in crushed mammalian spinal cord with polyethylene glycol. J. Neurocytol. 2000, 29, 633–643. [Google Scholar] [CrossRef]
- Regev, R.; Katzir, H.; Yeheskely-Hayon, D.; Eytan, G.D. Modulation of P-glycoprotein-mediated multidrug resistance by acceleration of passive drug permeation across the plasma membrane. FEBS J. 2007, 274, 6204–6214. [Google Scholar] [CrossRef]
- Carugo, D.; Aron, M.; Sezgin, E.; Bernardino de la Serna, J.; Kuimova, M.K.; Eggeling, C.; Stride, E. Modulation of the molecular arrangement in artificial and biological membranes by phospholipid-shelled microbubbles. Biomaterials 2017, 113, 105–117. [Google Scholar] [CrossRef]
- Quinn, P.J. Lipid-lipid interactions in bilayer membranes: Married couples and casual liaisons. Prog. Lipid Res. 2012, 51, 179–198. [Google Scholar] [CrossRef]
- Goñi, F.M.; Alonso, A. Effects of ceramide and other simple sphingolipids on membrane lateral structure. Biochim. Biophys. Acta Biomembr. 2009, 1788, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Stover, T.C.; Sharma, A.; Robertson, G.P.; Kester, M. Systemic delivery of liposomal short-chain ceramide limits solid tumor growth in murine models of breast adenocarcinoma. Clin. Cancer Res. 2005, 11, 3465–3474. [Google Scholar] [CrossRef] [Green Version]
- Escribá, P.V.; Busquets, X.; Inokuchi, J.I.; Balogh, G.; Török, Z.; Horváth, I.; Harwood, J.L.; Vígh, L. Membrane lipid therapy: Modulation of the cell membrane composition and structure as a molecular base for drug discovery and new disease treatment. Prog. Lipid Res. 2015, 59, 38–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, R.H.; Gordon, J.M.; Woodman, O.L.; Cao, A.H.; Dusting, G.J. Annexin-1 peptide Anx-12-26 protects adult rat cardiac myocytes from cellular injury induced by simulated ischaemia. Br. J. Pharmacol. 2005, 145, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.J.; Xia, C.J.; Wei, Y.; Yao, Y.; Dong, M.W.; Lin, K.Z.; Yu, L.S.; Gao, Y.; Fan, Y.Y. Annexin A1-derived peptide Ac2-26 facilitates wound healing in diabetic mice. Wound Repair Regen. 2020, 28, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Leoni, G.; Neumann, P.A.; Kamaly, N.; Quiros, M.; Nishio, H.; Jones, H.R.; Sumagin, R.; Hilgarth, R.S.; Alam, A.; Fredman, G.; et al. Annexin A1′containing extracellular vesicles and polymeric nanoparticles promote epithelial wound repair. J. Clin. Investig. 2015, 125, 1215–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purvis, G.S.D.; Solito, E.; Thiemermann, C. Annexin-A1: Therapeutic potential in microvascular disease. Front. Immunol. 2019, 10, 938. [Google Scholar] [CrossRef] [Green Version]
