
Skin-on-a-Chip Technology: Microengineering Physiologically Relevant In Vitro Skin Models



Abstract
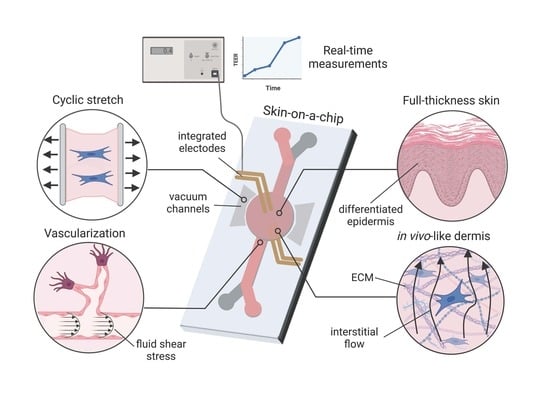
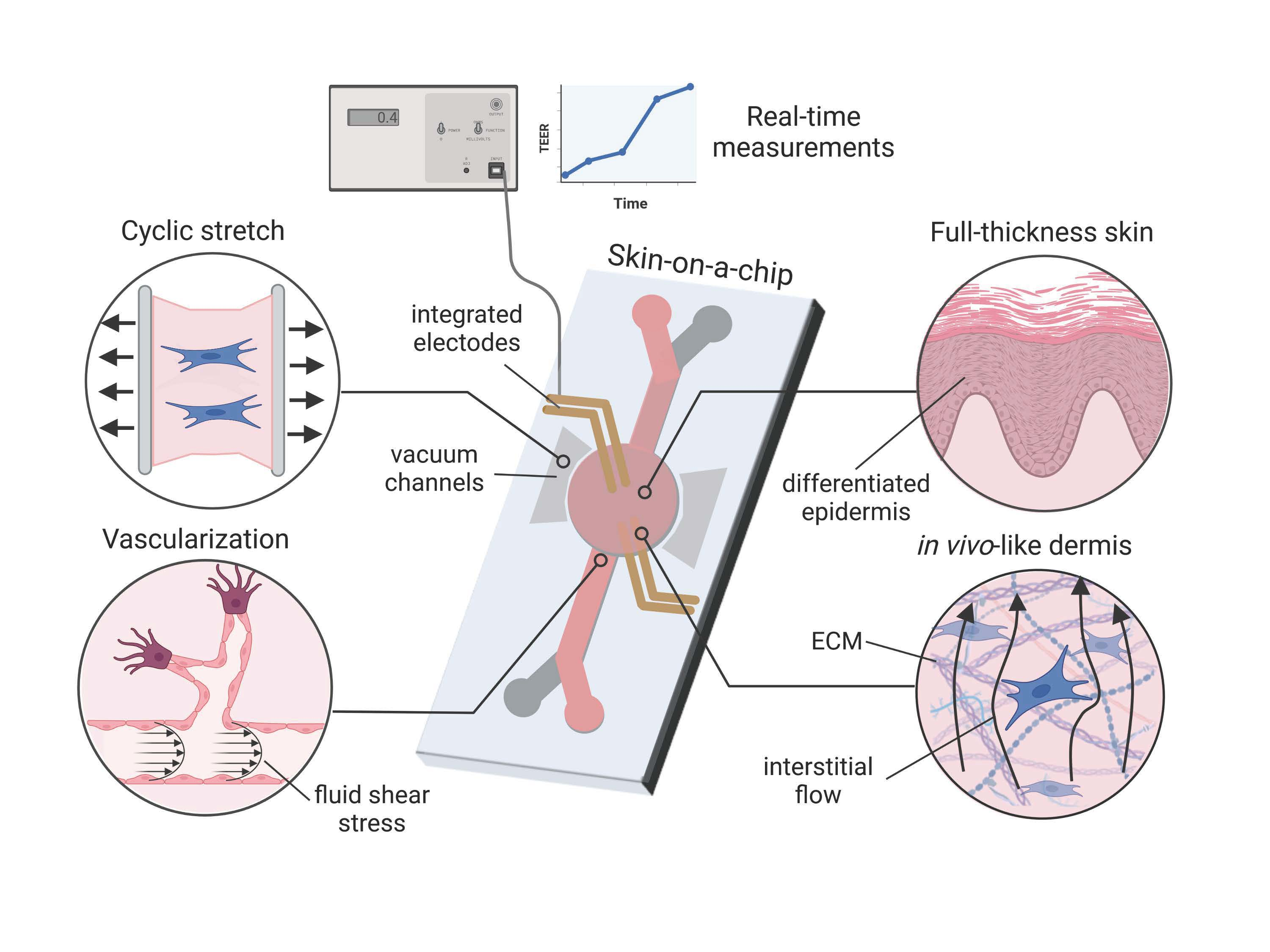
:
1. Introduction
2. Organ-on-a-Chip Technology
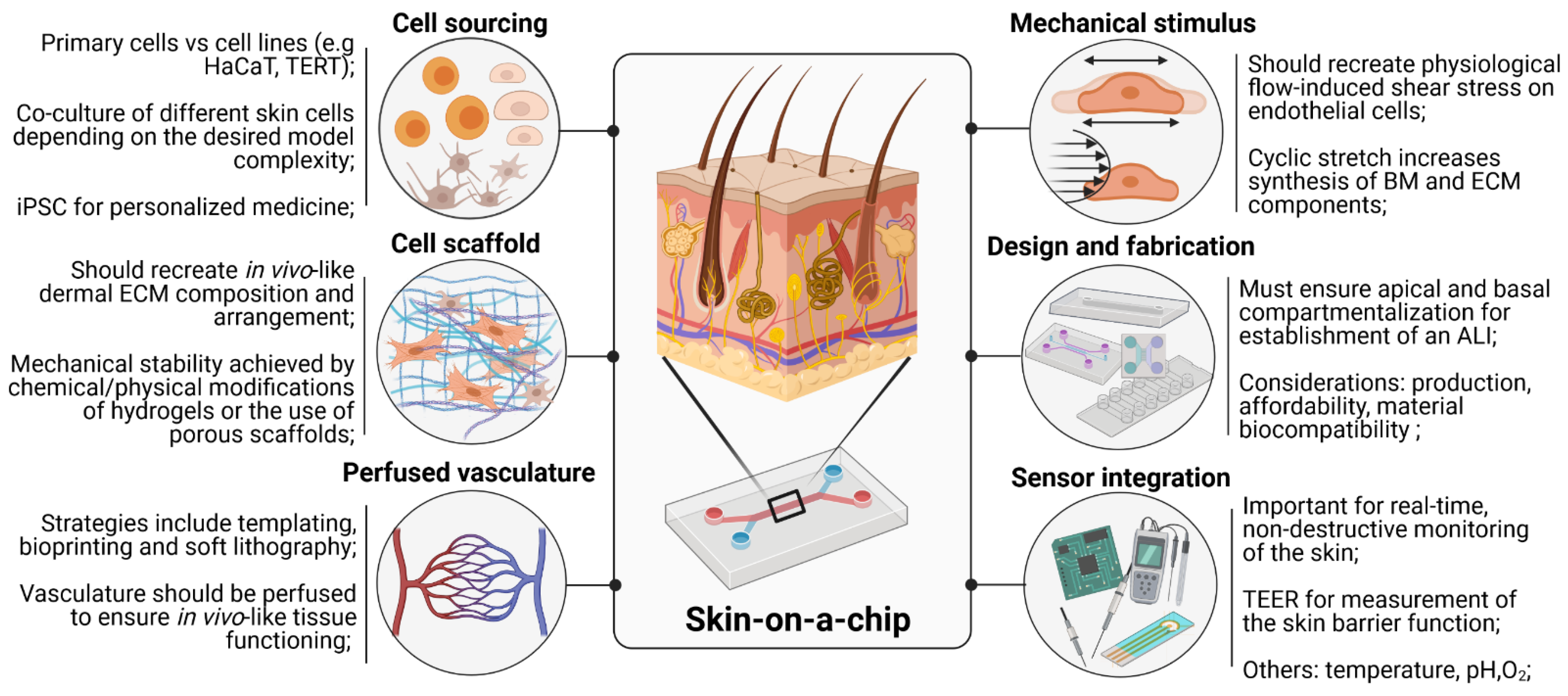
3. Key Requirements for the Development of Skin-on-a-Chip Devices
3.1. Cell Sourcing
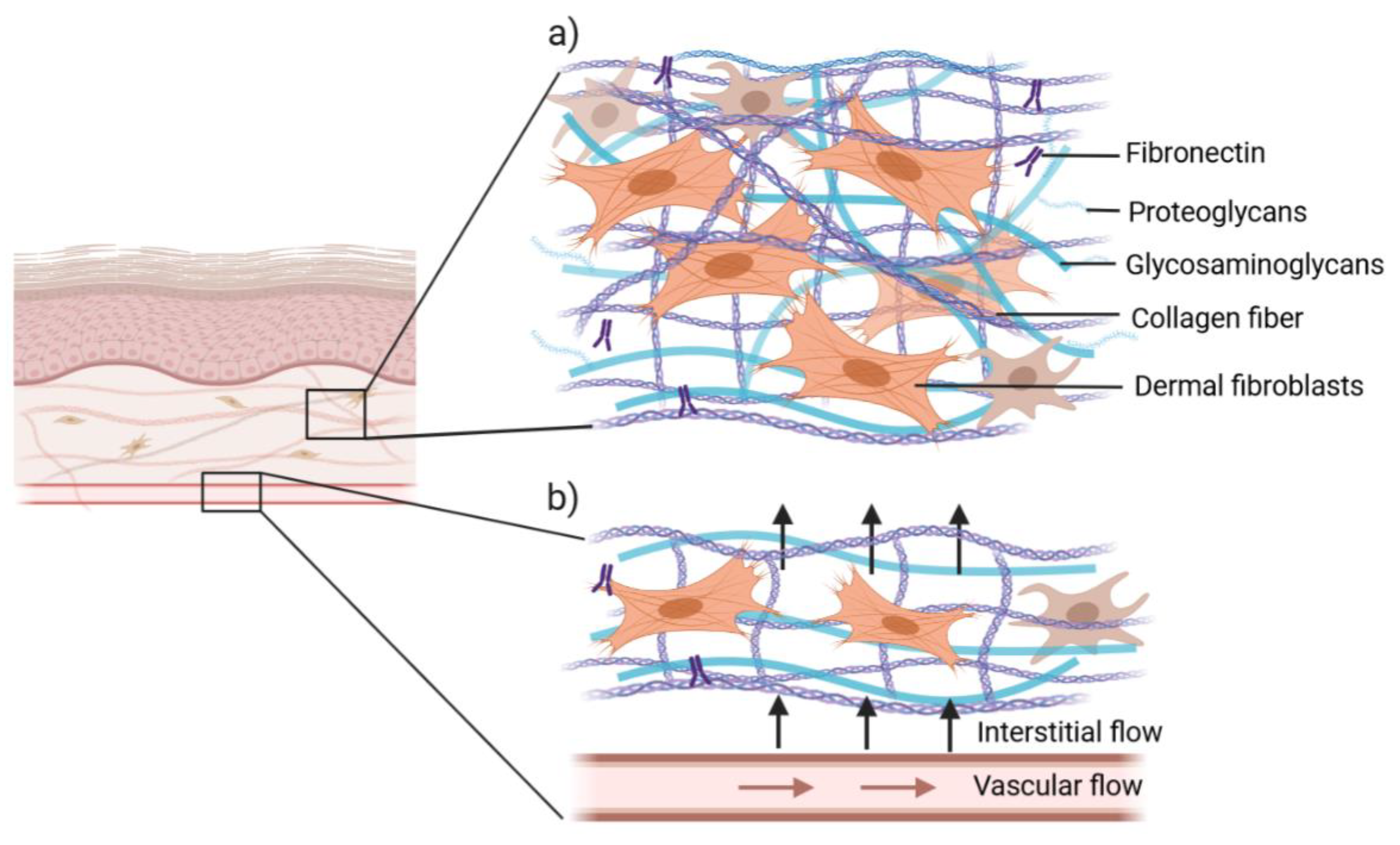
3.2. Cell Scaffolds
3.3. Vascularization
3.4. Mechanical Stimulus
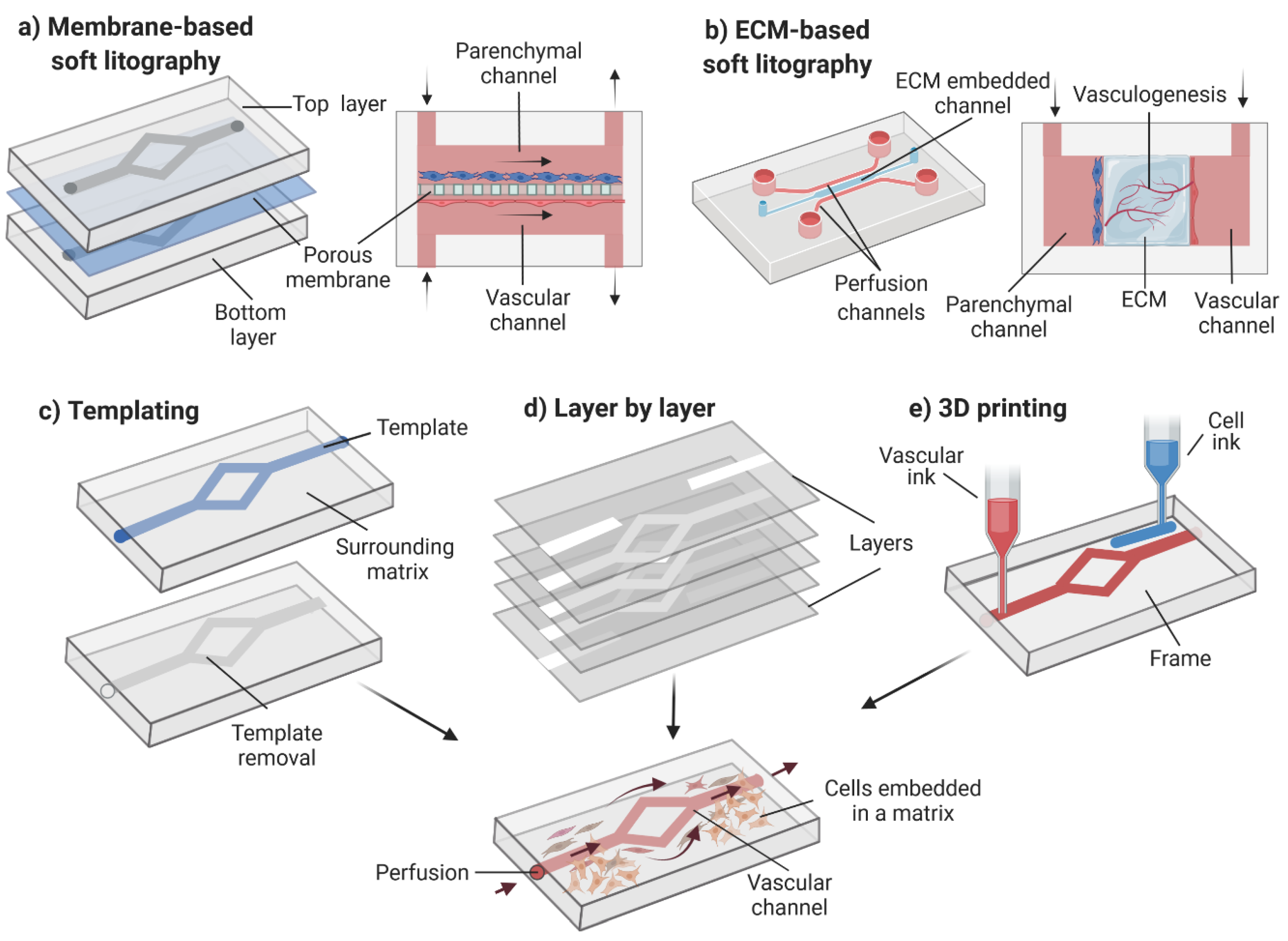
3.5. Design and Fabrication
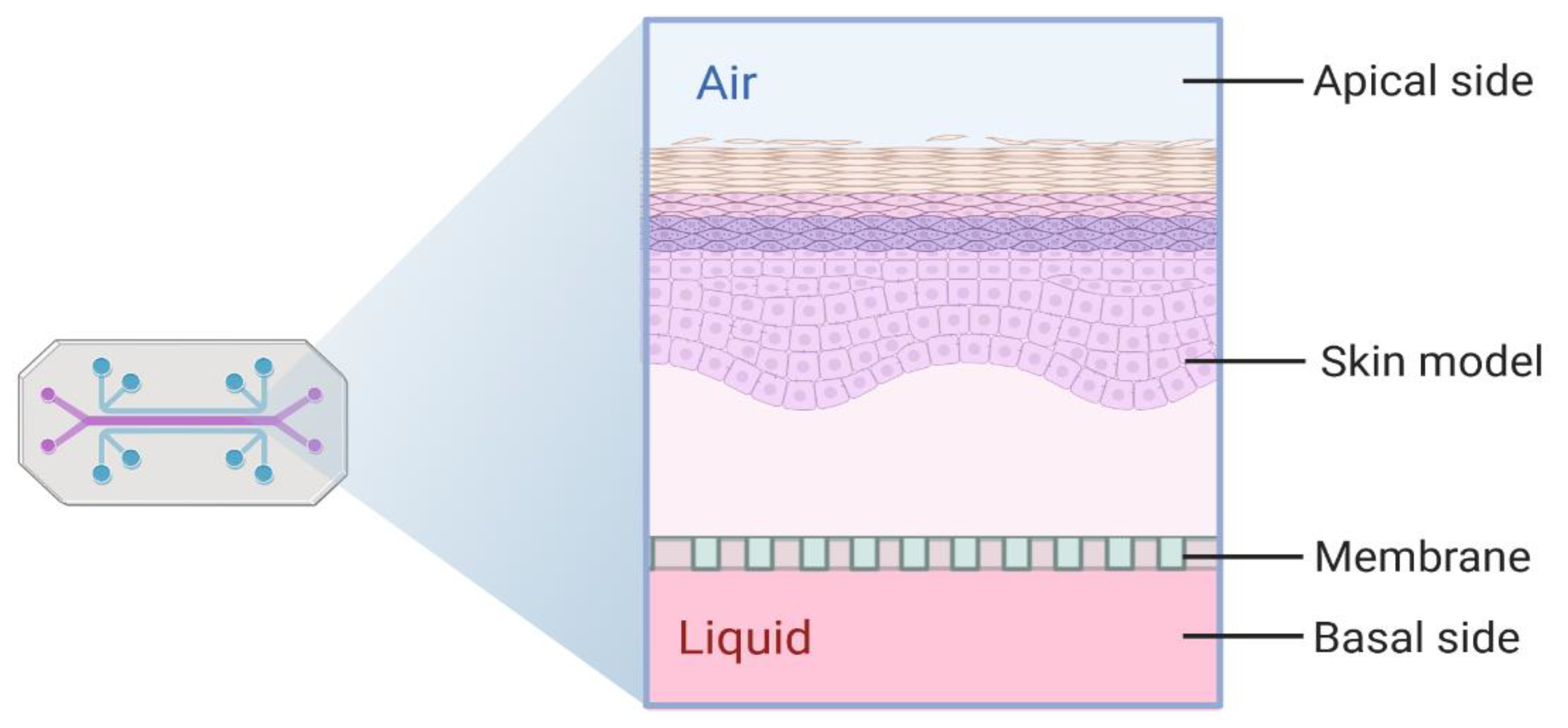
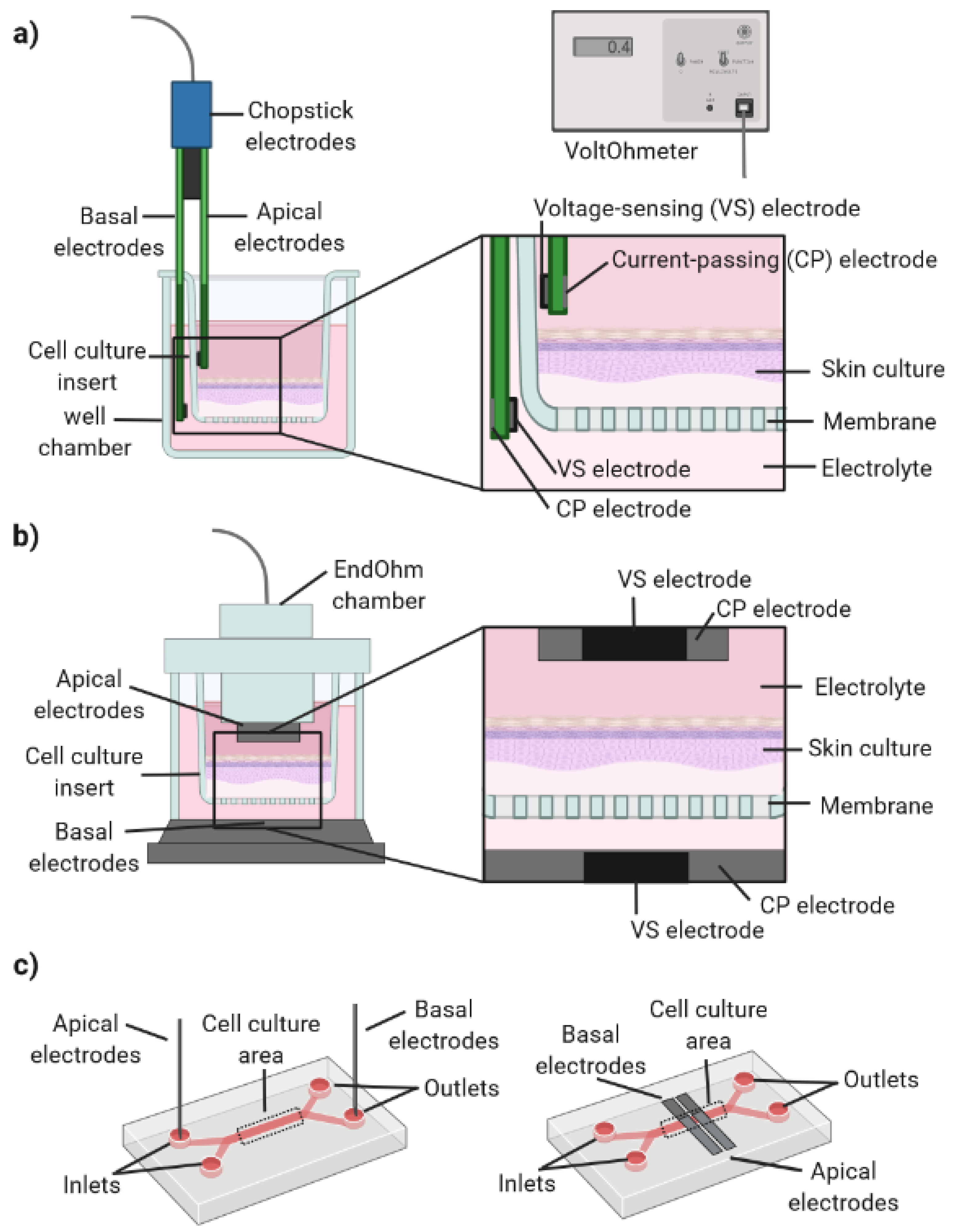
3.6. Sensor Integration
TEER: Applicability and Challenges
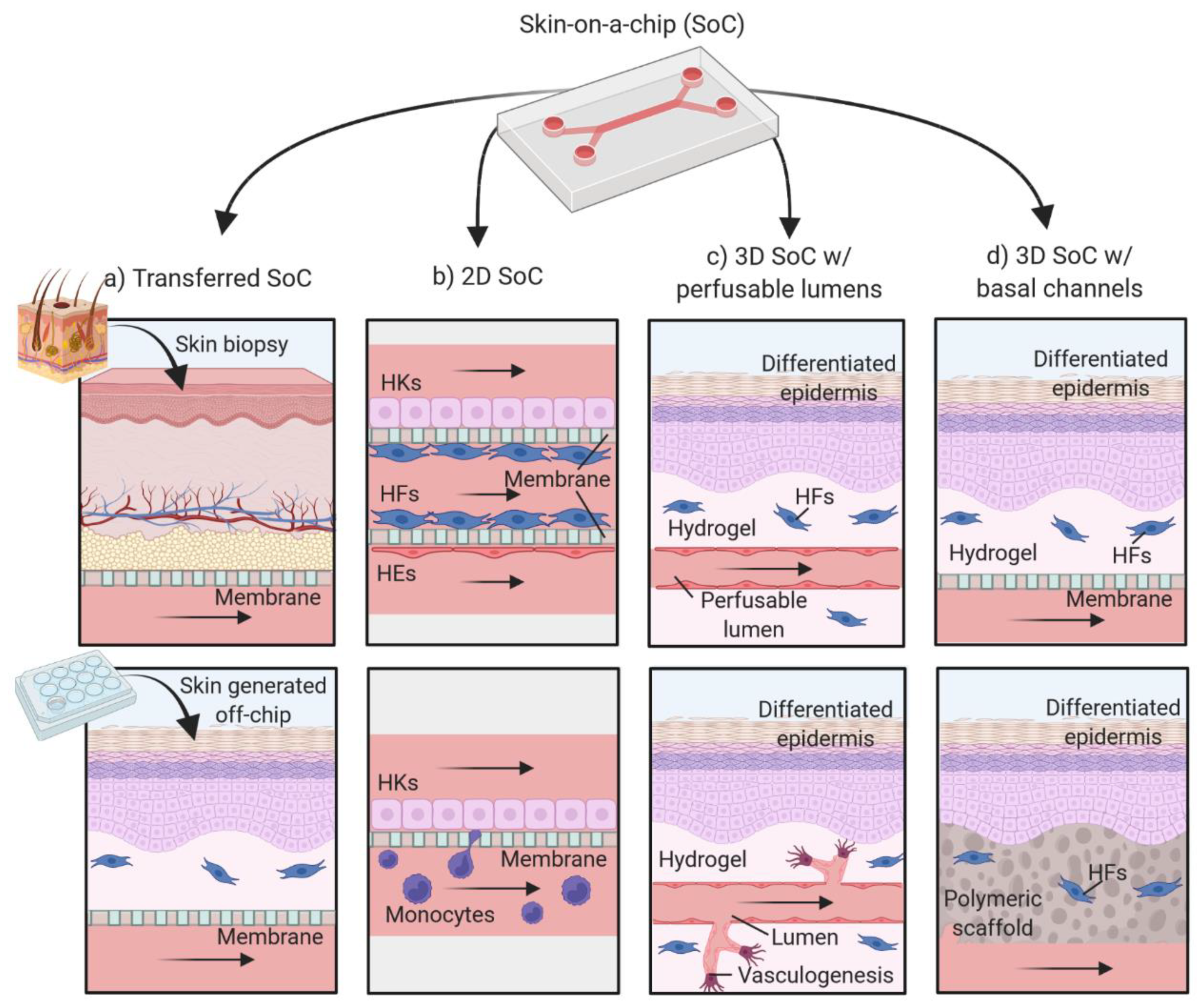
4. The Evolution of Skin-on-a-Chip Platforms
4.1. Two-Dimensional Skin-on-a-Chip Models
4.2. SoC Models Based on 3D Cell Cultures
4.2.1. Models with Perfusable Lumens
4.2.2. Models with Basal Perfusion
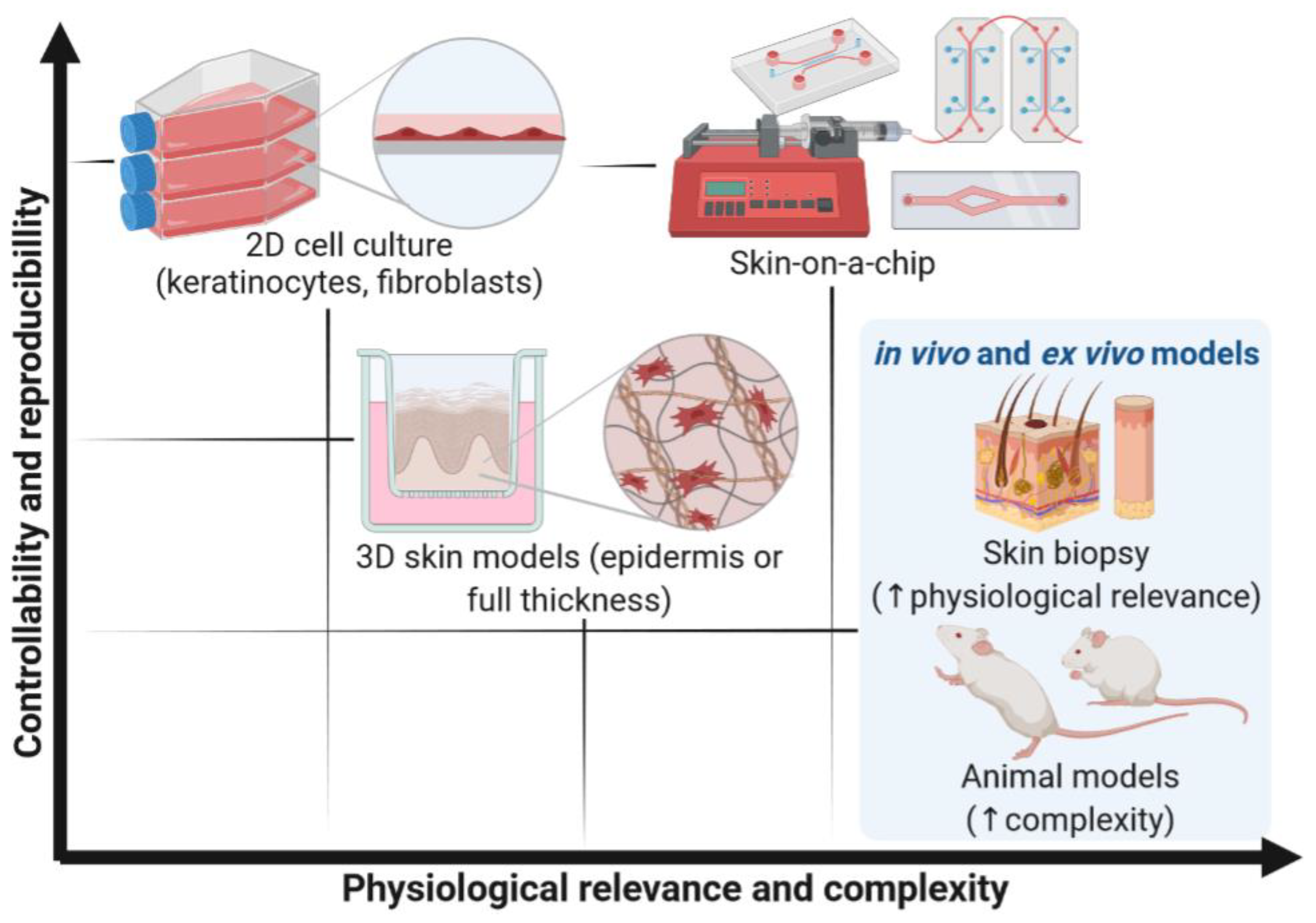
5. Comparative Analysis of SoC Devices
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Seth, D.; Cheldize, K.; Brown, D.; Freeman, E.F. Global Burden of Skin Disease: Inequities and Innovations. Curr. Dermatol. Rep. 2017, 6, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Mehrmal, S.; Uppal, P.; Giesey, R.L.; Delost, M.E.; Delost, G.R. Burden of skin disease and associated socioeconomic status in Europe: An ecologic study from the Global Burden of Disease Study 2017. JAAD Int. 2020, 1, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Brodersen, J.; Gøtzsche, P.C.; Jørgensen, K.J. Screening for reducing morbidity and mortality in malignant melanoma. Cochrane Database Syst. Rev. 2019, 6, CD012352. [Google Scholar] [CrossRef] [PubMed]
- Raeder, V.; Boura, I.; Leta, V.; Jenner, P.; Reichmann, H.; Trenkwalder, C.; Klingelhoefer, L.; Chaudhuri, K.R. Rotigotine Transdermal Patch for Motor and Non-motor Parkinson’s Disease: A Review of 12 Years’ Clinical Experience. CNS Drugs 2021, 35, 215–231. [Google Scholar] [CrossRef]
- Korkmaz, E.; Balmert, S.C.; Sumpter, T.L.; Carey, C.D.; Erdos, G.; Falo, L.D., Jr. Microarray patches enable the development of skin-targeted vaccines against COVID-19. Adv. Drug Deliv. Rev. 2021, 171, 164–186. [Google Scholar] [CrossRef]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning from 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Moniz, T.; Costa Lima, S.A.; Reis, S. Human skin models: From healthy to disease-mimetic systems; characteristics and applications. Br. J. Pharmacol. 2020, 177, 4314–4329. [Google Scholar] [CrossRef]
- Yun, Y.E.; Jung, Y.J.; Choi, Y.J.; Choi, J.S.; Cho, Y.W. Artificial skin models for animal-free testing. J. Pharm. Investig. 2018, 48, 215–223. [Google Scholar] [CrossRef]
- Franzen, N.; van Harten, W.H.; Retèl, V.P.; Loskill, P.; van den Eijnden-van Raaij, J.; IJzerman, M. Impact of organ-on-a-chip technology on pharmaceutical R&D costs. Drug Discov. Today 2019, 24, 1720–1724. [Google Scholar] [CrossRef]
- Taylor, K.; Rego Alvarez, L. Regulatory drivers in the last 20 years towards the use of in silico techniques as replacements to animal testing for cosmetic-related substances. Comput. Toxicol. 2020, 13, 100112. [Google Scholar] [CrossRef]
- Zuang, V.; Worth, A.P.; Balls, M. Chapter 2.10The Role of ECVAM. In History of Toxicology and Environmental Health; Balls, M., Combes, R., Worth, A., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 95–107. ISBN 978-0-12-813697-3. [Google Scholar]
- Langhans, S.A. Three-Dimensional In Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Peng, Y.; Li, H.; Chen, W. Organ-on-a-Chip: A New Paradigm for Drug Development. Trends Pharmacol. Sci. 2021, 42, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Sutterby, E.; Thurgood, P.; Baratchi, S.; Khoshmanesh, K.; Pirogova, E. Microfluidic Skin-on-a-Chip Models: Toward Biomimetic Artificial Skin. Small 2020, 16, 1–17. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, J.; Wang, X.; Feng, L.; Wu, J.; Zhu, X.; Wen, W.; Gong, X. Organ-on-a-chip: Recent breakthroughs and future prospects. Biomed. Eng. Online 2020, 19, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, C.L.; Fu, S.; Knight, M.M.; Thorpe, S.D. Mechanical Stimulation: A Crucial Element of Organ-on-Chip Models. Front. Bioeng. Biotechnol. 2020, 8, 602646. [Google Scholar] [CrossRef] [PubMed]
- Kilic, T.; Navaee, F.; Stradolini, F.; Renaud, P.; Carrara, S. Organs-on-chip monitoring: Sensors and other strategies. Microphysiol. Syst. 2018, 2, 1–32. [Google Scholar] [CrossRef]
