
Two Beats One: Osteosarcoma Therapy with Light-Activated and Chemo-Releasing Keratin Nanoformulation in a Preclinical Mouse Model

 , ,
, ,  , , , , , ,
, , , , , ,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents
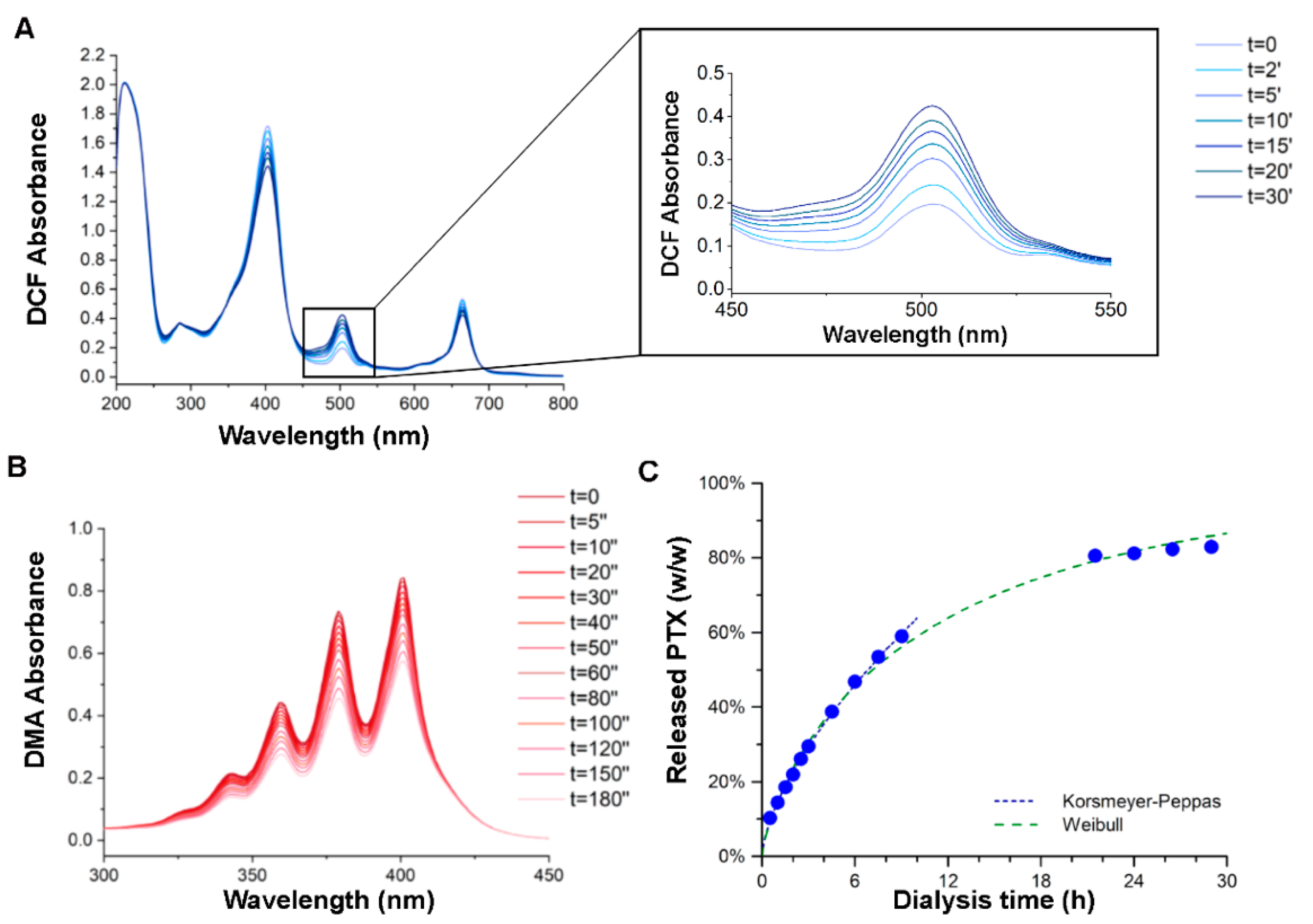
2.2. PTX-Ce6@ker Synthesis and Characterization
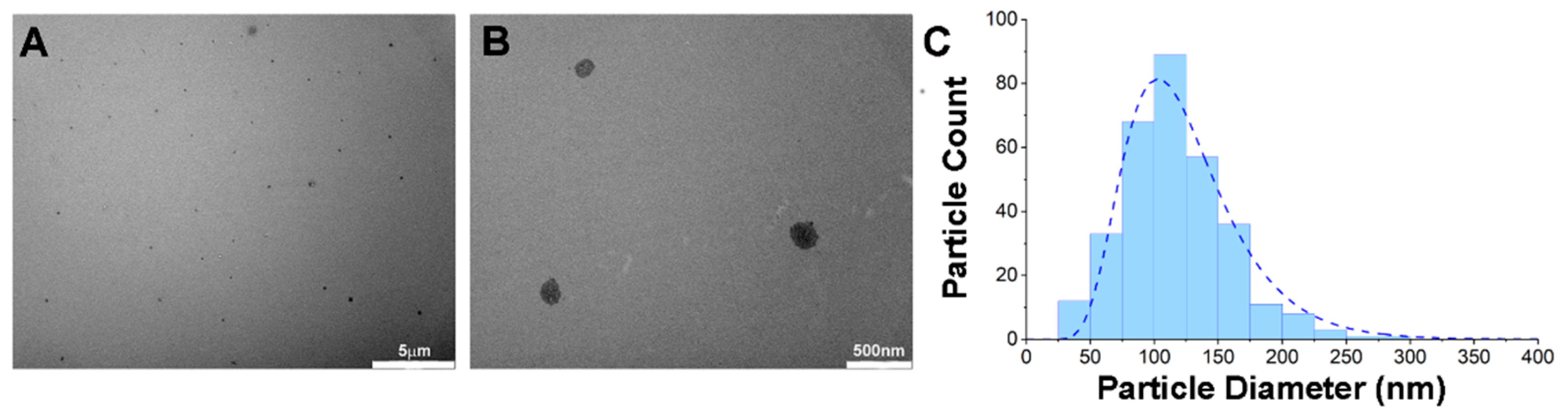
2.3. Transmission Electron Microscopy (TEM)
2.4. PTX Release Kinetics
2.5. Cell Culture
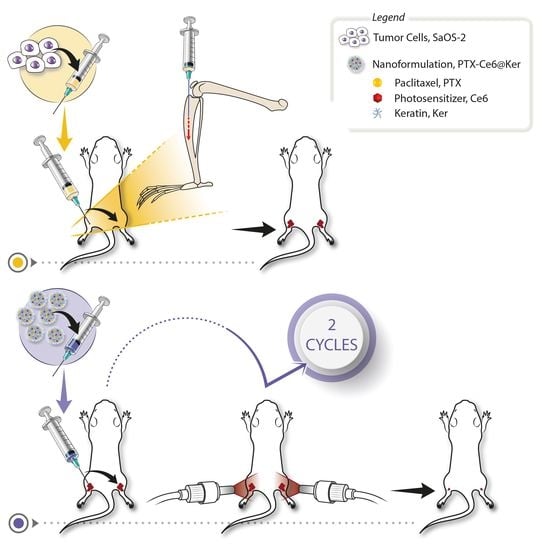
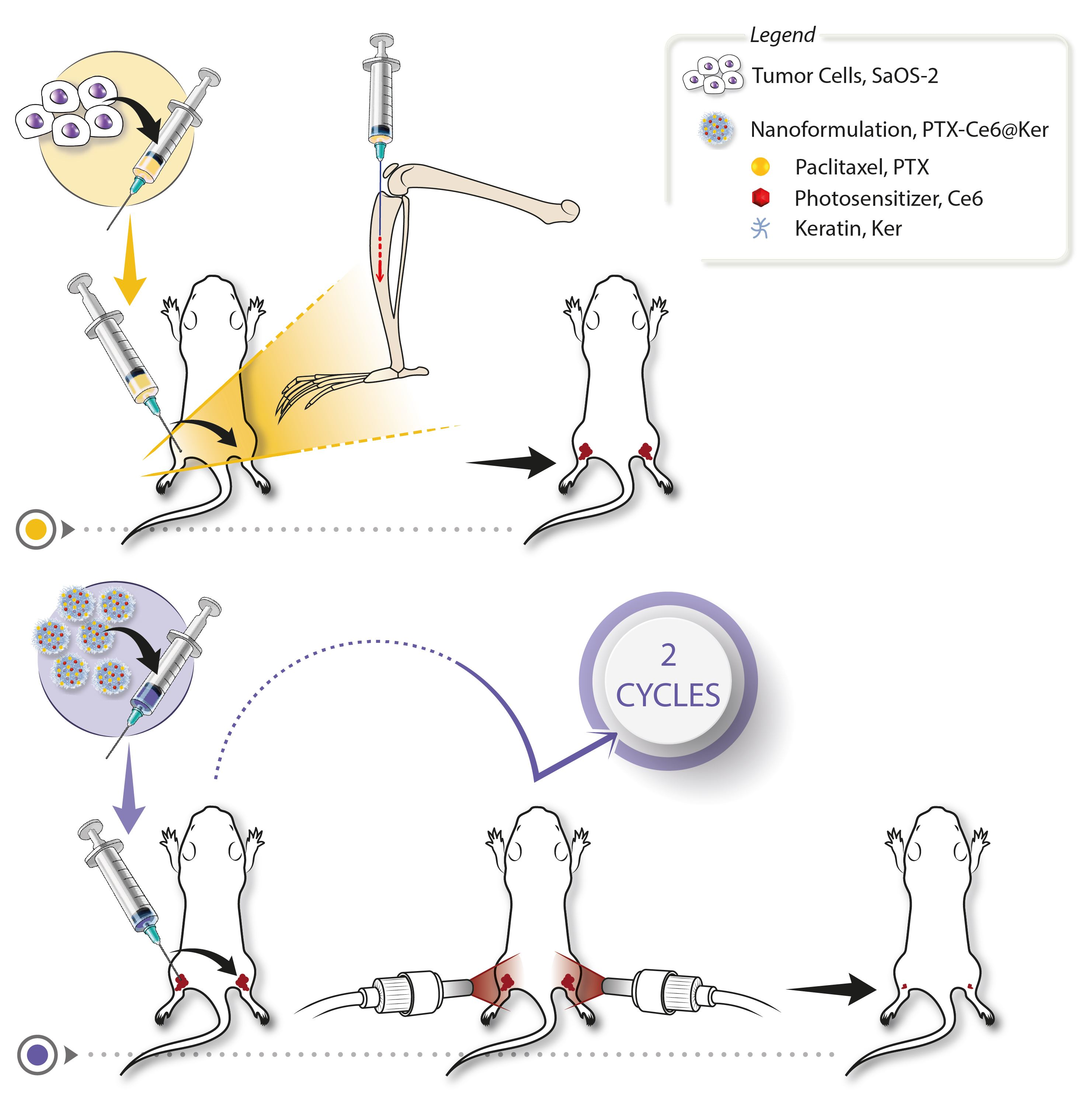
2.6. Orthotopic OS Model
2.7. PDT Device
2.8. Histological Analyses
2.9. Confocal Laser Scanning Microscopy and Fluorescent Lifetime Imaging
2.10. Statistical Analysis
3. Results and Discussion
3.1. Chemical and Physical Characterizations of PTX-Ce6@ker Nanoformulation
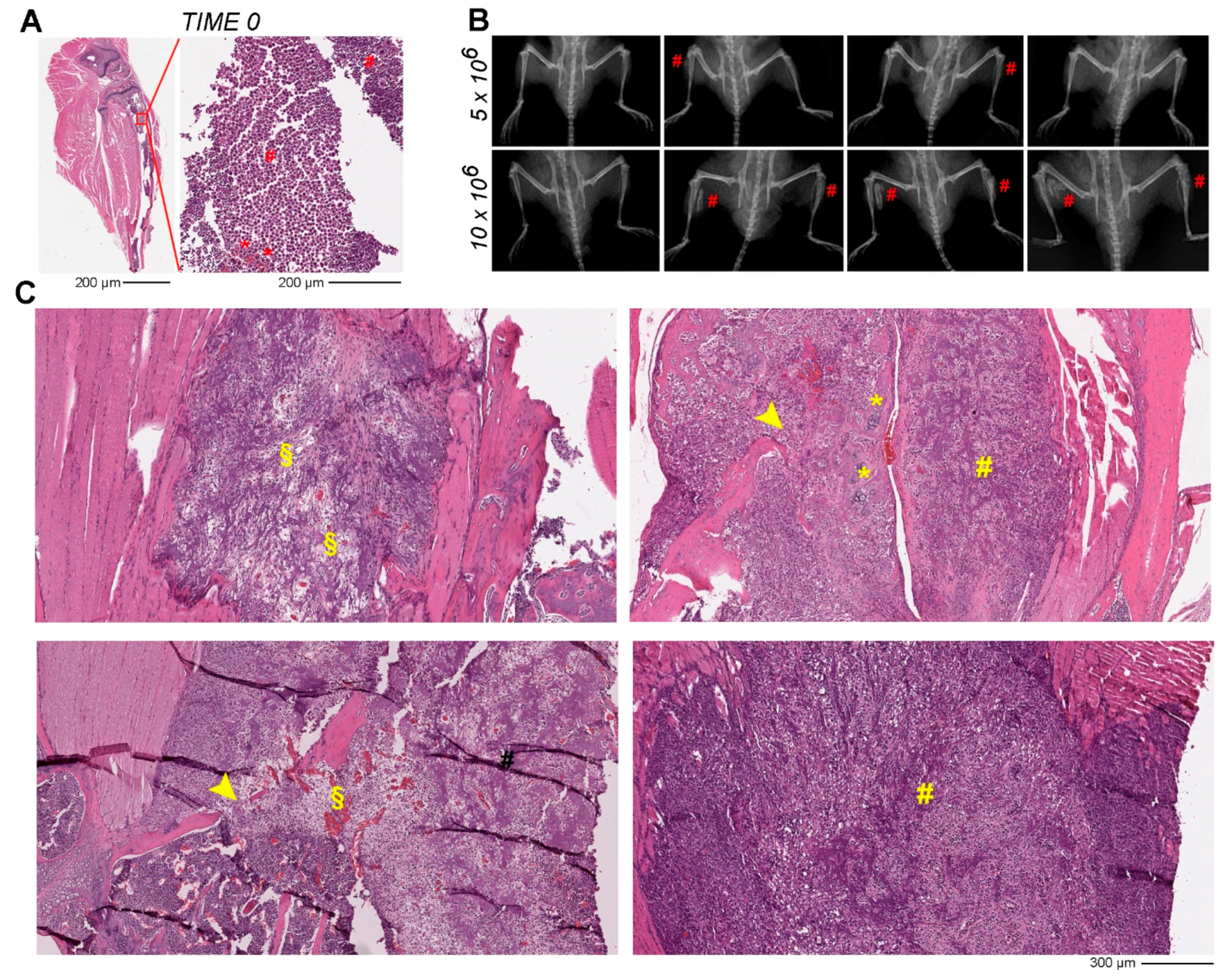
3.2. Orthotopic Osteosarcoma Mouse model Set Up
3.3. Treatment Regime Set Up
3.3.1. Light-Delivery Device
3.3.2. Nanoformulation Dosage and Treatment’s Schedule
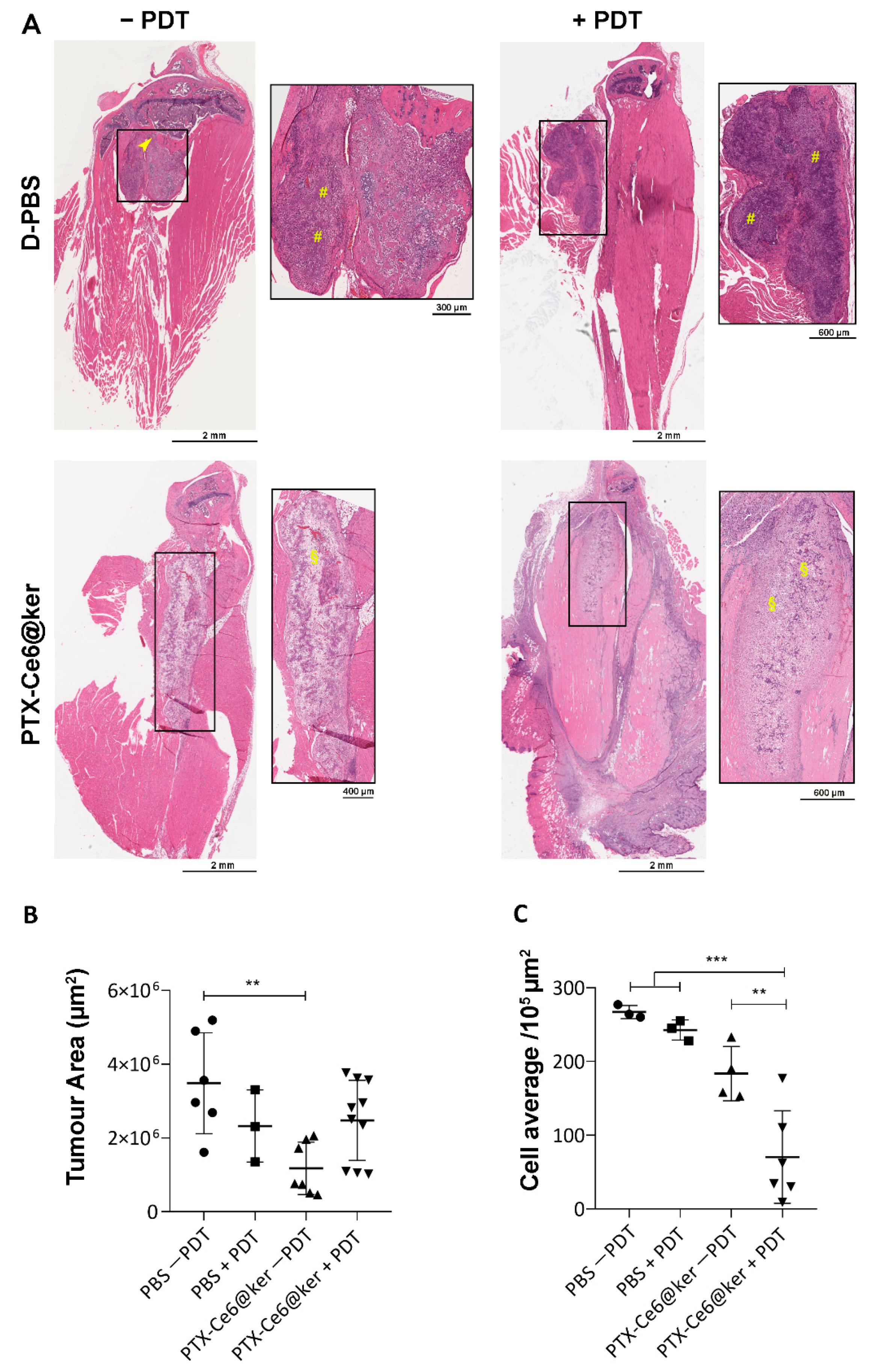
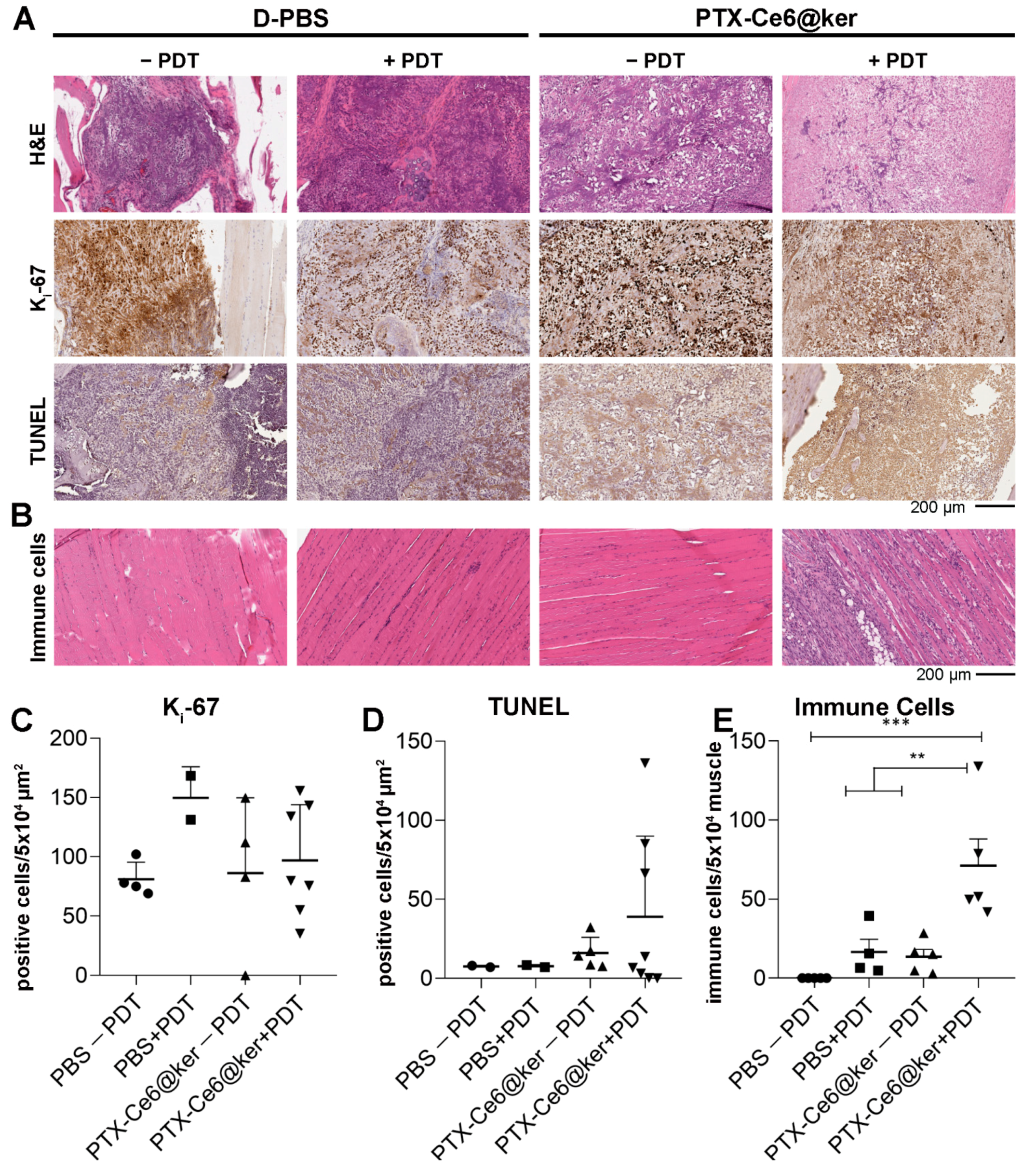
3.4. Preclinical Evaluation of the Therapy Efficacy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smeland, S.; Bielack, S.S.; Whelan, J.; Bernstein, M.; Hogendoorn, P.; Krailo, M.D.; Gorlick, R.; Janeway, K.A.; Ingleby, F.C.; Anninga, J.; et al. Survival and prognosis with osteosarcoma: Outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur. J. Cancer 2019, 109, 36–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakoff, M.S.; Bielack, S.S.; Meltzer, P.; Gorlick, R. Osteosarcoma: Current Treatment and a Collaborative Pathway to Success. J. Clin. Oncol. 