
The Importance of Endoplasmic Reticulum Stress as a Novel Antidepressant Drug Target and Its Potential Impact on CNS Disorders

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. MTT Test
2.4. Gene Expression Analysis
2.5. Annexin V Labelling
2.6. CHOP Protein Expression Analysis
2.7. ELISA Assay
2.8. Data Analysis
3. Results
3.1. Effect of Tunicamycin and Antidepressants on the Viability of Astrocytes
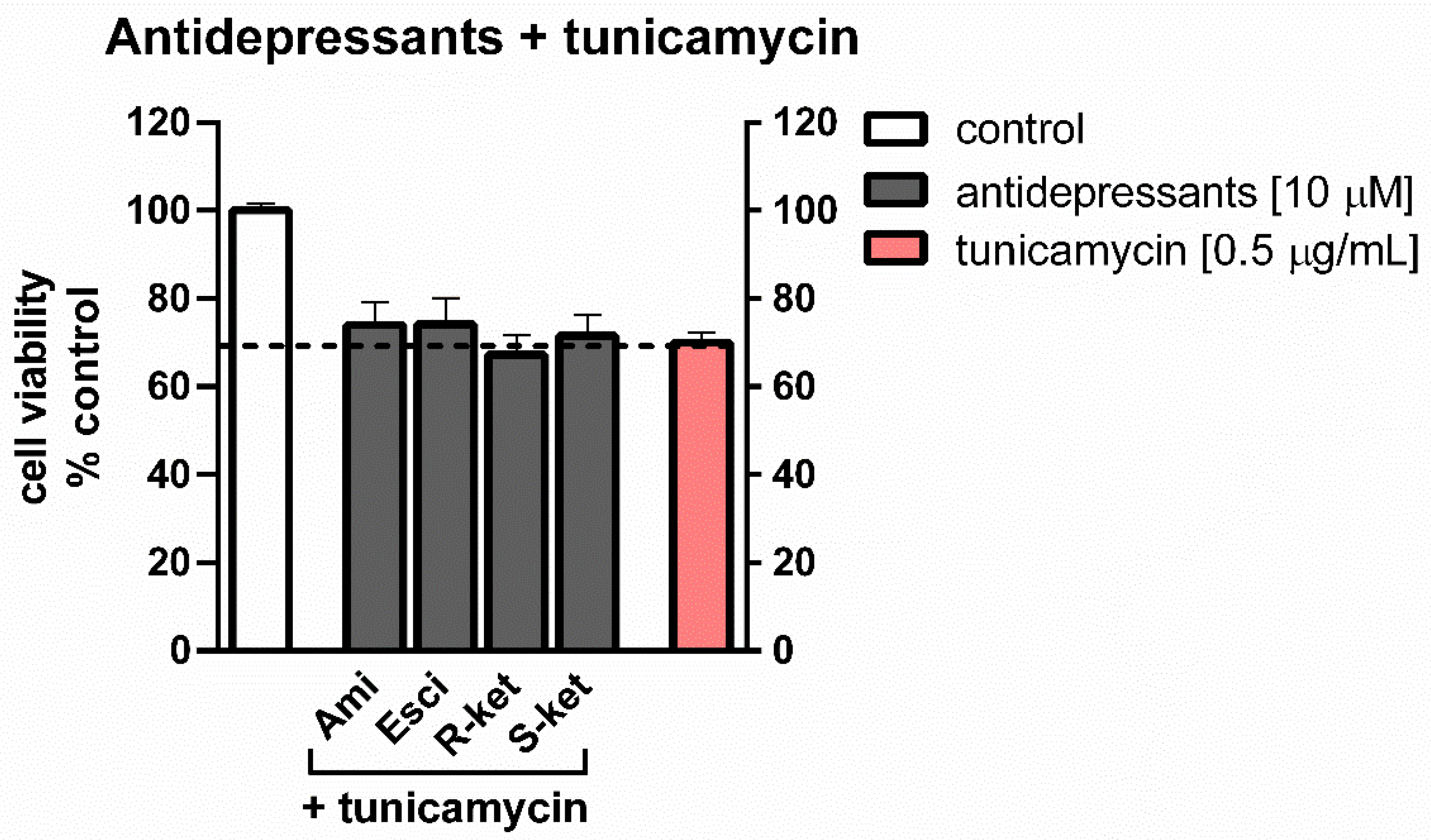
3.2. Effects of Tunicamycin and Antidepressants on the Viability of Astrocytes Undergoing ER Stress
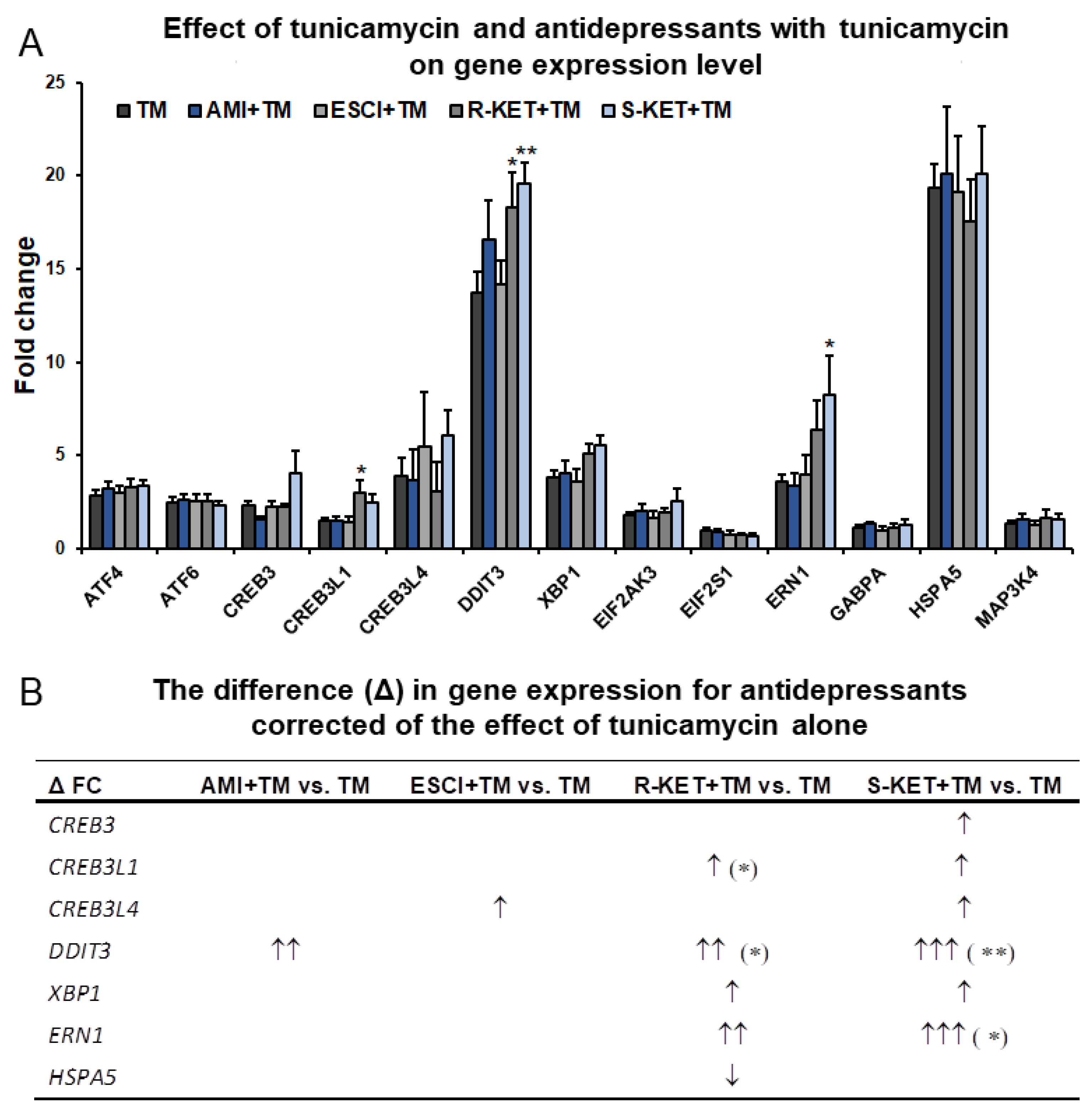
3.3. Effects of Antidepressants on Gene Expression Related to ER Stress in Astrocytes
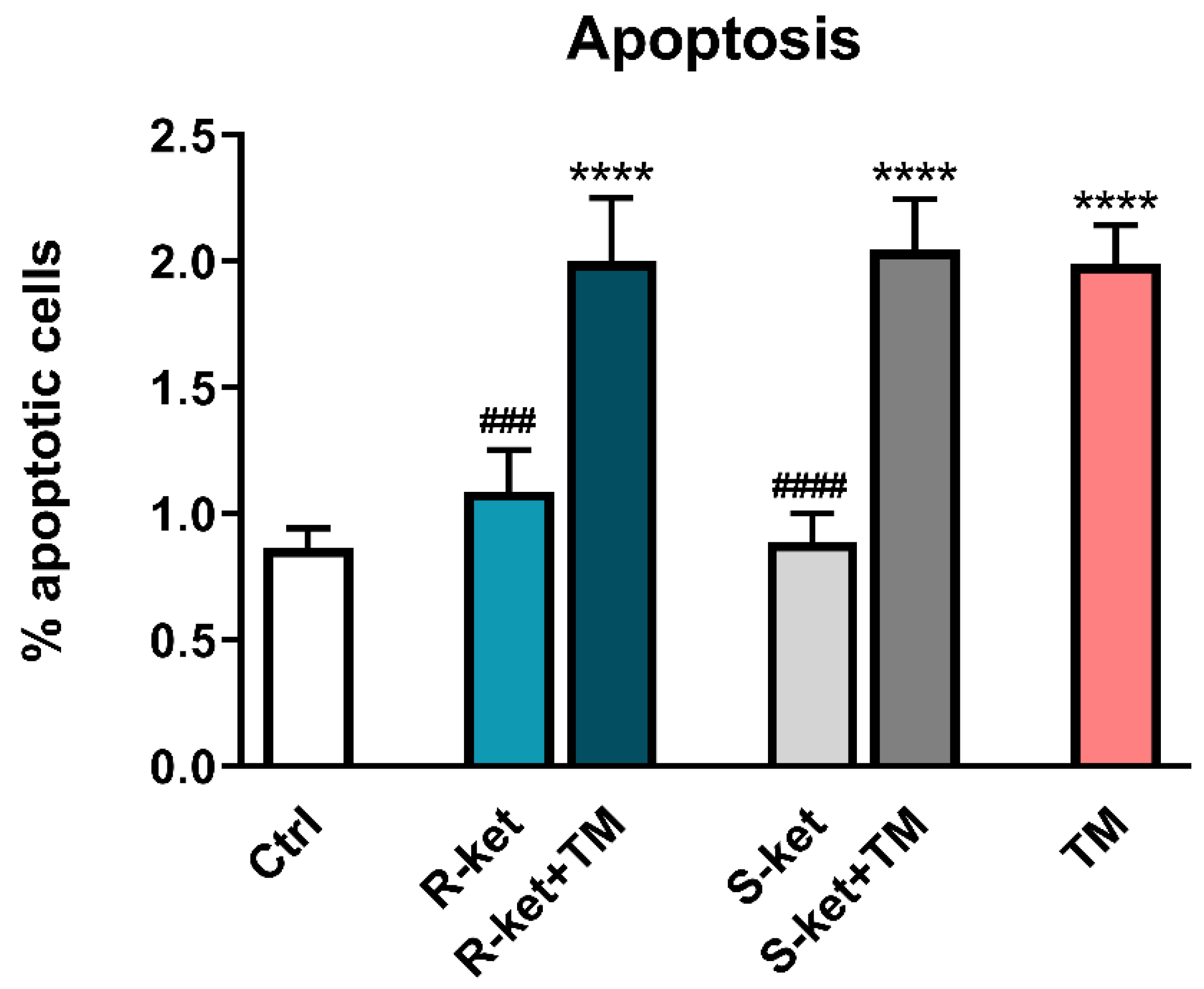
3.4. Apoptosis Induction
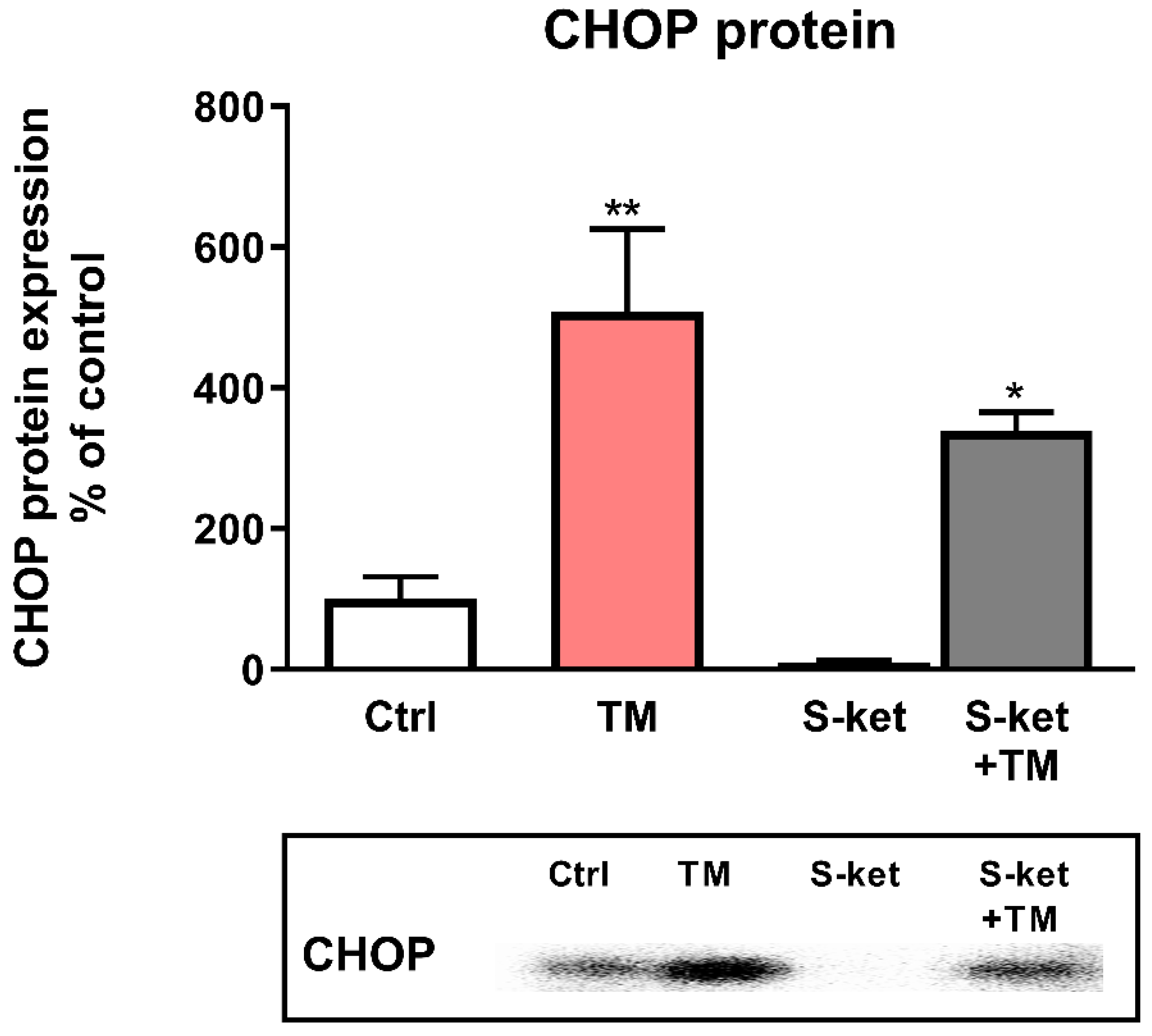
3.5. CHOP Protein Expression
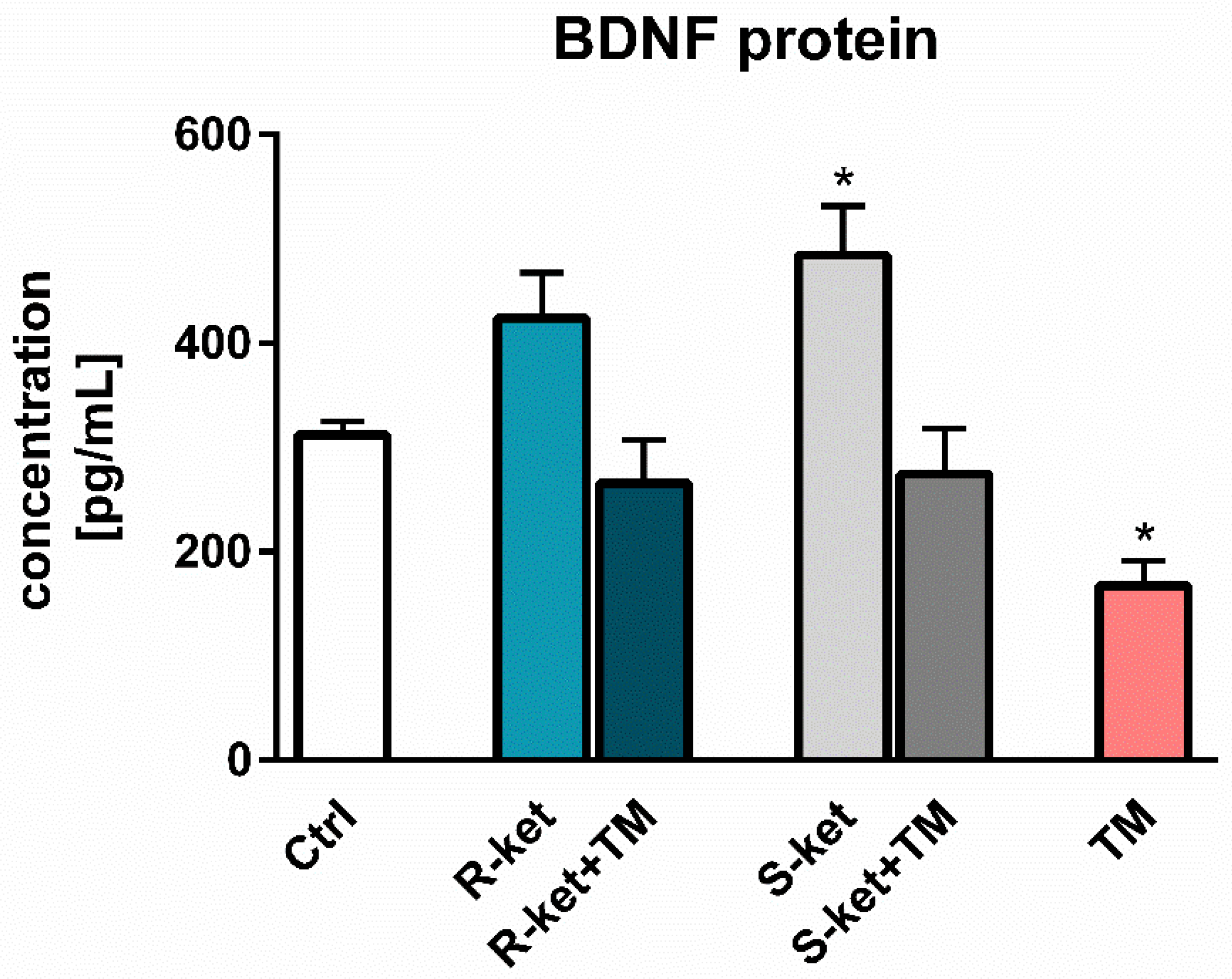
3.6. BDNF Release
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chlebowska, J. Endoplasmic reticulum stress and oxidative stress in acute myeloid leukemia. Acta Hematol. Pol. 2016, 47, 197–204. [Google Scholar] [CrossRef]
- Wandtke, T.; Wędrowska, E.; Goede, A.; Owczarska, P.; Piskorska, E.; Kopiński, P. Role of endoplasmicreticulum-associated protein degradation pathway in the virus infection cycle. J. Educ. Health Sport 2017, 7, 607–635. [Google Scholar]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Mori, K.; Ma, W.; Gething, M.J.; Sambrook, J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 1993, 74, 743–756. [Google Scholar] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Lee, J.; Liem, D.; Ping, P. HSPA5 Gene encoding Hsp70 chaperone BiP in the endoplasmic reticulum. Gene 2017, 618, 14–23. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef]
- Hetz, C. The biological meaning of the UPR. Nat. Rev. Mol. Cell Biol. 2013, 14, 404. [Google Scholar] [CrossRef]
- Kowalczyk, M.; Kowalczyk, E.; Kwiatkowski, P.; Łopusiewicz, Ł.; Sienkiewicz, M.; Talarowska, M. Ketamine-New Possibilities in the Treatment of Depression: A Narrative Review. Life 2021, 11, 1186. [Google Scholar] [CrossRef]