- Ronaldson-Bouchard, K.; Vunjak-Novakovic, G. Organs-on-a-Chip: A Fast Track for Engineered Human Tissues in Drug Development. Cell Stem Cell 2018, 22, 310–324. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, J.; Razavi Bazaz, S.; Aboulkheyr Es, H.; Yaghobian Azari, D.; Thierry, B.; Ebrahimi Warkiani, M.; Ghadiri, M. Lung-on-a-chip: The future of respiratory disease models and pharmacological studies. Crit. Rev. Biotechnol. 2020, 40, 213–230. [Google Scholar] [CrossRef]
- Deng, J.; Wei, W.; Chen, Z.; Lin, B.; Zhao, W.; Luo, Y.; Zhang, X. Engineered Liver-On-A-Chip Platform to Mimic Liver Functions and Its Biomedical Applications: A Review. Micromachines 2019, 10, 676. [Google Scholar] [CrossRef] [Green Version]
- Kitsara, M.; Kontziampasis, D.; Agbulut, O.; Chen, Y. Heart on a chip: Micro-nanofabrication and microfluidics steering the future of cardiac tissue engineering. Microelectron. Eng. 2019, 203–204, 44–62. [Google Scholar] [CrossRef]
- Mittal, R.; Woo, F.W.; Castro, C.S.; Cohen, M.A.; Karanxha, J.; Mittal, J.; Chhibber, T.; Jhaveri, V.M. Organ-on-chip models: Implications in drug discovery and clinical applications. J. Cell. Physiol. 2019, 234, 8352–8380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yesil-Celiktas, O.; Hassan, S.; Miri, A.K.; Maharjan, S.; Al-kharboosh, R.; Quiñones-Hinojosa, A.; Zhang, Y.S. Mimicking Human Pathophysiology in Organ-on-Chip Devices. Adv. Biosyst. 2018, 2, 1800109. [Google Scholar] [CrossRef]
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer 2019, 19, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Van den Broek, L.J.; Bergers, L.I.J.C.; Reijnders, C.M.A.; Gibbs, S. Progress and Future Prospectives in Skin-on-Chip Development with Emphasis on the use of Different Cell Types and Technical Challenges. Stem Cell Rev. Rep. 2017, 13, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Hosseini, M.; Vainio, S.; Taïeb, A.; Cario-André, M.; Rezvani, H.R. Skin equivalents: Skin from reconstructions as models to study skin development and diseases. Br. J. Dermatol. 2015, 173, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Zoio, P.; Ventura, S.; Leite, M.; Oliva, A. Pigmented full-thickness human skin model based on a fibroblast-derived matrix for long-term studies. Tissue Eng. Part C Methods 2021, 27, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Niehues, H.; van den Bogaard, E.H. Past, present and future of in vitro 3D reconstructed inflammatory skin models to study psoriasis. Exp. Dermatol. 2018, 27, 512–519. [Google Scholar] [CrossRef] [Green Version]
- Rioux, G.; Simard, M.; Morin, S.; Lorthois, I.; Guérin, S.L.; Pouliot, R. Development of a 3D psoriatic skin model optimized for infiltration of IL-17A producing T cells: Focus on the crosstalk between T cells and psoriatic keratinocytes. Acta Biomater. 2021, 136, 210–222. [Google Scholar] [CrossRef]
- Brohem, C.A.; da Silva Cardeal, L.B.; Tiago, M.; Soengas, M.S.; de Moraes Barros, S.B.; Maria-Engler, S.S. Artificial skin in perspective: Concepts and applications. Pigment Cell Melanoma Res. 2011, 24, 35–50. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.H.; Jung, S.M.; Shin, H.S. Co-stimulation of HaCaT keratinization with mechanical stress and air-exposure using a novel 3D culture device. Sci. Rep. 2016, 6, 33889. [Google Scholar] [CrossRef]
- Reijnders, C.M.A.; Van Lier, A.; Roffel, S.; Kramer, D.; Scheper, R.J.; Gibbs, S. Development of a Full-Thickness Human Skin Equivalent in Vitro Model Derived from TERT-Immortalized Keratinocytes and Fibroblasts. Tissue Eng.-Part A 2015, 21, 2448–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astashkina, A.; Mann, B.; Grainger, D.W. A critical evaluation of in vitro cell culture models for high-throughput drug screening and toxicity. Pharmacol. Ther. 2012, 134, 82–106. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.G.; Daley, G.Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Khurana, P.; Kolundzic, N.; Flohr, C.; Ilic, D. Human pluripotent stem cells: An alternative for 3D in vitro modelling of skin disease. Exp. Dermatol. 2021, 30, 1572–1587. [Google Scholar] [CrossRef]
- Gledhill, K.; Guo, Z.; Umegaki-Arao, N.; Higgins, C.A.; Itoh, M.; Christiano, A.M. Melanin transfer in human 3D skin equivalents generated exclusively from induced pluripotent stem cells. PLoS ONE 2015, 10, e0136713. [Google Scholar] [CrossRef]