2015, 33, 3029–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Zhang, S.; Feng, A.; Xu, D.; Zhu, Q.; Mao, Y.; Zhao, Y.; Lv, Y.; Han, C.; Liu, R.; et al. Methotrexate, doxorubicin, and cisplatinum regimen is still the preferred option for osteosarcoma chemotherapy: A meta-analysis and clinical observation. Medicine 2019, 98, e15582. [Google Scholar] [CrossRef] [PubMed]
- Prudowsky, Z.D.; Yustein, J.T. Recent insights into therapy resistance in osteosarcoma. Cancers 2021, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Marchandet, L.; Lallier, M.; Charrier, C.; Baud’huin, M.; Ory, B.; Lamoureux, F. Mechanisms of resistance to conventional therapies for osteosarcoma. Cancers 2021, 13, 683. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Kahen, E.; Cubitt, C.L.; McGuire, J.; Kreahling, J.; Lee, J.; Altiok, S.; Lynch, C.C.; Sullivan, D.M.; Reed, D.R. Identification of Synergistic, Clinically Achievable, Combination Therapies for Osteosarcoma. Sci. Rep. 2015, 5, 16991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saputra, E.C.; Huang, L.; Chen, Y.; Tucker-Kellogg, L. Combination therapy and the evolution of resistance: The theoretical merits of synergism and antagonism in cancer. Cancer Res. 2018, 78, 2419–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Kohli, M.; Smith, A. Nanoparticles