- Rozpędek, W.; Pytel, D.; Mucha, B.; Leszczyńska, H.; Diehl, J.A.; Majsterek, I. The role of the PERK/eIF2α/ATF4/CHOP signaling pathway in tumor progression during Endoplasmic Reticulum stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Sharma, D.; Kalia, K.; Tiwari, V. Crosstalk between endoplasmic reticulum stress and oxidative stress in schizophrenia: The dawn of new therapeutic approaches. Neurosci. Biobehav. Rev. 2017, 83, 589–603. [Google Scholar] [CrossRef]
- Hayashi, A.; Kasahara, T.; Kametani, M.; Toyota, T.; Yoshikawa, T.; Kato, T. Aberrant endoplasmic reticulum stress response in lymphoblastoid cells from patients with bipolar disorder. Int. J. Neuropsychopharmacol. 2009, 12, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Bown, C.; Wang, J.F.; MacQueen, G.; Young, L.T. Increased temporal cortex ER stress proteins in depressed subjects who died by suicide. Neuropsychopharmacology 2000, 22, 327–332. [Google Scholar] [CrossRef]
- Nevell, L.; Zhang, K.; Aiello, A.E.; Koenen, K.; Galea, S.; Soliven, R.; Zhang, C.; Wildman, D.E.; Uddin, M. Elevated systemic expression of ER stress related genes is associated with stress-related mental disorders in the Detroit Neighborhood Health Study. Psychoneuroendocrinology 2014, 43, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.F.; Sharma, M.S.; Brunoni, A.R.; Vieta, E.; Fava, G.A. The Safety, Tolerability and Risks Associated with the Use of Newer Generation Antidepressant Drugs: A Critical Review of the Literature. Psychother. Psychosom. 2016, 85, 270–288. [Google Scholar] [CrossRef]
- McLachlan, G. Treatment resistant depression: What are the options? BMJ 2018, 363, k5354. [Google Scholar] [CrossRef]
- Marathe, S.V.; D’Almeida, P.L.; Virmani, G.; Bathini, P.; Alberi, L. Effects of Monoamines and Antidepressants on Astrocyte Physiology: Implications for Monoamine Hypothesis of Depression. J. Exp. Neurosci. 2018, 12, 1179069518789149. [Google Scholar] [CrossRef] [Green Version]