- Sivarapatna, A.; Ghaedi, M.; Xiao, Y.; Han, E.; Aryal, B.; Zhou, J.; Fernandez-Hernando, C.; Qyang, Y.; Hirschi, K.K.; Niklason, L.E. Engineered Microvasculature in PDMS Networks Using Endothelial Cells Derived from Human Induced Pluripotent Stem Cells. Cell Transplant. 2017, 26, 1365–1379. [Google Scholar] [CrossRef]
- Hussey, G.S.; Dziki, J.L.; Badylak, S.F. Extracellular matrix-based materials for regenerative medicine. Nat. Rev. Mater. 2018, 3, 159–173. [Google Scholar] [CrossRef]
- Urbanczyk, M.; Layland, S.L.; Schenke-Layland, K. The role of extracellular matrix in biomechanics and its impact on bioengineering of cells and 3D tissues. Matrix Biol. 2020, 85–86, 1–14. [Google Scholar] [CrossRef]
- Grinnell, F. Fibroblast–collagen-matrix contraction: Growth-factor signalling and mechanical loading. Trends Cell Biol. 2000, 10, 362–365. [Google Scholar] [CrossRef]
- Lotz, C.; Schmid, F.F.; Oechsle, E.; Monaghan, M.G.; Walles, H.; Groeber-Becker, F. Cross-linked Collagen Hydrogel Matrix Resisting Contraction to Facilitate Full-Thickness Skin Equivalents. ACS Appl. Mater. Interfaces 2017, 9, 20417–20425. [Google Scholar] [CrossRef]
- El Ghalbzouri, A.; Commandeur, S.; Rietveld, M.H.; Mulder, A.A.; Willemze, R. Replacement of animal-derived collagen matrix by human fibroblast-derived dermal matrix for human skin equivalent products. Biomaterials 2009, 30, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Roger, M.; Fullard, N.; Costello, L.; Bradbury, S.; Markiewicz, E.; O’Reilly, S.; Darling, N.; Ritchie, P.; Määttä, A.; Karakesisoglou, I.; et al. Bioengineering the microanatomy of human skin. J. Anat. 2019, 234, 438–455. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A.; Fleury, M.E. Interstitial Flow and Its Effects in Soft Tissues. Annu. Rev. Biomed. Eng. 2007, 9, 229–256. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.P.; Swartz, M.A. Fibroblast alignment under interstitial fluid flow using a novel 3-D tissue culture model. Am. J. Physiol. Circ. Physiol. 2003, 284, H1771–H1777. [Google Scholar] [CrossRef] [PubMed]
- Low, D.A.; Jones, H.; Cable, N.T.; Alexander, L.M.; Kenney, W.L. Historical reviews of the assessment of human cardiovascular function: Interrogation and understanding of the control of skin blood flow. Eur. J. Appl. Physiol. 2020, 120, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huggenberger, R.; Detmar, M. The Cutaneous Vascular System in Chronic Skin Inflammation. J. Investig. Dermatol. Symp. Proc. 2011, 15, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, D.S.; Jones, R.E.; Ransom, R.C.; Longaker, M.T.; Norton, J.A. The evolving relationship of wound healing and tumor stroma. JCI Insight 2018, 3, e99911. [Google Scholar] [CrossRef] [Green Version]
- Kashani-Sabet, M.; Sagebiel, R.W.; Ferreira, C.M.M.; Nosrati, M.; Miller III, J.R. Vascular Involvement in the Prognosis of Primary Cutaneous Melanoma. Arch. Dermatol. 2001, 137, 1169–1173. [Google Scholar] [CrossRef] [Green Version]
- Cevc, G.; Vierl, U. Spatial distribution of cutaneous microvasculature and local drug clearance after drug application on the skin. J. Control. Release 2007, 118, 18–26. [Google Scholar] [CrossRef]
- Magliaro, C.; Mattei, G.; Iacoangeli, F.; Corti, A.; Piemonte, V.; Ahluwalia, A. Oxygen Consumption Characteristics in 3D Constructs Depend on Cell Density. Front. Bioeng. Biotechnol. 2019, 7, 251. [Google Scholar] [CrossRef]
- Lee, M.; Choi, J.S.; Eom, M.R.; Jeong, E.J.; Kim, J.; Park, S.A.; Kwon, S.K. Prevascularized Tracheal Scaffolds Using the Platysma Flap for Enhanced Tracheal Regeneration. Laryngoscope 2021, 131, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Shafiee, S.; Shariatzadeh, S.; Zafari, A.; Majd, A.; Niknejad, H. Recent Advances on Cell-Based Co-Culture Strategies for Prevascularization in Tissue Engineering. Front. Bioeng. Biotechnol. 2021, 9, 745314. [Google Scholar] [CrossRef] [PubMed]
- Dellaquila, A.; Le Bao, C.; Letourneur, D.; Simon-Yarza, T. In Vitro Strategies to Vascularize 3D Physiologically Relevant Models. Adv. Sci. 2021, 8, 2100798. [Google Scholar] [CrossRef] [PubMed]
- Martino, F.; Perestrelo, A.R.; Vinarský, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef] [PubMed]