for Combination Drug Therapy. ACS Nano 2013, 7, 9518–9525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira-Silva, M.; Alvarez-Lorenzo, C.; Concheiro, A.; Santos, A.C.; Veiga, F.; Figueiras, A. Nanomedicine in osteosarcoma therapy: Micelleplexes for delivery of nucleic acids and drugs toward osteosarcoma-targeted therapies. Eur. J. Pharm. Biopharm. 2020, 148, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Martella, E.; Ferroni, C.; Guerrini, A.; Ballestri, M.; Columbaro, M.; Santi, S.; Sotgiu, G.; Serra, M.; Donati, D.M.; Lucarelli, E.; et al. Functionalized keratin as nanotechnology-based drug delivery system for the pharmacological treatment of osteosarcoma. Int. J. Mol. Sci. 2018, 19, 3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Hong, S.P.; Oh, T.Y.; Bang, S.; Chung, J.B.; Song, S.Y. Paclitaxel augments cytotoxic effect of photodynamic therapy using verteporfin in gastric and bile duct cancer cells. Photochem. Photobiol. Sci. 2008, 7, 769–774. [Google Scholar] [CrossRef]
- Thapa, P.; Li, M.; Bio, M.; Rajaputra, P.; Nkepang, G.; Sun, Y.; Woo, S.; You, Y. Far-Red Light-Activatable Prodrug of Paclitaxel for the Combined Effects of Photodynamic Therapy and Site-Specific Paclitaxel Chemotherapy. J. Med. Chem. 2016, 59, 3204–3214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.; Bu, J.; Ding, L.; Lou, J.W.H.; Valic, M.S.; Cheng, M.H.Y.; Rosilio, V.; Chen, J.; Zheng, G. Porphyrin-lipid stabilized paclitaxel nanoemulsion for combined photodynamic therapy and chemotherapy. J. Nanobiotechnol. 2021, 19, 154. [Google Scholar] [CrossRef]
