
3,4-Dihydroxyphenylethanol (DPE or Hydroxytyrosol) Counteracts ERK1/2 and mTOR Activation, Pro-Inflammatory Cytokine Release, Autophagy and Mitophagy Reduction Mediated by Benzo[a]pyrene in Primary Human Colonic Epithelial Cells

 , and
, and 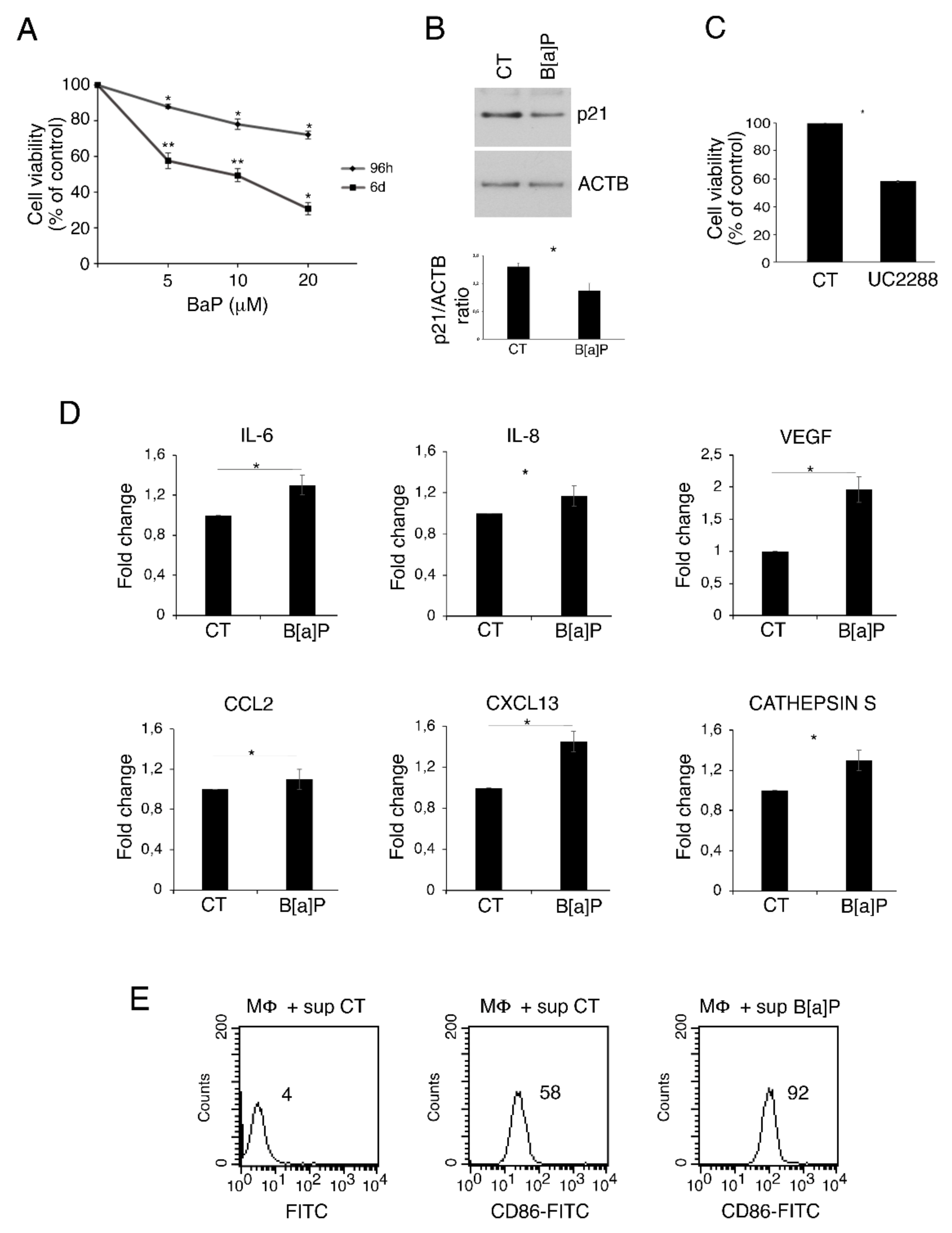{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures, Reagents and Treatments
- −
- Cells were grown for 96 h and 6 days, adding Benzo[a]pyrene every other day;
- −
- Pre-treatments with 1 µM Hydroxytyrosol (H) (MedChemExpress, Cat# HY-N0570, Monmouth Junction, NJ 08852, USA) were performed for 45 min before adding B[a]P;
- −
- HCoEpC were incubated with 1 µM UC2288 (Calbiochem- Sigma-Aldrich, Cat# 532813, St. Louis, MO, USA), a p21 inhibitor, for 6 days, refreshing medium supplemented with the inhibitor every other day;
- −
- In order to evaluate autophagy, cells treated or not (CT) with Benzo[a]pyrene for 6 days were incubated or not with 20 nM Bafilomycin A1 (MedChemExpress, Cat# HY-100558, Monmouth Junction, NJ 08852, USA) for the last 4 h.