- Silvani, G.; Romanov, V.; Cox, C.D.; Martinac, B. Biomechanical Characterization of Endothelial Cells Exposed to Shear Stress Using Acoustic Force Spectroscopy. Front. Bioeng. Biotechnol. 2021, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, T.; Narayana, G.H.; Banerjee, I. Keratinocytes are mechanoresponsive to the microflow-induced shear stress. Cytoskeleton 2019, 76, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Liu, B.; Wu, H.; Wu, X.; Wang, X.-L.; Song, Y.; Zhang, S.-S.; Li, J.-Q.; Bi, L.; Pei, G.-X. The effect of fluid shear stress on fibroblasts and stem cells on plane and groove topographies. Cell Adh. Migr. 2020, 14, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Roux, E.; Bougaran, P.; Dufourcq, P.; Couffinhal, T. Fluid Shear Stress Sensing by the Endothelial Layer. Front. Physiol. 2020, 11, 861. [Google Scholar] [CrossRef]
- Tsvirkun, D.; Grichine, A.; Duperray, A.; Misbah, C.; Bureau, L. Microvasculature on a chip: Study of the Endothelial Surface Layer and the flow structure of Red Blood Cells. Sci. Rep. 2017, 7, 45036. [Google Scholar] [CrossRef]
- Kaarj, K.; Yoon, J.Y. Methods of delivering mechanical stimuli to Organ-on-a-Chip. Micromachines 2019, 10, 700. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.I.; Shuler, M.L. UniChip enables long-term recirculating unidirectional perfusion with gravity-driven flow for microphysiological systems. Lab Chip 2018, 18, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.-C.; Thampatty, B.P.; Lin, J.-S.; Im, H.-J. Mechanoregulation of gene expression in fibroblasts. Gene 2007, 391, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Shelton, L.; Rada, J.S. Effects of cyclic mechanical stretch on extracellular matrix synthesis by human scleral fibroblasts. Exp. Eye Res. 2007, 84, 314–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuyama, E.; Nagai, Y.; Takahashi, K.; Kimata, Y.; Naruse, K. Mechanical stretch on human skin equivalents increases the epidermal thickness and develops the basement membrane. PLoS ONE 2015, 10, e0141989. [Google Scholar] [CrossRef] [Green Version]
- Lü, D.; Liu, X.; Gao, Y.; Huo, B.; Kang, Y.; Chen, J.; Sun, S.; Chen, L.; Luo, X.; Long, M. Asymmetric migration of human keratinocytes under mechanical stretch and cocultured fibroblasts in a wound repair model. PLoS ONE 2013, 8, e74563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Huang, G.; Xu, F. Engineering Biomaterials and Approaches for Mechanical Stretching of Cells in Three Dimensions. Front. Bioeng. Biotechnol. 2020, 8, 1151. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.D. A human breathing lung-on-a-chip. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. S1), S42–S44. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Leslie, D.C.; Matthews, B.D.; Fraser, J.P.; Jurek, S.; Hamilton, G.A.; Thorneloe, K.S.; McAlexander, M.A.; Ingber, D.E. A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Sci. Transl. Med. 2012, 4, 159ra147. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Cole, T.; Zhang, Y.; Tang, S.-Y. Mechanical Strain-Enabled Reconstitution of Dynamic Environment in Organ-on-a-Chip Platforms: A Review. Micromachines 2021, 12, 765. [Google Scholar] [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Yuan Hsin, H.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [Green Version]
- Sakolish, C.M.; Esch, M.B.; Hickman, J.J.; Shuler, M.L.; Mahler, G.J. Modeling Barrier Tissues In Vitro: Methods, Achievements, and Challenges. EBioMedicine 2016, 5, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasman, T.; Grijpma, D.; Stamatialis, D.; Poot, A. Flat and microstructured polymeric membranes in organs-on-chips. J. R. Soc. Interface 2018, 15, 20180351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadan, Q.; Zourob, M. Organ-on-a-chip engineering: Toward bridging the gap between lab and industry. Biomicrofluidics 2020, 14, 41501. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, A.; Khan, S.; Didar, T.F. Conventional and emerging strategies for the fabrication and functionalization of PDMS-based microfluidic devices. Lab Chip 2021, 21, 3053–3075. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Gruner, D.; Richter, A.; Loskill, P. Membrane integration into PDMS-free microfluidic platforms for organ-on-chip and analytical chemistry applications. Lab Chip 2021, 21, 1866–1885. [Google Scholar] [CrossRef]
- Abhyankar, V.V.; Wu, M.; Koh, C.Y.; Hatch, A.V. A reversibly sealed, easy access, modular (SEAM) microfluidic architecture to establish in vitro tissue interfaces. PLoS ONE 2016, 11, e0156341. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Fu, Y.; Song, Y. Recent advances of microneedles for biomedical applications: Drug delivery and beyond. Acta Pharm. Sin. B 2019, 9, 469–483. [Google Scholar] [CrossRef]