- Kowalczyk, M.; Kowalczyk, E.; Kwiatkowski, P.; Łopusiewicz, Ł.; Talarowska, M.; Sienkiewicz, M. Cellular Response to Unfolded Proteins in Depression. Life 2021, 11, 1376. [Google Scholar] [CrossRef]
- Liu, W.; Ge, T.; Leng, Y.; Pan, Z.; Fan, J.; Yang, W.; Cui, R. The Role of Neural Plasticity in Depression: From Hippocampus to Prefrontal Cortex. Neural Plast. 2017, 2017, 6871089. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, P.; Reid, K.S.; Ferrier, I.N. Neuropsychological functioning in health and mood disorder: Modulation by glucocorticoids and their receptors. Psychoneuroendocrinology 2009, 34, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Sanacora, G.; Treccani, G.; Popoli, M. Towards a glutamate hypothesis of depression: An emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 2012, 62, 63–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.C.; Yao, W.; Hashimoto, K. Brain-derived Neurotrophic Factor (BDNF)-TrkB Signaling in Inflammation-related Depression and Potential Therapeutic Targets. Curr. Neuropharmacol. 2016, 14, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troubat, R.; Barone, P.; Leman, S.; Desmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; et al. Neuroinflammation and depression: A review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Rajkowska, G.; Miguel-Hidalgo, J.J. Gliogenesis and glial pathology in depression. CNS Neurol. Disord. Drug Targets 2007, 6, 219–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Zuo, Y.X.; Jiang, R.T. Astrocyte morphology: Diversity, plasticity, and role in neurological diseases. CNS Neurosci. Ther. 2019, 25, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, W.; Zhoud, Y.; Ma, C.; Li, S.; Cong, B. Endoplasmic reticulum stress is involved in restraint stress-induced hippocampal apoptosis and cognitive impairments in rats. Physiol. Behav. 2014, 131, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhao, Z.; Lu, L.; Liu, J.L.; Sun, J.; Wu, X.; Dong, J. Icariin and icaritin ameliorated hippocampus neuroinflammation via inhibiting HMGB1-related pro-inflammatory signals in lipopolysaccharide-induced inflammation model in C57BL/6J mice. Int. Immunopharmacol. 2019, 68, 95–105. [Google Scholar] [CrossRef]