2.2. Cell Viability Assay
2.3. Chemiluminescent Immunometric Assay
2.4. Monocyte Isolation and Macrophage Differentiation
2.5. Immunofluorescence Staining and FACS Analysis
2.6. Western Blotting
2.7. Antibodies
2.7.1. Indirect Immunofluorescence Assay (IFA)
2.7.2. MitoTrackerTM Green-FM Staining
2.8. Statistical Analysis
3. Results
3.1. B[a]P Exerts a Cytotoxic Effect in Primary Colonic Epithelial Cells, Increases the Release of IL-6, VEGF, IL-8, CXCL13, Cathepsin S and Activates Macrophages
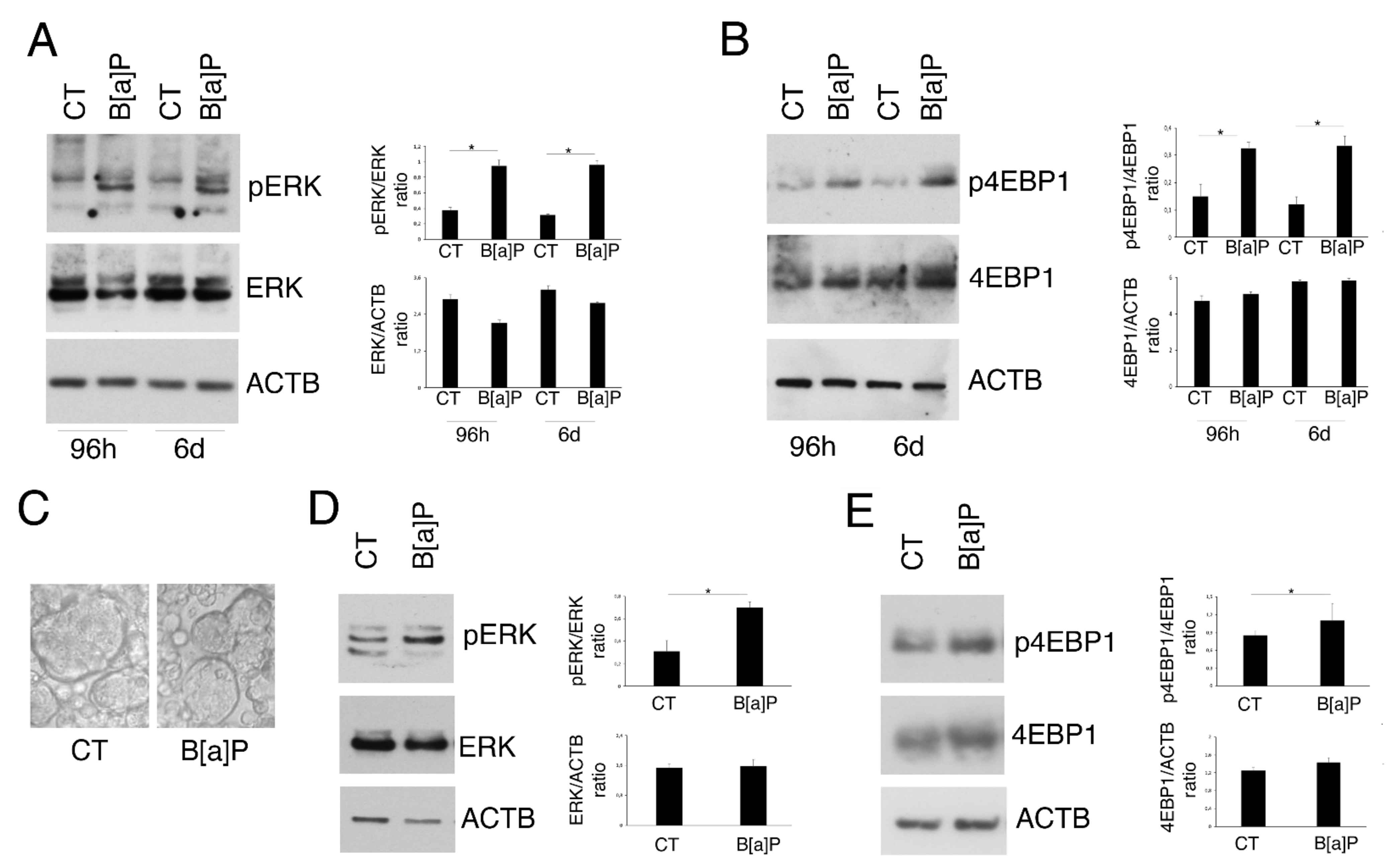
3.2. B[a]P Activates ERK1/2 and mTOR in 2D and 3D Culture Models of HCoEpC
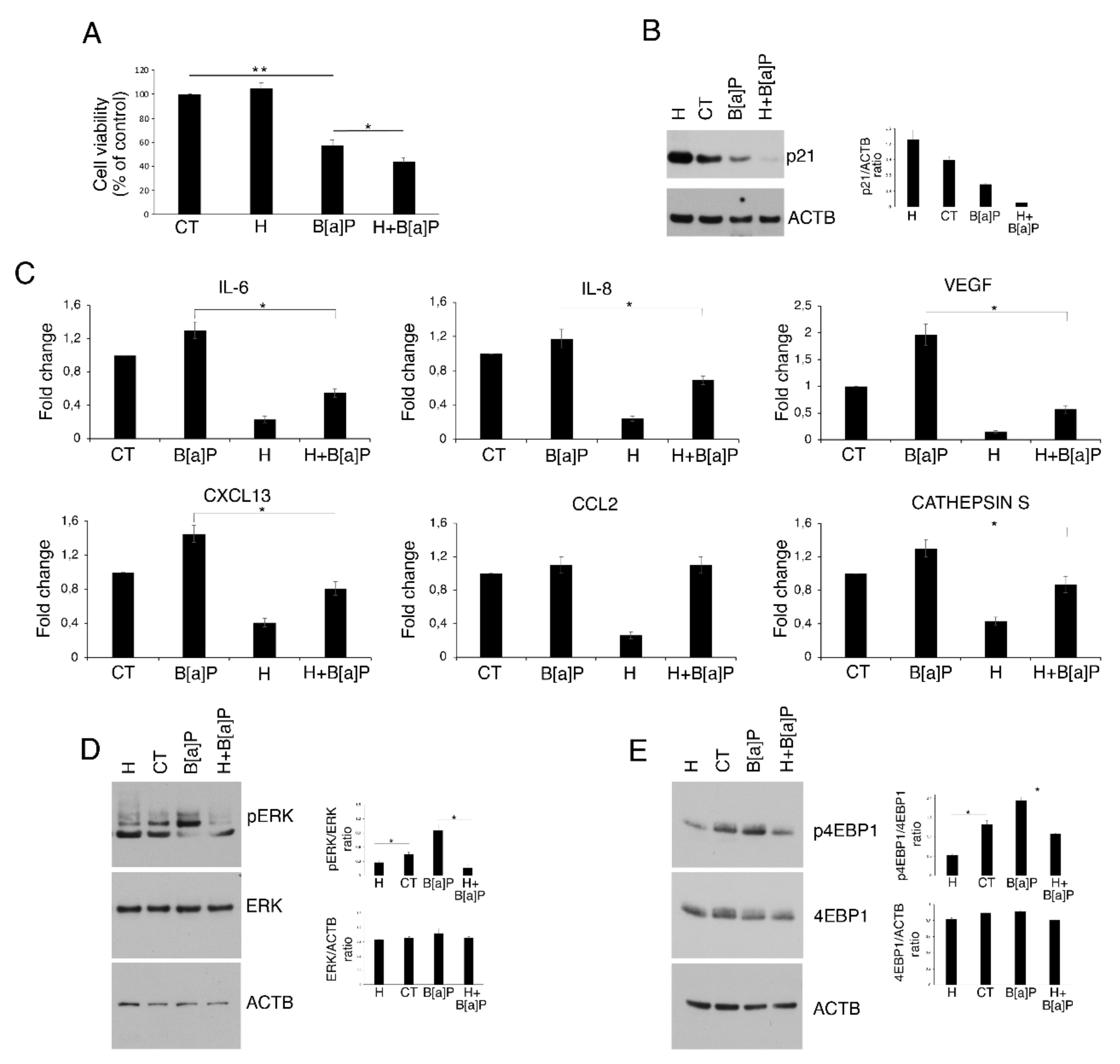
3.3. Hydroxytyrosol Slightly Increases Cell Death and Counteracts Cytokine and Chemokine Release as Well as ERK1/2 and mTOR Activation in B[a]P-treated HCoEpC
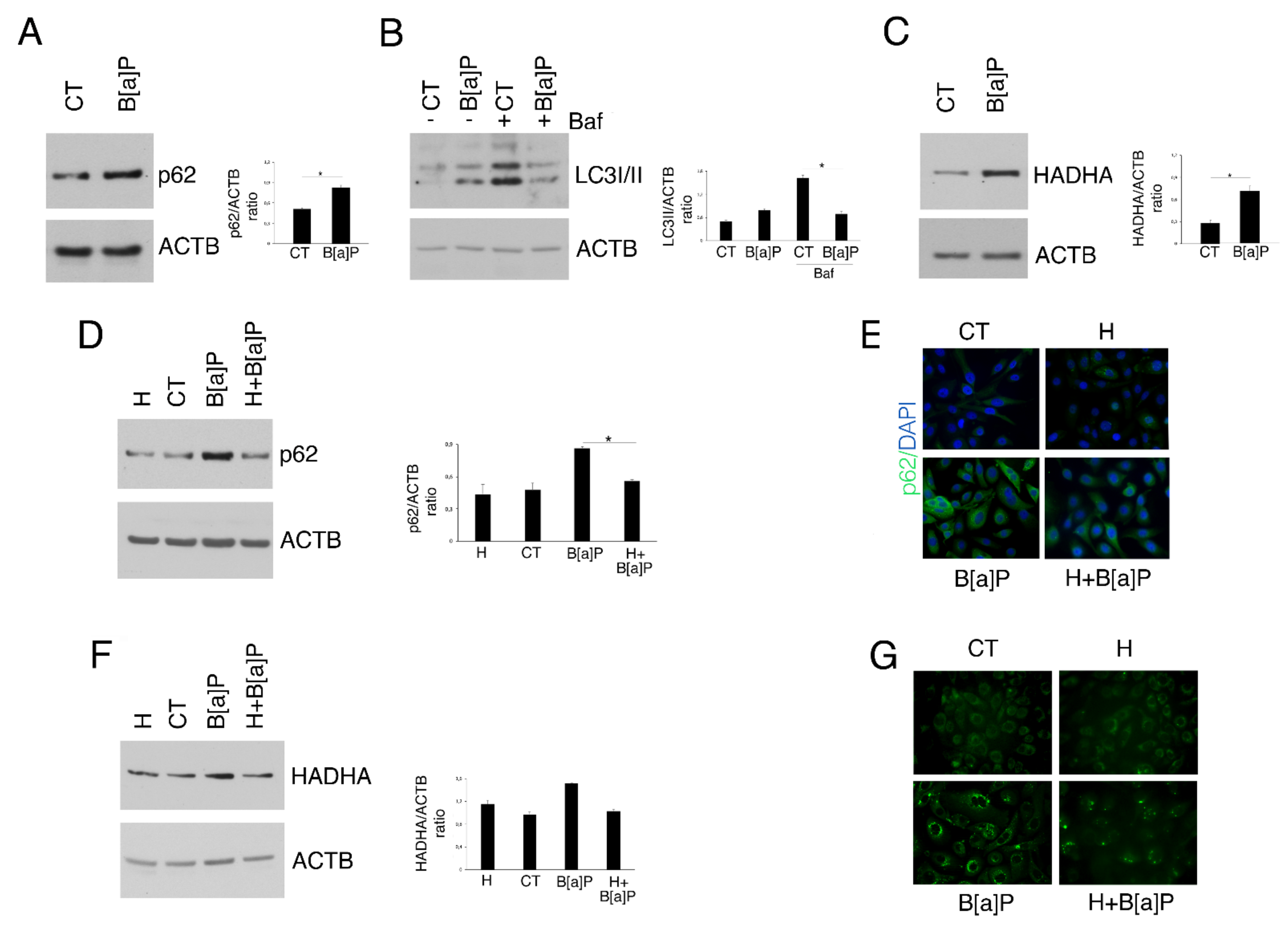
3.4. Hydroxytyrosol Restores B[a]P-mediated Reduction in Autophagy and Mitophagy in HCoEpC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Korniluk, A.; Koper, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Ir. J. Med. Sci. 2017, 186, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stidham, R.W.; Higgins, P.D.R. Colorectal Cancer in Inflammatory Bowel Disease. Clin. Colon. Rectal. Surg. 2018, 31, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Zhunussova, G.; Afonin, G.; Abdikerim, S.; Jumanov, A.; Perfilyeva, A.; Kaidarova, D.; Djansugurova, L. Mutation Spectrum of Cancer-Associated Genes in Patients with Early Onset of Colorectal Cancer. Front. Oncol. 2019, 9, 673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattray, N.J.W.; Charkoftaki, G.; Rattray, Z.; Hansen, J.E.; Vasiliou, V.; Johnson, C.H. Environmental influences in the etiology of colorectal cancer: The premise of metabolomics. Curr. Pharmacol. Rep. 2017, 3, 114–125. [Google Scholar] [CrossRef] [PubMed]
- IARC. IARC Monographs on the Identification of Carcinogenic Hazards to Humans; IARC: Lyon, France, 2021. [Google Scholar]