- Demonbreun, A.R.; Fallon, K.S.; Oosterbaan, C.C.; Bogdanovic, E.; Warner, J.L.; Sell, J.J.; Page, P.G.; Quattrocelli, M.; Barefield, D.Y.; McNally, E.M. Recombinant annexin A6 promotes membrane repair and protects against muscle injury. J. Clin. Investig. 2019, 129, 4657–4670. [Google Scholar] [CrossRef]
- Mui, L.; Martin, C.M.; Tschirhart, B.J.; Feng, Q. Therapeutic Potential of Annexins in Sepsis and COVID-19. Front. Pharmacol. 2021, 12, 2377. [Google Scholar] [CrossRef] [PubMed]
- Burkel, B.M.; Benink, H.A.; Vaughan, E.M.; von Dassow, G.; Bement, W.M. A Rho GTPase signal treadmill backs a contractile array. Dev. Cell 2013, 23, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, R.; Kamei, M.; Sage, P.T.; Massol, R.; Varghese, L.; Sciuto, T.; Toporsian, M.; Dvorak, A.M.; Kirchhausen, T.; Springer, T.A.; et al. Release of cellular tension signals self-restorative ventral lamellipodia to heal barrier micro-wounds. J. Cell Biol. 2013, 201, 449–465. [Google Scholar] [CrossRef] [Green Version]
- Ñeco, P.; Fernández-Peruchena, C.; Navas, S.; Gutiérrez, L.M.; Álvarez De Toledo, G.; Alés, E. Myosin II contributes to fusion pore expansion during exocytosis. J. Biol. Chem. 2008, 283, 10949–10957. [Google Scholar] [CrossRef] [Green Version]


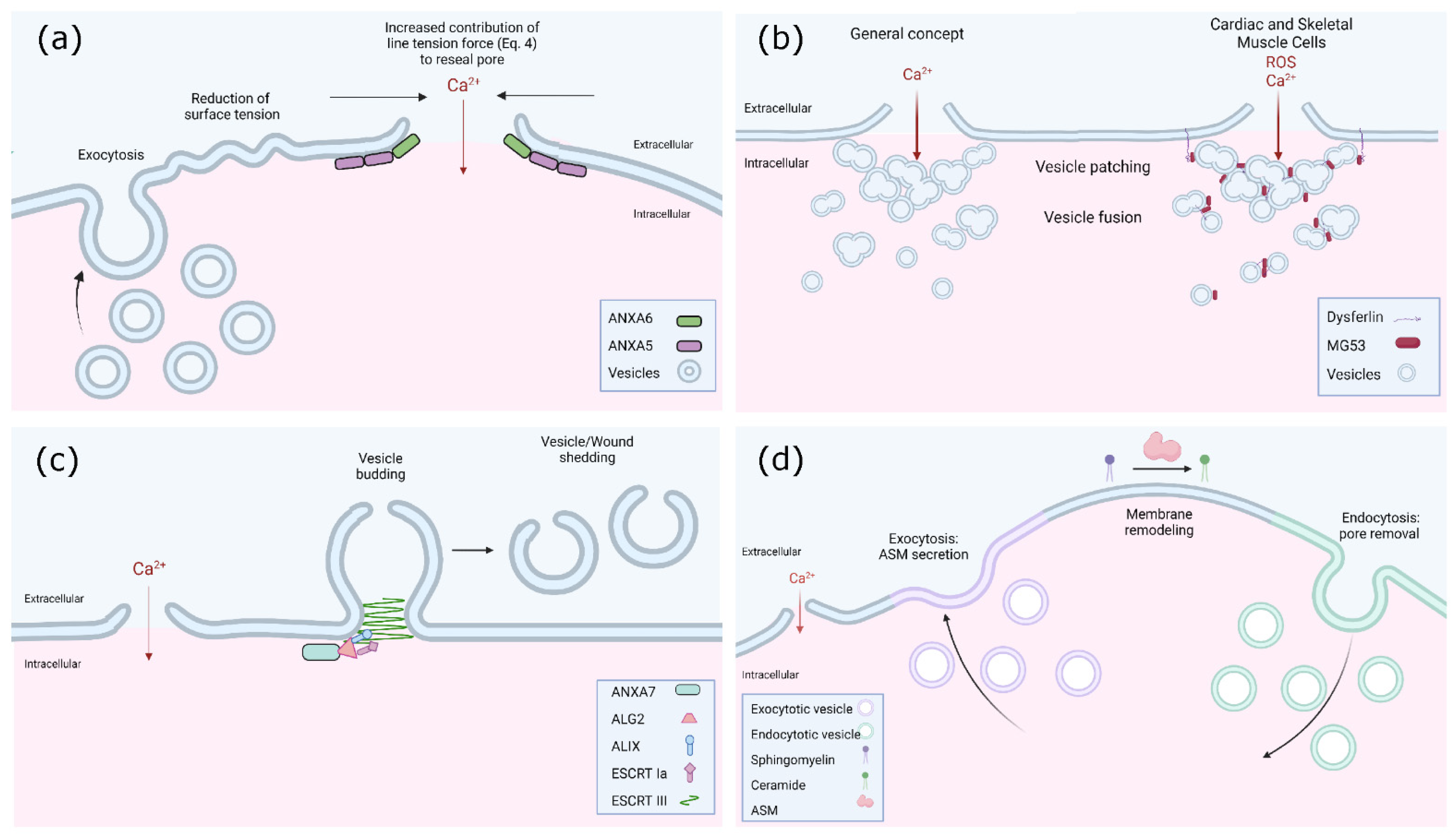{kind=link}
| Method | Pore Features | Relative Advantages | Relative Disadvantages |
|---|---|---|---|
| Microinjection | Single pore of similar size to the fine-tipped glass micropipette (~200–1000 nm). |
|
|
| Sonoporation | Pore radii ranging from sub-micron to ~10 µm. Single pore per bubble, with the possibility of multiple pores per cell. |
|
|
| Electroporation | Pore radii generally < 1nm, with up to 109 pores per cm2. |
|
|
| Photoporation | Pore radii ranging from ~10–2000 nm. |
|
|
| Protein Family | Role | Ca2+ Binding | Estimated Pore Sizes in Which Proteins Have Been Observed | Suggested Plasma Membrane Repair Mechanism(s) |
|---|---|---|---|---|
| Annexins | Play a role in membrane patching, fusion, reshaping, reducing membrane tension, removing damaged membrane, limiting pore expansion | Yes | m [71,72,73] | Patch, Tension Reduction, Exocytosis/Endocytosis, Membrane Budding (A7 [74]) |