- Kashaninejad, N.; Munaz, A.; Moghadas, H.; Yadav, S.; Umer, M.; Nguyen, N.-T. Microneedle Arrays for Sampling and Sensing Skin Interstitial Fluid. Chemosensors 2021, 9, 83. [Google Scholar] [CrossRef]
- Ballesteros Hernando, J.; Ramos Gómez, M.; Díaz Lantada, A. Modeling Living Cells Within Microfluidic Systems Using Cellular Automata Models. Sci. Rep. 2019, 9, 14886. [Google Scholar] [CrossRef] [Green Version]
- Zahorodny-Burke, M.; Nearingburg, B.; Elias, A.L. Finite element analysis of oxygen transport in microfluidic cell culture devices with varying channel architectures, perfusion rates, and materials. Chem. Eng. Sci. 2011, 66, 6244–6253. [Google Scholar] [CrossRef]
- Kheiri, S.; Kumacheva, E.; Young, E.W.K. Computational Modelling and Big Data Analysis of Flow and Drug Transport in Microfluidic Systems: A Spheroid-on-a-Chip Study. Front. Bioeng. Biotechnol. 2021, 9, 781566. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, A.; Joseph, A. Porous Medium Modeling of Combined Effects of Cell Migration and Anisotropicity of Stratum Corneum on Transdermal Drug Delivery. J. Heat Transf. 2015, 137, 121007. [Google Scholar] [CrossRef]
- Ponmozhi, J.; Dhinakaran, S.; Varga-medveczky, Z.; Fónagy, K.; Bors, L.A.; Iván, K.; Erdő, F. Development of skin-on-a-chip platforms for different utilizations: Factors to be considered. Micromachines 2021, 12, 294. [Google Scholar] [CrossRef] [PubMed]
- Graf, B.W.; Boppart, S.A. Imaging and Analysis of Three-Dimensional Cell Culture Models BT—Live Cell Imaging: Methods and Protocols; Papkovsky, D.B., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 211–227. ISBN 978-1-60761-404-3. [Google Scholar]
- Arlk, Y.B.; Van Der Helm, M.W.; Odijk, M.; Segerink, L.I.; Passier, R.; Van Den Berg, A.; Van Der Meer, A.D. Barriers-on-chips: Measurement of barrier function of tissues in organs-on-chips. Biomicrofluidics 2018, 12, 042218. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.S.; Aleman, J.; Shin, S.R.; Kilic, T.; Kim, D.; Shaegh, S.A.M.; Massa, S.; Riahi, R.; Chae, S.; Hu, N.; et al. Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc. Natl. Acad. Sci. USA 2017, 114, E2293–E2302. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER Measurement Techniques for In Vitro Barrier Model Systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef] [Green Version]
- Petrova, A.; Celli, A.; Jacquet, L.; Dafou, D.; Crumrine, D.; Hupe, M.; Arno, M.; Hobbs, C.; Cvoro, A.; Karagiannis, P.; et al. 3D In vitro model of a functional epidermal permeability barrier from human embryonic stem cells and induced pluripotent stem cells. Stem Cell Rep. 2014, 2, 675–689. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-C.; Hsu, H.-C.; Lin, C.-H.; Wu, C.-Y.; Chen, W.; Lai, H.-M. Testing Method Development and Validation for In Vitro Skin Irritation Testing (SIT) by Using Reconstructed Human Epidermis (RhE) Skin Equivalent—EPiTRI® BT—Alternatives to Animal Testing; Kojima, H., Seidle, T., Spielmann, H., Eds.; Springer: Singapore, 2019; pp. 8–19. [Google Scholar]
- Groeber, F.; Engelhardt, L.; Egger, S.; Werthmann, H.; Monaghan, M.; Walles, H.; Hansmann, J. Impedance Spectroscopy for the Non-Destructive Evaluation of In Vitro Epidermal Models. Pharm. Res. 2015, 32, 1845–1854. [Google Scholar] [CrossRef] [Green Version]
- Gorzelanny, C.; Mess, C.; Schneider, S.W.; Huck, V.; Brandner, J.M. Skin barriers in dermal drug delivery: Which barriers have to be overcome and how can we measure them? Pharmaceutics 2020, 12, 684. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, X.; Ooka, M.; Zhang, L.; Song, M.J.; Huang, R.; Kleinstreuer, N.C.; Simeonov, A.; Xia, M.; Ferrer, M. Two-Dimensional Cellular and Three-Dimensional Bio-Printed Skin Models to Screen Topical-Use Compounds for Irritation Potential. Front. Bioeng. Biotechnol. 2020, 8, 109. [Google Scholar] [CrossRef] [Green Version]
- Abdayem, R.; Callejon, S.; Portes, P.; Kirilov, P.; Demarne, F.; Pirot, F.; Jannin, V.; Haftek, M. Modulation of transepithelial electric resistance (TEER) in reconstructed human epidermis by excipients known to permeate intestinal tight junctions. Exp. Dermatol. 2015, 24, 686–691. [Google Scholar] [CrossRef]
- Zoio, P.; Lopes-Ventura, S.; Marto, J.; Oliva, A. Open-source human skin model with an in vivo-like barrier for drug testing. ALTEX, 2022; accepted. [Google Scholar]