- Vazquez-Gomez, G.; Rocha-Zavaleta, L.; Rodriguez-Sosa, M.; Petrosyan, P.; Rubio-Lightbourn, J. Benzo[a]pyrene activates an AhR/Src/ERK axis that contributes to CYP1A1 induction and stable DNA adducts formation in lung cells. Toxicol. Lett. 2018, 289, 54–62. [Google Scholar] [CrossRef]
- Sinha, R.; Kulldorff, M.; Gunter, M.J.; Strickland, P.; Rothman, N. Dietary benzo[a]pyrene intake and risk of colorectal adenoma. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 2030–2034. [Google Scholar] [CrossRef] [Green Version]
- John, K.; Pratt, M.M.; Beland, F.A.; Churchwell, M.I.; McMullen, G.; Olivero, O.A.; Pogribny, I.P.; Poirier, M.C. Benzo[a]pyrene (BP) DNA adduct formation in DNA repair-deficient p53 haploinsufficient [Xpa(−/−)p53(+/−)] and wild-type mice fed BP and BP plus chlorophyllin for 28 days. Carcinogenesis 2012, 33, 2236–2241. [Google Scholar] [CrossRef]
- Shi, Q.; Boots, A.W.; Maas, L.; Veith, C.; van Kuijk, K.; Haenen, G.R.; Godschalk, R.W.; Van Schooten, F.J. Effect of interleukin (IL)-8 on benzo[a]pyrene metabolism and DNA damage in human lung epithelial cells. Toxicology 2017, 381, 64–74. [Google Scholar] [CrossRef]
- Khalil, A.; Villard, P.H.; Dao, M.A.; Burcelin, R.; Champion, S.; Fouchier, F.; Savouret, J.F.; Barra, Y.; Seree, E. Polycyclic aromatic hydrocarbons potentiate high-fat diet effects on intestinal inflammation. Toxicol. Lett. 2010, 196, 161–167. [Google Scholar] [CrossRef]
- Alzohairy, M.A.; Khan, A.A.; Alsahli, M.A.; Almatroodi, S.A.; Rahmani, A.H. Protective Effects of Thymoquinone, an Active Compound of Nigella sativa, on Rats with Benzo(a)pyrene-Induced Lung Injury through Regulation of Oxidative Stress and Inflammation. Molecules 2021, 26, 3218. [Google Scholar] [CrossRef]
- Banks, L.D.; Hamoah, P.; Niaz, M.S.; Washington, M.K.; Anduyah, S.E.; Ramesh, A. Olive oil prevents benzo(a)pyrene [B(a)P]-induced colon carcinogenesis through altered [B(a)P] metabolism and decreased oxidative damage in ApcMin mouse model. J. Nutr. Biochem. 2016, 28, 37–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Sun, Q.; Zhou, L.; He, M.; Dong, X.; Lai, M.; Liu, M.; Su, Y.; Jia, C.; Han, Z.; et al. Colonic epithelial mTORC1 promotes ulcerative colitis through COX-2-mediated Th17 responses. Mucosal. Immunol. 2018, 11, 1663–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broom, O.J.; Widjaya, B.; Troelsen, J.; Olsen, J.; Nielsen, O.H. Mitogen activated protein kinases: A role in inflammatory bowel disease? Clin. Exp. Immunol. 2009, 158, 272–280. [Google Scholar] [CrossRef]