- Guijarro, M.V.; Ghivizzani, S.C.; Gibbs, C.P. Animal models in osteosarcoma. Front. Oncol. 2014, 4, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ek, E.T.H.; Dass, C.R.; Choong, P.F.M. Commonly used mouse models of osteosarcoma. Crit. Rev. Oncol. Hematol. 2006, 60, 1–8. [Google Scholar] [CrossRef]
- Jacques, C.; Renema, N.; Lezot, F.; Ory, B.; Walkley, C.R.; Grigoriadis, A.E.; Heymann, D. Small animal models for the study of bone sarcoma pathogenesis:characteristics, therapeutic interests and limitations. J. Bone Oncol. 2018, 12, 7–13. [Google Scholar] [CrossRef]
- Frese, K.K.; Tuveson, D.A. Maximizing mouse cancer models. Nat. Rev. Cancer 2007, 7, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Landuzzi, L.; Manara, M.C.; Lollini, P.L.; Scotlandi, K. Patient Derived Xenografts for Genome-Driven Therapy of Osteosarcoma. Cells 2021, 10, 416. [Google Scholar] [CrossRef] [PubMed]
- Imle, R.; Kommoss, F.K.F.; Banito, A. Preclinical in vivo modeling of pediatric sarcoma—Promises and limitations. J. Clin. Med. 2021, 10, 1578. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, Q.; Gong, X.; Liu, J.; Ma, Y. Osteosarcoma: A review of current and future therapeutic approaches. Biomed. Eng. Online 2021, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Uluçkan, Ö.; Segaliny, A.; Botter, S.; Santiago, J.M.; Mutsaers, A.J. Preclinical