| SNARE proteins | Mediates fusion of membranes | No | m [75,76] | Patch, Tension Reduction, Exocytosis/Endocytosis |
| SYT7 | Helps activate SNAREs | Yes | ||
| S100A11 | Implicated in membrane and cytoskeletal dynamics, interacts with A2 | Yes | m [71,77] | Tension Reduction [78] |
| Dysferlin | In muscle cells, accumulates at the site of membrane damage, interacts with some annexins, MG53, BIN1, EHD1/2 | Yes | nm scale [79], μm scale [72] | Patch, Exocytosis/Endocytosis |
| MG53 | In muscle cells, it is tethered to plasma membrane and intracellular vesicles and, upon ROS stimulus, oligomerizes and accumulates at wound sites | No | nm scale [79] | Patch [80], Tension Reduction [81] |
| ESCRT-III | Involved in membrane budding | No | <100 nm [82], >1 μm [83] | Membrane Budding |
| ESCRT-I | Recruits ESCRT-III | No | ||
| ALIX | Recruits ESCRT machinery | Yes | ||
| ALG-2 | Recruits ALIX | Yes | ||
| ASM | Outer plasma membrane remodeling to initiate inward vesicle budding. Converts sphingomyelin into ceramide | No | nm scale [84] | Exocytosis/Endocytosis |
| Cytoskeletal Remodeling | Main Proteins Involved | Estimated Pore Sizes in Which Proteins Have Been Observed |
|---|---|---|
| Contractile Ring [108] | Actin, myosin II, GTPases (Cdc42, Rho), Arp2/3, | m scale [109,110] |
| S100A11-A2 [71] | S100A11, annexins A1, A2 | m [71,77] |
| Repair Cap [111] | Annexins A1, A2, A5 and A6, dysferlin, EHD1/2, MG53, BIN1 | m scale [72] |
| Exocytosis/Endocytosis (e.g., [112]) | Myosin family, kinesin, actin, microtubules, GTPases, formins, SNARE complexes | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, S.; Singh, D.; Helfield, B. An Overview of Cell Membrane Perforation and Resealing Mechanisms for Localized Drug Delivery. Pharmaceutics 2022, 14, 886. https://doi.org/10.3390/pharmaceutics14040886
He S, Singh D, Helfield B. An Overview of Cell Membrane Perforation and Resealing Mechanisms for Localized Drug Delivery. Pharmaceutics. 2022; 14(4):886. https://doi.org/10.3390/pharmaceutics14040886
Chicago/Turabian StyleHe, Stephanie, Davindra Singh, and Brandon Helfield. 2022. "An Overview of Cell Membrane Perforation and Resealing Mechanisms for Localized Drug Delivery" Pharmaceutics 14, no. 4: 886. https://doi.org/10.3390/pharmaceutics14040886