- Stefani, C.; Miricescu, D.; Stanescu, S., II; Nica, R.I.; Greabu, M.; Totan, A.R.; Jinga, M. Growth Factors, PI3K/AKT/mTOR and MAPK Signaling Pathways in Colorectal Cancer Pathogenesis: Where Are We Now? Int. J. Mol. Sci 2021, 22, 10260. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.R.; Chang, D.K. Colorectal cancer in inflammatory bowel disease: The risk, pathogenesis, prevention and diagnosis. World J. Gastroenterol. 2014, 20, 9872–9881. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Fang, X.; Wang, X. Autophagy and inflammation. Clin. Transl. Med. 2017, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Adil, M.A.A.; Ameenudeen, S.; Kumar, A.; Hemalatha, S.; Ahmed, N.; Ali, N.; AlAsmari, A.F.; Aashique, M.; Waseem, M. Emerging Role of Mitophagy in Inflammatory Diseases: Cellular and Molecular Episodes. Curr. Pharm. Des. 2020, 26, 485–491. [Google Scholar] [CrossRef]
- Cirone, M. Cancer cells dysregulate PI3K/AKT/mTOR pathway activation to ensure their survival and proliferation: Mimicking them is a smart strategy of gammaherpesviruses. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 500–509. [Google Scholar] [CrossRef]
- Santarelli, R.; Pompili, C.; Gilardini Montani, M.S.; Romeo, M.A.; Gonnella, R.; D’Orazi, G.; Cirone, M. Lovastatin reduces PEL cell survival by phosphorylating ERK1/2 that blocks the autophagic flux and engages a cross-talk with p53 to activate p21. IUBMB Life 2021, 73, 968–977. [Google Scholar] [CrossRef]
- Shinojima, N.; Yokoyama, T.; Kondo, Y.; Kondo, S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy 2007, 3, 635–637. [Google Scholar] [CrossRef] [Green Version]