- Yeste, J.; Illa, X.; Gutiérrez, C.; Solé, M.; Guimerà, A.; Villa, R. Geometric correction factor for transepithelial electrical resistance measurements in transwell and microfluidic cell cultures. J. Phys. D Appl. Phys. 2016, 49, 375401. [Google Scholar] [CrossRef] [Green Version]
- Zoio, P.; Lopes-Ventura, S.; Oliva, A. Barrier-on-a-Chip with a Modular Architecture and Integrated Sensors for Real-Time Measurement of Biological Barrier Function. Micromachines 2021, 12, 816. [Google Scholar] [CrossRef]
- Henry, O.Y.F.; Villenave, R.; Cronce, M.J.; Leineweber, W.D.; Benz, M.A.; Ingber, D.E. Organs-on-chips with integrated electrodes for trans-epithelial electrical resistance (TEER) measurements of human epithelial barrier function. Lab Chip 2017, 17, 2264–2271. [Google Scholar] [CrossRef]
- van der Helm, M.W.; Odijk, M.; Frimat, J.-P.; van der Meer, A.D.; Eijkel, J.C.T.; van den Berg, A.; Segerink, L.I. Direct quantification of transendothelial electrical resistance in organs-on-chips. Biosens. Bioelectron. 2016, 85, 924–929. [Google Scholar] [CrossRef] [Green Version]
- Odijk, M.; van der Meer, A.D.; Levner, D.; Kim, H.J.; van der Helm, M.W.; Segerink, L.I.; Frimat, J.-P.; Hamilton, G.A.; Ingber, D.E.; van den Berg, A. Measuring direct current trans-epithelial electrical resistance in organ-on-a-chip microsystems. Lab Chip 2015, 15, 745–752. [Google Scholar] [CrossRef]
- Grimnes, S.; Martinsen, Ø.G. Sources of error in tetrapolar impedance measurements on biomaterials and other ionic conductors. J. Phys. D Appl. Phys. 2007, 40, 9–14. [Google Scholar] [CrossRef]
- Ataç, B.; Wagner, I.; Horland, R.; Lauster, R.; Marx, U.; Tonevitsky, A.G.; Azar, R.P.; Lindner, G. Skin and hair on-a-chip: In vitro skin models versus ex vivo tissue maintenance with dynamic perfusion. Lab Chip 2013, 13, 3555–3561. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.J.; Ellett, F.; Thomas, C.N.; Jalali, F.; Anderson, R.R.; Irimia, D.; Raff, A.B. A microscale, full-thickness, human skin on a chip assay simulating neutrophil responses to skin infection and antibiotic treatments. Lab Chip 2019, 19, 3094–3103. [Google Scholar] [CrossRef]
- Wagner, I.; Materne, E.M.; Brincker, S.; Süßbier, U.; Frädrich, C.; Busek, M.; Sonntag, F.; Sakharov, D.A.; Trushkin, E.V.; Tonevitsky, A.G.; et al. A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab Chip 2013, 13, 3538–3547. [Google Scholar] [CrossRef] [Green Version]
- Maschmeyer, I.; Lorenz, A.K.; Schimek, K.; Hasenberg, T.; Ramme, A.P.; Hübner, J.; Lindner, M.; Drewell, C.; Bauer, S.; Thomas, A.; et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip 2015, 15, 2688–2699. [Google Scholar] [CrossRef] [Green Version]
- Abaci, H.E.; Gledhill, K.; Guo, Z.; Christiano, A.M.; Shuler, M.L. Pumpless microfluidic platform for drug testing on human skin equivalents. Lab Chip 2015, 15, 882–888. [Google Scholar] [CrossRef] [Green Version]
- Kühnl, J.; Tao, T.P.; Brandmair, K.; Gerlach, S.; Rings, T.; Müller-Vieira, U.; Przibilla, J.; Genies, C.; Jaques-Jamin, C.; Schepky, A.; et al. Characterization of application scenario-dependent pharmacokinetics and pharmacodynamic properties of permethrin and hyperforin in a dynamic skin and liver multi-organ-chip model. Toxicology 2021, 448, 152637. [Google Scholar] [CrossRef]
- Tavares, R.S.N.; Phuong-Tao, T.; Maschmeyer, I.; Maria-Engler, S.S.; Schäfer-Korting, M.; Winter, A.; Zoschke, C.; Lauster, R.; Marx, U.; Gaspar, L.R. Toxicity of topically applied drugs beyond skin irritation: Static skin model vs. Two organs-on-a-chip. Int. J. Pharm. 2020, 589, 119788. [Google Scholar] [CrossRef]
- Risueño, I.; Valencia, L.; Jorcano, J.L.; Velasco, D. Skin-on-a-chip models: General overview and future perspectives. APL Bioeng. 2021, 5, 30901. [Google Scholar] [CrossRef]
- Wufuer, M.; Lee, G.H.; Hur, W.; Jeon, B.; Kim, B.J.; Choi, T.H.; Lee, S.H. Skin-on-a-chip model simulating inflammation, edema and drug-based treatment. Sci. Rep. 2016, 6, 37471. [Google Scholar] [CrossRef] [Green Version]
- Ramadan, Q.; Ting, F.C.W. In vitro micro-physiological immune-competent model of the human skin. Lab Chip 2016, 16, 1899–1908. [Google Scholar] [CrossRef]
- Sasaki, N.; Tsuchiya, K.; Kobayashi, H. Photolithography-free skin-on-a-chip for parallel permeation assays. Sens. Mater. 2019, 31, 107–115. [Google Scholar] [CrossRef]