mouse models of osteosarcoma. Bonekey Rep. 2015, 4, 670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferroni, C.; Sotgiu, G.; Sagnella, A.; Varchi, G.; Guerrini, A.; Giuri, D.; Polo, E.; Orlandi, V.T.; Marras, E.; Gariboldi, M.; et al. Wool Keratin 3D Scaffolds with Light-Triggered Antimicrobial Activity. Biomacromolecules 2016, 17, 2882–2890. [Google Scholar] [CrossRef] [PubMed]
- Moret, F.; Menilli, L.; Battan, M.; Tedesco, D.; Columbaro, M.; Guerrini, A.; Avancini, G.; Ferroni, C.; Varchi, G. Pheophorbide a and paclitaxel bioresponsive nanoparticles as double-punch platform for cancer therapy. Pharmaceutics 2021, 13, 1130. [Google Scholar] [CrossRef] [PubMed]
- Pyrz, W.D.; Buttrey, D.J. Particle size determination using TEM: A discussion of image acquisition and analysis for the novice microscopist. Langmuir 2008, 24, 11350–11360. [Google Scholar] [CrossRef]
- Mircioiu, C.; Voicu, V.; Anuta, V.; Tudose, A.; Celia, C.; Paolino, D.; Fresta, M.; Sandulovici, R.; Mircioiu, I. Mathematical modeling of release kinetics from supramolecular drug delivery systems. Pharmaceutics 2019, 11, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Du, L.; Xu, W.; Fan, Q.; Tu, B.; Shen, Y.; Yan, W.; Tang, T.; Wang, Y. Tumorigenesis and spontaneous metastasis by luciferase-labeled human xenograft osteosarcoma cells in nude mice. Chin. Med. J. 2012, 125, 4022–4030. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abouelmagd, S.A.; Sun, B.; Chang, A.C.; Ku, Y.J.; Yeo, Y. Release Assay of Non-Soluble Drugs. Mol. Pharm. 2015, 12, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruschi, M.L. 5—Mathematical models of drug release. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Woodhead Publishing: Cambridge, UK, 2015; pp. 63–86. ISBN 978-0-08-100092-2. [Google Scholar]
- Shimizu, T.; Ishikawa, T.; Iwai, S.; Ueki, A.; Sugihara, E.; Onishi, N.; Kuninaka, S.; Miyamoto, T.; Toyama, Y.; Ijiri, H.; et al. Fibroblast growth factor-2 is an important factor that maintains cellular immaturity and contributes to aggressiveness of osteosarcoma. Mol. Cancer Res. 2012, 10, 454–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavirayani, A.M.; Sundberg, J.P.; Foreman, O. Primary neoplasms of bones in mice: Retrospective study and review of literature. Vet. Pathol. 2012, 49, 182–205. [Google Scholar] [CrossRef]
- Miwa, S.; Otsuka, T. Practical use of imaging technique for management of bone and soft tissue tumors. J. Orthop. Sci. 2017, 22, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Taki, J.; Yamamoto, N.; Shirai, T.; Nishida, H.; Hayashi, K.; Tanzawa, Y.; Kimura, H.; Takeuchi, A.; Igarashi, K.; et al. A novel combined radiological method for evaluation of the response to chemotherapy for primary bone sarcoma. J. Surg. Oncol. 2012, 106, 273–279. [Google Scholar] [CrossRef]