- Rajkowska, G.; Stockmeier, C.A. Astrocyte pathology in major depressive disorder: Insights from human postmortem brain tissue. Curr. Drug Targets 2013, 14, 1225–1236. [Google Scholar] [CrossRef] [Green Version]
- Timberlake, M.; Dwivedi, Y. Altered Expression of Endoplasmic Reticulum Stress Associated Genes in Hippocampus of Learned Helpless Rats: Relevance to Depression Pathophysiology. Front. Pharmacol. 2016, 6, 319. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Hu, Y.; Ruan, L.; Ji, Y.; Lou, Z. Role of endoplasmic reticulum stress in depression (Review). Mol. Med. Rep. 2019, 20, 4774–4780. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Yang, Y.R.; Chen, W.; Chen, M.H.; Wang, H.; Wang, X.D.; Sun, L.L.; Wang, F.Z.; Wang, D.C. Fluoxetine synergizes with temozolomide to induce the CHOP-dependent endoplasmic reticulum stress-related apoptosis pathway in glioma cells. Oncol. Rep. 2016, 36, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Peng, T.; Liu, X.; Wang, J.; Liu, Y.; Fu, Z.; Ma, X.; Li, J.; Sun, G.; Ji, Y.; Lu, J.; et al. Fluoxetine-mediated inhibition of endoplasmic reticulum stress is involved in the neuroprotective effects of Parkinson’s disease. Aging 2018, 10, 4188–4196. [Google Scholar] [CrossRef]
- Abdullahi, A.; Stanojcic, M.; Parousis, A.; Patsouris, D.; Jeschke, M.G. Modeling Acute ER stress in vivo and in vitro. Shock 2017, 47, 506–513. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Xiao, Q.; Xie, L.; Yang, F.; Wang, L.; Tu, J. Astrocyte, a Promising Target for Mood Disorder Interventions. Front Mol. Neurosci. 2019, 12, 136. [Google Scholar] [CrossRef]