- Groeber, F.; Engelhardt, L.; Lange, J.; Kurdyn, S.; Schmid, F.F.; Rücker, C.; Mielke, S.; Walles, H.; Hansmann, J. A first vascularized skin equivalent as an alternative to animal experimentation. ALTEX-Altern. Anim. Exp. 2016, 33, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Abaci, H.E.; Guo, Z.; Coffman, A.; Gillette, B.; Lee, W.H.; Sia, S.K.; Christiano, A.M. Human Skin Constructs with Spatially Controlled Vasculature Using Primary and iPSC-Derived Endothelial Cells. Adv. Healthc. Mater. 2016, 5, 1800–1807. [Google Scholar] [CrossRef] [Green Version]
- Mori, N.; Morimoto, Y.; Takeuchi, S. Skin integrated with perfusable vascular channels on a chip. Biomaterials 2017, 116, 48–56. [Google Scholar] [CrossRef]
- Kim, B.S.; Gao, G.; Kim, J.Y.; Cho, D.W. 3D Cell Printing of Perfusable Vascularized Human Skin Equivalent Composed of Epidermis, Dermis, and Hypodermis for Better Structural Recapitulation of Native Skin. Adv. Healthc. Mater. 2019, 8, 1801019. [Google Scholar] [CrossRef]
- Salameh, S.; Tissot, N.; Cache, K.; Lima, J.; Suzuki, I.; Marinho, P.A.; Rielland, M.; Soeur, J.; Takeuchi, S.; Germain, S.; et al. A perfusable vascularized full-thickness skin model for potential topical and systemic applications. Biofabrication 2021, 13, 35042. [Google Scholar] [CrossRef]
- Lee, S.; Jin, S.P.; Kim, Y.K.; Sung, G.Y.; Chung, J.H.; Sung, J.H. Construction of 3D multicellular microfluidic chip for an in vitro skin model. Biomed. Microdevices 2017, 19, 22. [Google Scholar] [CrossRef]
- Song, H.J.; Lim, H.Y.; Chun, W.; Choi, K.C.; Lee, T.Y.; Sung, J.H.; Sung, G.Y. Development of 3D skin-equivalent in a pump-less microfluidic chip. J. Ind. Eng. Chem. 2018, 60, 355–359. [Google Scholar] [CrossRef]
- Lim, H.Y.; Kim, J.; Song, H.J.; Kim, K.; Choi, K.C.; Park, S.; Sung, G.Y. Development of wrinkled skin-on-a-chip (WSOC) by cyclic uniaxial stretching. J. Ind. Eng. Chem. 2018, 68, 238–245. [Google Scholar] [CrossRef]
- Strüver, K.; Friess, W.; Hedtrich, S. Development of a Perfusion Platform for Dynamic Cultivation of in vitro Skin Models. Skin Pharmacol. Physiol. 2017, 30, 180–189. [Google Scholar] [CrossRef]
- Sriram, G.; Alberti, M.; Dancik, Y.; Wu, B.; Wu, R.; Feng, Z.; Ramasamy, S.; Bigliardi, P.L.; Bigliardi-Qi, M.; Wang, Z. Full-thickness human skin-on-chip with enhanced epidermal morphogenesis and barrier function. Mater. Today 2018, 21, 326–340. [Google Scholar] [CrossRef]
- Valencia, L.; Tejero, V.C.; Clemente, M.; Fernaud, I.; Holgado, M. OPEN A new microfluidic method enabling the generation of Multi—Layered Tissues—On—Chips using skin cells as a proof of concept. Sci. Rep. 2021, 11, 13160. [Google Scholar] [CrossRef]
- Zoio, P.; Lopes-Ventura, S.; Oliva, A. Biomimetic Full-Thickness Skin-on-a-Chip Based on a Fibroblast-Derived Matrix. Micro 2022, 2, 191–211. [Google Scholar] [CrossRef]
- Rimal, R.; Marquardt, Y.; Nevolianis, T.; Djeljadini, S.; Marquez, A.B.; Huth, S.; Chigrin, D.N.; Wessling, M.; Baron, J.M.; Möller, M.; et al. Dynamic flow enables long-term maintenance of 3-D vascularized human skin models. Appl. Mater. Today 2021, 25, 101213. [Google Scholar] [CrossRef]
- Allwardt, V.; Ainscough, A.J.; Viswanathan, P.; Sherrod, S.D.; McLean, J.A.; Haddrick, M.; Pensabene, V. Translational Roadmap for the Organs-on-a-Chip Industry toward Broad Adoption. Bioengineering 2020, 7, 112. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Cell Type | Flow Type; Velocity | Fabrication Method; Materials | Main Features |
|---|---|---|---|---|
| Wufuer et al. (2016) [110] | HaCaT, immortalized HS27 and HUVECS | Gravity driven flow; Not stated | Photolithography; PDMS and PET membrane | TNF-α induced skin inflammation; Simulation of skin edema |
| Ramadam et al. (2016) [111] | HaCaT and U937 dendritic cells | Syringe pumping; 0.6–1.2 µL/min | Rapid prototyping; PMMA, PDMS and PET membrane | Immune competent model; TEER measurements |
| Sasaki et al. (2019) [112] | HaCaT | Syringe pumping; 10 µL/min | Laser cutter; PMMA, PDMS and PET membrane | Irritation testing with potassium dichromate |
| Reference | Cell Type | Dermal Matrix | Flow Type; Velocity | Fabrication Method; Materials | Main Features |
|---|---|---|---|---|---|
| Groeber et al. (2016) [113] | Primary HEKs, primary HDFs and hDMECC | Decellularized porcine jejunum | Peristaltic pumping; Not stated | Rapid prototyping; PEEK, PC | TEER measurements, |