- Burada, F.; Nicoli, E.R.; Ciurea, M.E.; Uscatu, D.C.; Ioana, M.; Gheonea, D.I. Autophagy in colorectal cancer: An important switch from physiology to pathology. World J. Gastrointest. Oncol. 2015, 7, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Gilardini Montani, M.S.; Benedetti, R.; Piconese, S.; Pulcinelli, F.M.; Timperio, A.M.; Romeo, M.A.; Masuelli, L.; Mattei, M.; Bei, R.; DOrazi, G.; et al. PGE2 Released by Pancreatic Cancer Cells Undergoing ER Stress Transfers the Stress to DCs Impairing Their Immune Function. Mol. Cancer Ther. 2021, 20, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.N.; Harris, K.L.; Rekhadevi, P.V.; Pratap, S.; Ramesh, A. Benzo(a)pyrene-induced cytotoxicity, cell proliferation, DNA damage, and altered gene expression profiles in HT-29 human colon cancer cells. Cell Biol. Toxicol. 2021, 37, 891–913. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Liu, J.; Deng, H.; Gao, C. Benzo[a]pyrene Induces Autophagic and Pyroptotic Death Simultaneously in HL-7702 Human Normal Liver Cells. J. Agric. Food Chem. 2017, 65, 9763–9773. [Google Scholar] [CrossRef] [PubMed]
- Ajayi, B.O.; Adedara, I.A.; Farombi, E.O. Benzo(a)pyrene induces oxidative stress, pro-inflammatory cytokines, expression of nuclear factor-kappa B and deregulation of wnt/beta-catenin signaling in colons of BALB/c mice. Food Chem. Toxicol. 2016, 95, 42–51. [Google Scholar] [CrossRef]
- Khaket, T.P.; Kwon, T.K.; Kang, S.C. Cathepsins: Potent regulators in carcinogenesis. Pharmacol. Ther. 2019, 198, 1–19. [Google Scholar] [CrossRef]
- Burden, R.E.; Gormley, J.A.; Jaquin, T.J.; Small, D.M.; Quinn, D.J.; Hegarty, S.M.; Ward, C.; Walker, B.; Johnston, J.A.; Olwill, S.A.; et al. Antibody-mediated inhibition of cathepsin S blocks colorectal tumor invasion and angiogenesis. Clin. Cancer Res. 2009, 15, 6042–6051. [Google Scholar] [CrossRef] [Green Version]
- Parker, D. CD80/CD86 signaling contributes to the proinflammatory response of Staphylococcus aureus in the airway. Cytokine 2018, 107, 130–136. [Google Scholar] [CrossRef]
- Benedetti, R.; Gilardini Montani, M.S.; Romeo, M.A.; Arena, A.; Santarelli, R.; D’Orazi, G.; Cirone, M. Role of UPR Sensor Activation in Cell Death-Survival Decision of Colon Cancer Cells Stressed by DPE Treatment. Biomedicines 2021, 9, 1262. [Google Scholar] [CrossRef]