- Ma, J.; Qiu, Y.; Yang, L.; Peng, L.; Xia, Z.; Hou, L.N.; Fang, C.; Qi, H.; Chen, H.Z. Desipramine induces apoptosis in rat glioma cells via endoplasmic reticulum stress-dependent CHOP pathway. J. Neurooncol. 2011, 101, 41–48. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, D.; Li, Y.; Qiao, H.; Shan, Z. Ketamine enhances autophagy and endoplasmic reticulum stress in rats and SV-HUC-1 cells via activating IRE1-TRAF2-ASK1-JNK pathway. Cell Cycle 2021, 20, 1907–1922. [Google Scholar] [CrossRef]
- Chen, S.; Xuan, J.; Couch, L.; Iyer, A.; Wu, Y.; Li, Q.Z.; Guo, L. Sertraline induces endoplasmic reticulum stress in hepatic cells. Toxicology 2014, 322, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zheng, L.; Wan, Y.; Chen, Z.; Li, P.; Wang, Y. Metoprolol, N-Acetylcysteine, and Escitalopram Prevents Chronic Unpredictable Mild Stress-Induced Depression by Inhibition of Endoplasmic Reticulum Stress. Front. Psychiatry 2018, 9, 696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Chen, Z.; Li, J.; Ding, P.; Wang, Y. Effects of Escitalopram on Endoplasmic Reticulum Stress and Oxidative Stress Induced by Tunicamycin. Front. Neurosci. 2021, 15, 737509. [Google Scholar] [CrossRef]
- Saito, A. Physiological functions of endoplasmic reticulum stress transducer OASIS in central nervous system. Anat. Sci. Int. 2014, 89, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Hou, L.N.; Rong, Z.X.; Liang, P.; Fang, C.; Li, H.F.; Qi, H.; Chen, H.Z. Antidepressant desipramine leads to C6 glioma cell autophagy: Implication for the adjuvant therapy of cancer. Anticancer Agents Med. Chem. 2013, 13, 254–260. [Google Scholar] [CrossRef] [PubMed]
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, Regional, and National Incidence, Prevalence, and Years Lived with Disability for 354 Diseases and Injuries for 195 Countries and Territories, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- Bartoli, F.; Riboldi, I.; Crocamo, C.; Di Brita, C.; Clerici, M.; Carrà, G. Ketamine as a Rapid-Acting Agent for Suicidal Ideation: A Meta-Analysis. Neurosci. Biobehav. Rev. 2017, 77, 232–236. [Google Scholar] [CrossRef]
- Irifune, M.; Shimizu, T.; Nomoto, M.; Fukuda, T. Ketamine-Induced Anesthesia Involves the N-Methyl-D-Aspartate Receptor Channel Complex in Mice. Brain Res. 1992, 596, 1–9. [Google Scholar] [CrossRef]