- Mangham, D.C.; Athanasou, N.A. Guidelines for histopathological specimen examination and diagnostic reporting of primary bone tumours. Clin. Sarcoma Res. 2011, 1, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgia, F.; Giuffrida, R.; Caradonna, E.; Vaccaro, M.; Guarneri, F.; Cannavò, S.P. Early and Late Onset Side Effects of Photodynamic Therapy. Biomedicines 2018, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Fink, C.; Enk, A.; Gholam, P. Photodynamic therapy--aspects of pain management. J. Dtsch. Dermatol. Ges. 2015, 13, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Duchi, S.; Ramos-Romero, S.; Dozza, B.; Guerra-Rebollo, M.; Cattini, L.; Ballestri, M.; Dambruoso, P.; Guerrini, A.; Sotgiu, G.; Varchi, G.; et al. Development of near-infrared photoactivable phthalocyanine-loaded nanoparticles to kill tumor cells: An improved tool for photodynamic therapy of solid cancers. Nanomedicine 2016, 12, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Bisland, S.K.; Burch, S. Photodynamic therapy of diseased bone. Photodiagnosis Photodyn. Ther. 2006, 3, 147–155. [Google Scholar] [CrossRef]
- Reginato, E. Immune response after photodynamic therapy increases anti-cancer and anti-bacterial effects. World J. Immunol. 2014, 4, 1. [Google Scholar] [CrossRef]
- Alzeibak, R.; Mishchenko, T.A.; Shilyagina, N.Y.; Balalaeva, I.V.; Vedunova, M.V.; Krysko, D.V. Targeting immunogenic cancer cell death by photodynamic therapy: Past, present and future. J. Immunother. Cancer 2021, 9, e001926. [Google Scholar] [CrossRef] [PubMed]