- Guichard, C.; Pedruzzi, E.; Fay, M.; Marie, J.C.; Braut-Boucher, F.; Daniel, F.; Grodet, A.; Gougerot-Pocidalo, M.A.; Chastre, E.; Kotelevets, L.; et al. Dihydroxyphenylethanol induces apoptosis by activating serine/threonine protein phosphatase PP2A and promotes the endoplasmic reticulum stress response in human colon carcinoma cells. Carcinogenesis 2006, 27, 1812–1827. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Netea-Maier, R.T.; Plantinga, T.S.; van de Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, M.G.; Aguilar, A.G.; Burillo, J.; Oca, R.G.; Manca, M.A.; Novials, A.; Alcarraz-Vizan, G.; Guillen, C.; Benito, M. Pancreatic beta cells overexpressing hIAPP impaired mitophagy and unbalanced mitochondrial dynamics. Cell Death Dis. 2018, 9, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Thanikachalam, K.; Khan, G. Colorectal Cancer and Nutrition. Nutrients 2019, 11, 164. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Chan, A.T.; Sun, J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology 2020, 158, 322–340. [Google Scholar] [CrossRef]
- Lee, B.M.; Shim, G.A. Dietary exposure estimation of benzo[a]pyrene and cancer risk assessment. J. Toxicol. Environ. Health A 2007, 70, 1391–1394. [Google Scholar] [CrossRef]
- Harris, K.L.; Pulliam, S.R.; Okoro, E.; Guo, Z.; Washington, M.K.; Adunyah, S.E.; Amos-Landgraf, J.M.; Ramesh, A. Western diet enhances benzo(a)pyrene-induced colon tumorigenesis in a polyposis in rat coli (PIRC) rat model of colon cancer. Oncotarget 2016, 7, 28947–28960. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, Y.; Pei, X.H.; Takayama, K.; Bai, F.; Izumi, M.; Kimotsuki, K.; Inoue, K.; Minami, T.; Wataya, H.; Hara, N. Polycyclic aromatic hydrocarbon carcinogens increase ubiquitination of p21 protein after the stabilization of p53 and the expression of p21. Am. J. Respir. Cell Mol. Biol. 2000, 22, 747–754. [Google Scholar] [CrossRef]
- Schmitt, M.; Greten, F.R. The inflammatory pathogenesis of colorectal cancer. Nat. Rev. Immunol. 2021, 21, 653–667. [Google Scholar] [CrossRef]