- Miller, K.W. The Nature of Sites of General Anaesthetic Action. Br. J. Anaesth. 2002, 89, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Abelaira, H.M.; Réus, G.Z.; Ignácio, Z.M.; Dos Santos, M.A.; de Moura, A.B.; Matos, D.; Demo, J.P.; da Silva, J.B.; Michels, M.A.; Abatti, M.; et al. Effects of ketamine administration on mTOR and reticulum stress signaling pathways in the brain after the infusion of rapamycin into prefrontal cortex. J. Psychiatr. Res. 2017, 87, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Réus, G.Z.; Nacif, M.P.; Abelaira, H.M.; Tomaz, D.B.; dos Santos, M.A.B.; Carlessi, A.S.; Matias, B.I.; da Luz, J.R.; Steckert, A.V.; Jeremias, G.C.; et al. Ketamine Treatment Partly Reverses Alterations in Brain Derived- Neurotrophic Factor, Oxidative Stress and Energy Metabolism Parameters Induced by an Animal Model of Depression. Curr. Neurovasc. Res. 2015, 12, 73–84. [Google Scholar] [CrossRef]
- Deyama, S.; Duman, R.S. Neurotrophic mechanisms underlying the rapid and sustained antidepressant actions of ketamine. Pharmacol. Biochem. Behav. 2020, 188, 172837. [Google Scholar] [CrossRef]
- Matveychuk, D.; Thomas, R.K.; Swainson, J.; Khullar, A.; MacKay, M.A.; Baker, G.B.; Dursun, S.M. Ketamine as an antidepressant: Overview of its mechanisms of action and potential predictive biomarkers. Ther. Adv. Psychopharmacol. 2020, 10, 2045125320916657. [Google Scholar] [CrossRef]
- Zhang, J.C.; Li, S.X.; Hashimoto, K. R (-)-ketamine shows greater potency and longer lasting antidepressant effects than S(+)-ketamine. Pharmacol. Biochem. Behav. 2014, 116, 137–141. [Google Scholar] [CrossRef]
- Hashimoto, K. Rapid-acting antidepressant ketamine, its metabolites and other candidates: A historical overview and future perspective. Psychiatry Clin. Neurosci. 2019, 73, 613–627. [Google Scholar] [CrossRef]
- Yang, C.; Shirayama, Y.; Zhang, J.C.; Ren, Q.; Yao, W.; Ma, M.; Dong, C.; Hashimoto, K. R-ketamine: A rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl. Psychiatry 2015, 5, e632. [Google Scholar] [CrossRef]