- Falk-Mahapatra, R.; Gollnick, S.O. Photodynamic Therapy and Immunity: An Update. Photochem. Photobiol. 2020, 96, 550–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DOSE | I | II | III |
|---|---|---|---|
| PTX/tibia | 97.5 μg | 73.2 μg | 48.8 μg |
| PTX/mouse | 195 μg | 146.4 μg | 97.6 μg |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martella, E.; Dozza, B.; Ferroni, C.; Obeyok, C.O.; Guerrini, A.; Tedesco, D.; Manet, I.; Sotgiu, G.; Columbaro, M.; Ballestri, M.; et al. Two Beats One: Osteosarcoma Therapy with Light-Activated and Chemo-Releasing Keratin Nanoformulation in a Preclinical Mouse Model. Pharmaceutics 2022, 14, 677. https://doi.org/10.3390/pharmaceutics14030677
Martella E, Dozza B, Ferroni C, Obeyok CO, Guerrini A, Tedesco D, Manet I, Sotgiu G, Columbaro M, Ballestri M, et al. Two Beats One: Osteosarcoma Therapy with Light-Activated and Chemo-Releasing Keratin Nanoformulation in a Preclinical Mouse Model. Pharmaceutics. 2022; 14(3):677. https://doi.org/10.3390/pharmaceutics14030677
Chicago/Turabian StyleMartella, Elisa, Barbara Dozza, Claudia Ferroni, Clement Osuru Obeyok, Andrea Guerrini, Daniele Tedesco, Ilse Manet, Giovanna Sotgiu, Marta Columbaro, Marco Ballestri, and et al. 2022. "Two Beats One: Osteosarcoma Therapy with Light-Activated and Chemo-Releasing Keratin Nanoformulation in a Preclinical Mouse Model" Pharmaceutics 14, no. 3: 677. https://doi.org/10.3390/pharmaceutics14030677