- Day, S.D.; Enos, R.T.; McClellan, J.L.; Steiner, J.L.; Velazquez, K.T.; Murphy, E.A. Linking inflammation to tumorigenesis in a mouse model of high-fat-diet-enhanced colon cancer. Cytokine 2013, 64, 454–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Cirone, M.; Gilardini Montani, M.S.; Granato, M.; Garufi, A.; Faggioni, A.; D’Orazi, G. Autophagy manipulation as a strategy for efficient anticancer therapies: Possible consequences. J. Exp. Clin. Cancer Res. 2019, 38, 262. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Salesse, L.; Hoang, M.H.T.; Bonnet, M.; Sauvanet, P.; Larabi, A.; Godfraind, C.; Gagniere, J.; Pezet, D.; Rosenstiel, P.; et al. Autophagy of Intestinal Epithelial Cells Inhibits Colorectal Carcinogenesis Induced by Colibactin-Producing Escherichia coli in Apc(Min/+) Mice. Gastroenterology 2020, 158, 1373–1388. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Debnath, J. Autophagy and tumorigenesis. FEBS Lett. 2010, 584, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Debnath, J. The Evolving, Multifaceted Roles of Autophagy in Cancer. Adv. Cancer Res. 2016, 130, 1–53. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Wang, Y.; Li, B.; Shen, K.; Li, Q.; Ni, Y.; Huang, L. Mitophagy in carcinogenesis, drug resistance and anticancer therapeutics. Cancer Cell Int. 2021, 21, 350. [Google Scholar] [CrossRef]
- Mantovani, A. The inflammation—Cancer connection. FEBS J. 2018, 285, 638–640. [Google Scholar] [CrossRef] [Green Version]
- Van ’t Wout, E.F.; Dickens, J.A.; van Schadewijk, A.; Haq, I.; Kwok, H.F.; Ordonez, A.; Murphy, G.; Stolk, J.; Lomas, D.A.; Hiemstra, P.S.; et al. Increased ERK signalling promotes inflammatory signalling in primary airway epithelial cells expressing Z alpha1-antitrypsin. Hum. Mol. Genet. 2014, 23, 929–941. [Google Scholar] [CrossRef] [Green Version]
- Lu, N.; Malemud, C.J. Extracellular Signal-Regulated Kinase: A Regulator of Cell Growth, Inflammation, Chondrocyte and Bone Cell Receptor-Mediated Gene Expression. Int. J. Mol. Sci. 2019, 20, 3792. [Google Scholar] [CrossRef] [Green Version]