- Mansouri, S.; Agartz, I.; Ögren, S.O.; Patrone, C.; Lundberg, M. PACAP Protects Adult Neural Stem Cells from the Neurotoxic Effect of Ketamine Associated with Decreased Apoptosis, ER Stress and mTOR Pathway Activation. PLoS ONE 2017, 12, e0170496. [Google Scholar]
- Cui, L.; Jiang, X.; Zhang, C.; Li, D.; Yu, S.; Wan, F.; Ma, Y.; Guo, W.; Shan, Z. Ketamine induces endoplasmic reticulum stress in rats and SV-HUC-1 human uroepithelial cells by activating NLRP3/TXNIP aix. Biosci. Rep. 2019, 39, BSR20190595. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Cai, L.; Matsuhisa, K.; Ohtake, Y.; Kaneko, M.; Kanemoto, S.; Asada, R.; Imaizumi, K. Neuronal activity-dependent local activation of dendritic unfolded protein response promotes expression of brain-derived neurotrophic factor in cell soma. J. Neurochem. 2018, 144, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chihara, K.; Saito, A.; Murakami, T.; Hino, S.; Aoki, Y.; Sekiya, H.; Aikawa, Y.; Wanaka, A.; Imaizumi, K. Increased vulnerability of hippocampal pyramidal neurons to the toxicity of kainic acid in OASIS-deficient mice. J. Neurochem. 2009, 110, 956–965. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Studied Gene | Alternative Name | Encoded Protein * | Fold Change | |||
|---|---|---|---|---|---|---|
| Ami | Esci | R-ket | S-ket | |||
| ATF4 | CREB-2 | Activating transcription factor 4 | 0.98 ± 0.08 | 0.84 ± 0.13 | 1.10 ± 0.14 | 0.86 ± 0.14 |
| ATF6 | - | Activating transcription factor 6 | 1.01 ± 0.13 | 0.74 ± 0.05 | 0.98 ± 0.1 | 1.00 ± 0.2 |
| CREB3 | Luman | CAMP responsive element binding protein 3 | – | – | 0.82 + 0.01 | 1.19 ± 0.44 |
| CREB3L1 | Oasis | CAMP responsive element binding protein 3 like 1 | 1.30 ± 0.13 | 0.89 ± 0.08 | 2.11 ± 0.66 | 1.85 ± 0.5 |
| CREB3L4 | CREB4 | CAMP responsive element binding protein 3 like 4 | – | – | 1.21 ± 0.39 | 1.25 ± 0.53 |
| DDIT3 | CHOP # | DNA damage inducible transcript 3/C/EBP-homologous protein | 1.21 ± 0.22 | 0.99 ± 0.26 | 1.21 ± 0.12 | 1.08 ± 0.31 |