| Abaci et al. (2016) [114] | Primary HEKs, primary HDFs iPSC-derived endothelial cells | Collagen | Syringe pumping; Not stated | 3D printing, templating; Tranwell insets, PET membranes | Integration of iPSC; Promotion of neovascularization in a rat model |
| Mori et al. (2016) [115] | Primary HEKs, primary HDFs and HUVECs | Collagen | Peristaltic pumping; 33–50 µL/min | 3D printing, templating; Not stated | Vascular channels coated with endothelial cells; Permation testing caffeine and ISDN |
| Kim et al. (2019) [116] | Primary HEKs, primary HDFs, HUVECs and primary HPAs | Fibrinogen, dECM porcine skin | Peristaltic pumping; 50–100 µL/min | 3D printing; PCL | Bioprinting; Integration of hypodermis |
| Salameh (2021) [117] | Primary HEKs, primary HDFs and HUVECs | Collagen | Peristaltic pumping; 33–50 µL/min | 3D printing, templating; Not stated | Formation of angiogenic sprouts; systemic drug delivery studies |
| Reference | Cell Type | Dermal Matrix | Flow Type; Velocity | Fabrication Method; Materials | Main Features |
|---|---|---|---|---|---|
| Lee et al. (2017) [118] | HaCaT or primary HEKs, primary HDFs and HUVECs | Collagen | Gravity driven; 5–10 µL/min | Lithography; PDMS and PC membrane | Studies of mass transport with FITC-dextran |
| Song et al. (2017) [119] | Primary HEKs and primary HDFs | Collagen | Gravity driven; Not stated | Lithography; PDMS and PC membrane | Study of collagen contraction |
| Lim et al. (2018) [120] | Primary HEKs and primary HDFs | Collagen | Gravity driven; Not stated | Lithography; PDMS and PC membrane | Uniaxial stretch applied for modeling wrinkles |
| Strüver et al. (2017) [121] | Primary HEKs and primary HDFs | Collagen | Peristaltic pumping; 20–167 µL/min | Not stated; PTFE and PET membrane | Improved skin differentiation |
| Sriram et al. (2019) [122] | N/TERT and primary HDFs | Fibrin + PEG | Peristaltic pumping; 1 µL/min | Micromilling; PMMA and PC membrane | Improved skin differentiation and barrier function; stable dermis |
| Valencia et al. (2021) [123] | HaCaT and primary HDFs | Fibrin | Syringe pumping; 0.67 µL/min | Edge plotter; PMMA, PDMS, vinyl and PC membrane | Parallel flow method for bilayer tissue formation |
| Zoio et al. (2021, 2022) [97,124] | Primary HEKs and primary HDFs | FDM + PS scaffold | Syringe pumping; 1–2 µL/min | Rapid prototyping; PMMA | Improved barrier function; TEER measurements on-chip |
| Rimal et al. (2021) [125] | Primary HEKs, primary HDFs and HUVECS | ECM-coating of single cells (FN and G) | Peristatic pumping; 5 × 103 µL/min | 3D printing; Not stated | Scaffold-free; vascularized dermal tissue; 3D-wound healing assay |
| Reference | Epidermis and DEJ Markers | Dermis Markers | Thickness | Functional Studies | Others |
|---|---|---|---|---|---|
| Mori et al. (2016) [115] | =K10, =K15 | - | =epidermis (VE + SC) | =capacitance | ↑ cross-sectional area channels, ↑ cell density |
| Kim et al. (2019) [116] | ↑ K19 | - | - | - | ↑ p63-positive cells |
| Lee et al. (2017) [118] | =K5, =involucrin, =filaggrin | - | - | - | ↓ SC homogeneity |
| Song et al. (2017) [119] | ↓ collagen IV, =K10 | =fibronectin, ↓collagen | - | - | ↓ hydrogel contraction |
| Strüver et al. (2017) [121] | ↑ filaggrin, ↑ involucrin | - | ↑ SC, =VE, ↓ dermis | ↓ barrier function (increased testosterone permeability) | ↑ claudin 1, ↑ occludin |
| Sriram et al. (2019) [122] | ↑ collagen IV, ↑ involucrin, ↑ collagen VII, ↑collagen XVII, =filaggrin, =K10 | - | ↑ SC, ↑ VE | ↑ TEER ↑ Barrier function (decreased caffeine permeability) | ↓ SC water content |
| Zoio et al. (2022) [124] | =K10, =K14, ↑ filaggrin, ↑ involucrin | ↑ collagen I, ↑ fibronectin | =SC, ↑ VE | ↑ TEER ↑ barrier function (decreased FD permeability) | - |
| Rimal et al. (2021) [125] | ↑ filaggrin, =filaggrin 2 | ↑ fibronectin, =collagen I | - | ↑ TEER | ↑ wound healing ↑ ECM homeostasis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zoio, P.; Oliva, A. Skin-on-a-Chip Technology: Microengineering Physiologically Relevant In Vitro Skin Models. Pharmaceutics 2022, 14, 682. https://doi.org/10.3390/pharmaceutics14030682
Zoio P, Oliva A. Skin-on-a-Chip Technology: Microengineering Physiologically Relevant In Vitro Skin Models. Pharmaceutics. 2022; 14(3):682. https://doi.org/10.3390/pharmaceutics14030682
Chicago/Turabian StyleZoio, Patrícia, and Abel Oliva. 2022. "Skin-on-a-Chip Technology: Microengineering Physiologically Relevant In Vitro Skin Models" Pharmaceutics 14, no. 3: 682. https://doi.org/10.3390/pharmaceutics14030682