- Sedda, S.; Dinallo, V.; Marafini, I.; Franze, E.; Paoluzi, O.A.; Izzo, R.; Giuffrida, P.; Di Sabatino, A.; Corazza, G.R.; Monteleone, G. mTOR sustains inflammatory response in celiac disease. Sci. Rep. 2020, 10, 10798. [Google Scholar] [CrossRef] [PubMed]
- Lopez de Las Hazas, M.C.; Pinol, C.; Macia, A.; Motilva, M.J. Hydroxytyrosol and the Colonic Metabolites Derived from Virgin Olive Oil Intake Induce Cell Cycle Arrest and Apoptosis in Colon Cancer Cells. J. Agric. Food Chem. 2017, 65, 6467–6476. [Google Scholar] [CrossRef] [PubMed]
- Storniolo, C.E.; Martinez-Hovelman, N.; Martinez-Huelamo, M.; Lamuela-Raventos, R.M.; Moreno, J.J. Extra Virgin Olive Oil Minor Compounds Modulate Mitogenic Action of Oleic Acid on Colon Cancer Cell Line. J. Agric. Food Chem. 2019, 67, 11420–11427. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santarelli, R.; Pompili, C.; Gilardini Montani, M.S.; Evangelista, L.; Gonnella, R.; Cirone, M. 3,4-Dihydroxyphenylethanol (DPE or Hydroxytyrosol) Counteracts ERK1/2 and mTOR Activation, Pro-Inflammatory Cytokine Release, Autophagy and Mitophagy Reduction Mediated by Benzo[a]pyrene in Primary Human Colonic Epithelial Cells. Pharmaceutics 2022, 14, 663. https://doi.org/10.3390/pharmaceutics14030663
Santarelli R, Pompili C, Gilardini Montani MS, Evangelista L, Gonnella R, Cirone M. 3,4-Dihydroxyphenylethanol (DPE or Hydroxytyrosol) Counteracts ERK1/2 and mTOR Activation, Pro-Inflammatory Cytokine Release, Autophagy and Mitophagy Reduction Mediated by Benzo[a]pyrene in Primary Human Colonic Epithelial Cells. Pharmaceutics. 2022; 14(3):663. https://doi.org/10.3390/pharmaceutics14030663
Chicago/Turabian StyleSantarelli, Roberta, Chiara Pompili, Maria Saveria Gilardini Montani, Lorenzo Evangelista, Roberta Gonnella, and Mara Cirone. 2022. "3,4-Dihydroxyphenylethanol (DPE or Hydroxytyrosol) Counteracts ERK1/2 and mTOR Activation, Pro-Inflammatory Cytokine Release, Autophagy and Mitophagy Reduction Mediated by Benzo[a]pyrene in Primary Human Colonic Epithelial Cells" Pharmaceutics 14, no. 3: 663. https://doi.org/10.3390/pharmaceutics14030663