| EDEM1 | EDEM | ER degradation enhancing alpha-mannosidase like protein 1 | 1.27 ± 0.14 | 0.86 ± 0.01 | 1.02 ± 0.15 | 1.05 ± 0.23 |
| EIF2AK3 | PERK | Eukaryotic translation initiation factor 2 alpha kinase 3 | 1.38 ± 0.13 | 0.91 ± 0.07 | 1.06 ± 0.21 | 1.08 ± 0.23 |
| EIF2S1 | eIF2α | Eukaryotic translation initiation factor 2 subunit alpha | 1.17 ± 0.07 | 1.03 ± 0.00 | 0.89 ± 0.25 | 0.74 ± 0.12 |
| ERN1 | IRE1 | Endoplasmic reticulum to nucleus signalling 1/Inositol-requiring enzyme 1 | 1.38 ± 0.41 | 0.93 ± 0.07 | 2.06 ± 0.97 | 2.22 ± 0.66 |
| GABPA | NRF2 | GA binding protein transcription factor subunit alpha | 1.07 ± 0.07 | 0.98 ± 0.12 | 1.03 ± 0.25 | 0.95 ± 0.19 |
| HSPA5 | GRP78 | Heat shock protein family A (Hsp70) member 5 | 1.11 ± 0.03 | 0.98 ± 0.05 | 0.90 ± 0.08 | 0.86 ± 0.19 |
| MAP3K4 | MEKK4 | Mitogen-activated protein kinase kinase kinase 4 | 1.13 ± 0.14 | 0.89 ± 0.07 | 1.24 ± 0.71 | 1.09 ± 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jóźwiak-Bębenista, M.; Sokołowska, P.; Siatkowska, M.; Panek, C.A.; Komorowski, P.; Kowalczyk, E.; Wiktorowska-Owczarek, A. The Importance of Endoplasmic Reticulum Stress as a Novel Antidepressant Drug Target and Its Potential Impact on CNS Disorders. Pharmaceutics 2022, 14, 846. https://doi.org/10.3390/pharmaceutics14040846
Jóźwiak-Bębenista M, Sokołowska P, Siatkowska M, Panek CA, Komorowski P, Kowalczyk E, Wiktorowska-Owczarek A. The Importance of Endoplasmic Reticulum Stress as a Novel Antidepressant Drug Target and Its Potential Impact on CNS Disorders. Pharmaceutics. 2022; 14(4):846. https://doi.org/10.3390/pharmaceutics14040846
Chicago/Turabian StyleJóźwiak-Bębenista, Marta, Paulina Sokołowska, Małgorzata Siatkowska, Cecilia Analia Panek, Piotr Komorowski, Edward Kowalczyk, and Anna Wiktorowska-Owczarek. 2022. "The Importance of Endoplasmic Reticulum Stress as a Novel Antidepressant Drug Target and Its Potential Impact on CNS Disorders" Pharmaceutics 14, no. 4: 846. https://doi.org/10.3390/pharmaceutics14040846