
Long-Acting Thioredoxin Ameliorates Doxorubicin-Induced Cardiomyopathy via Its Anti-Oxidative and Anti-Inflammatory Action

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation and Purification of HSA-Trx
2.3. Mice Model of Dox-Induced Heart Failure
2.4. Measurement of Myocardial Damage
2.5. Histological Analysis
2.6. qRT-PCR
2.7. Western Blotting
2.8. Isolation and Cell Culture of Neonatal Rat Cardiomyocytes (NRCMs)
2.9. Evaluation of Cell Size in NRCMs
2.10. Measurement of Intracellular ROS Production
2.11. Statistical Analysis
3. Results
3.1. Effect of HSA-Trx on Myocardial Damage and Fibrosis in the Dox-Induced Cardiomyopathy Model
3.2. Anti-Oxidative and Anti-Inflammatory Effect of HSA-Trx on the Dox-Induced Cardiomyopathy Model
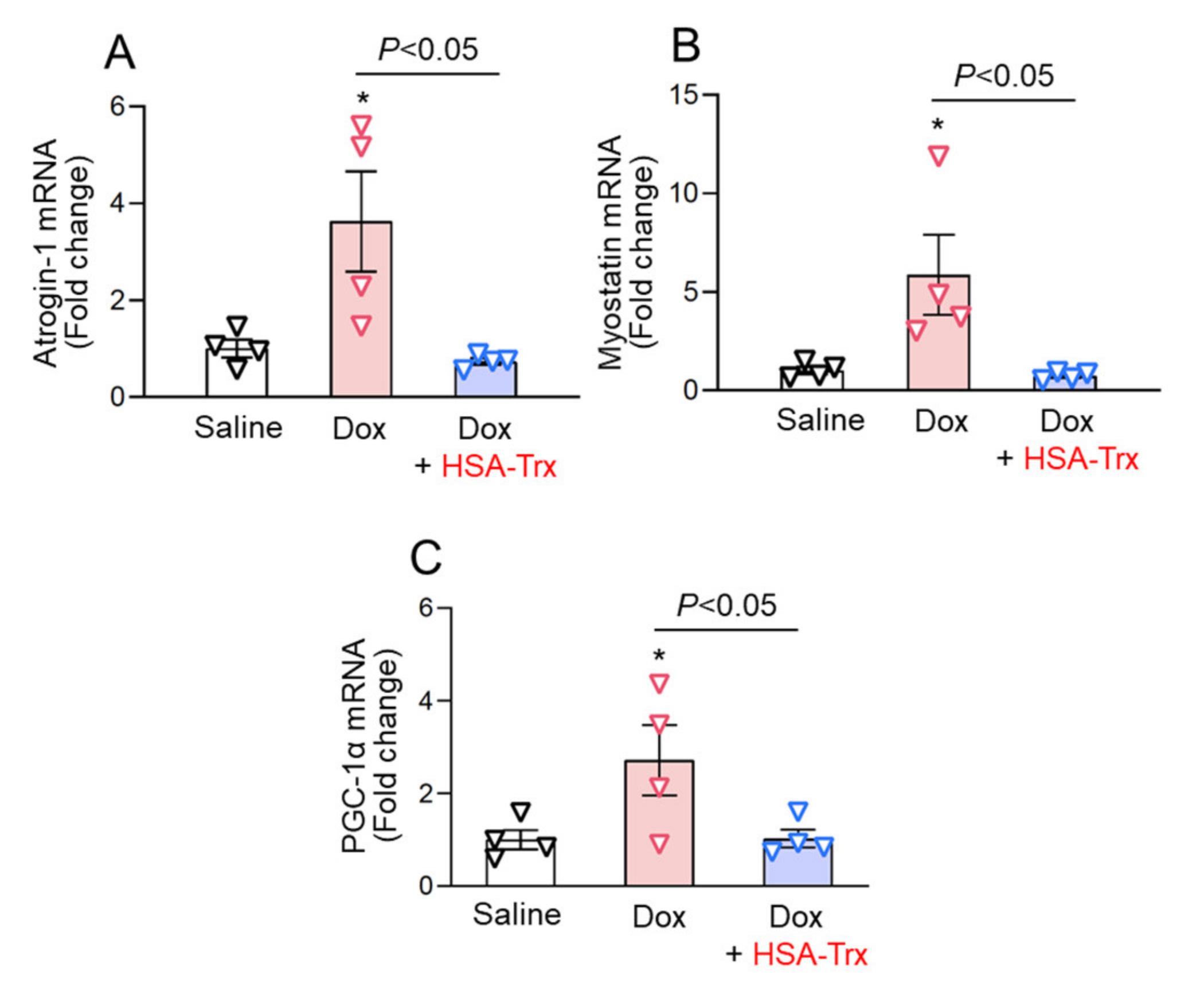
3.3. Anti-Myocardial Atrophy Effect of HSA-Trx on the Dox-Induced Cardiomyopathy Model
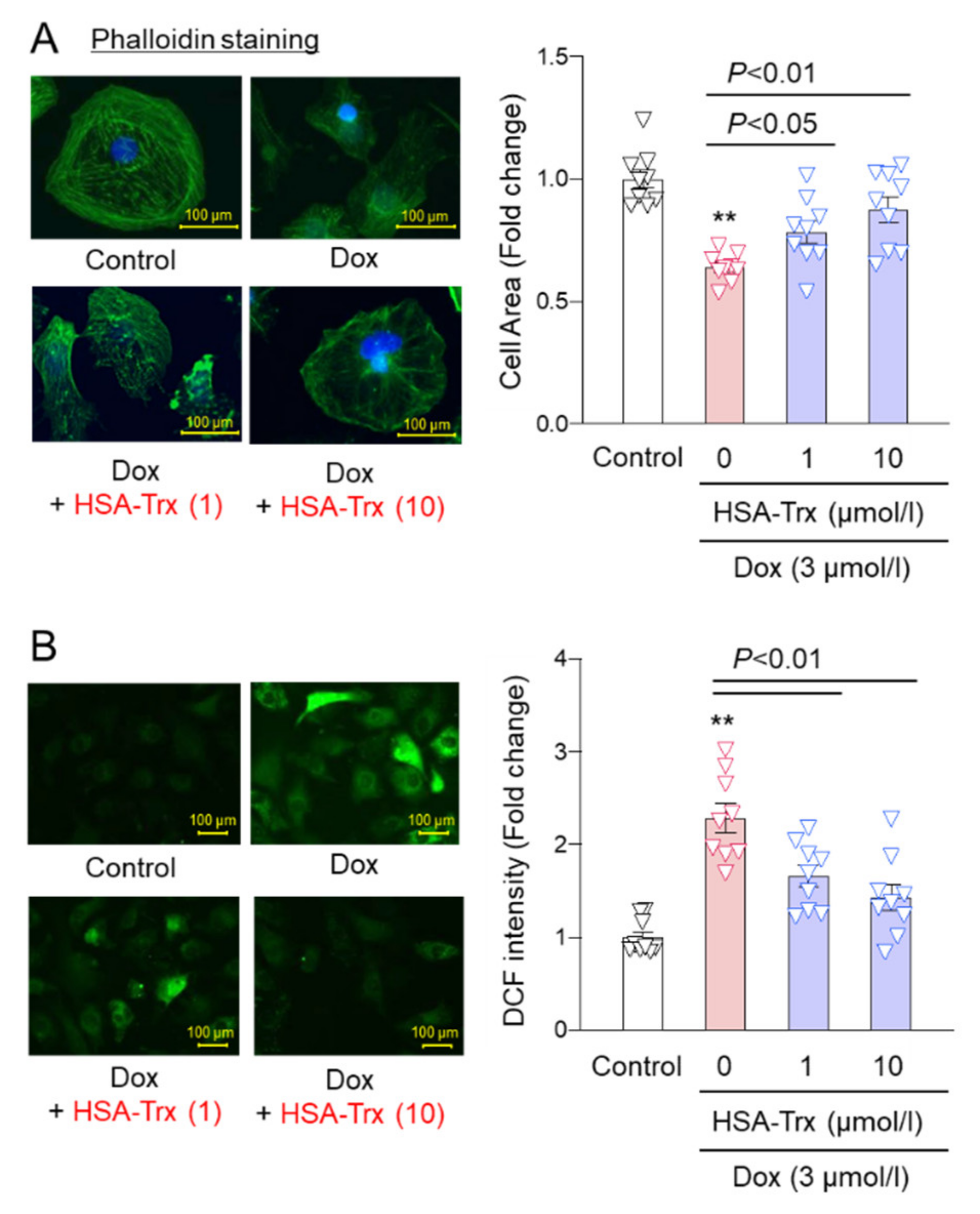
3.4. Effect of HSA-Trx on Dox-Induced Cardiomyocyte Atrophy and ROS Production
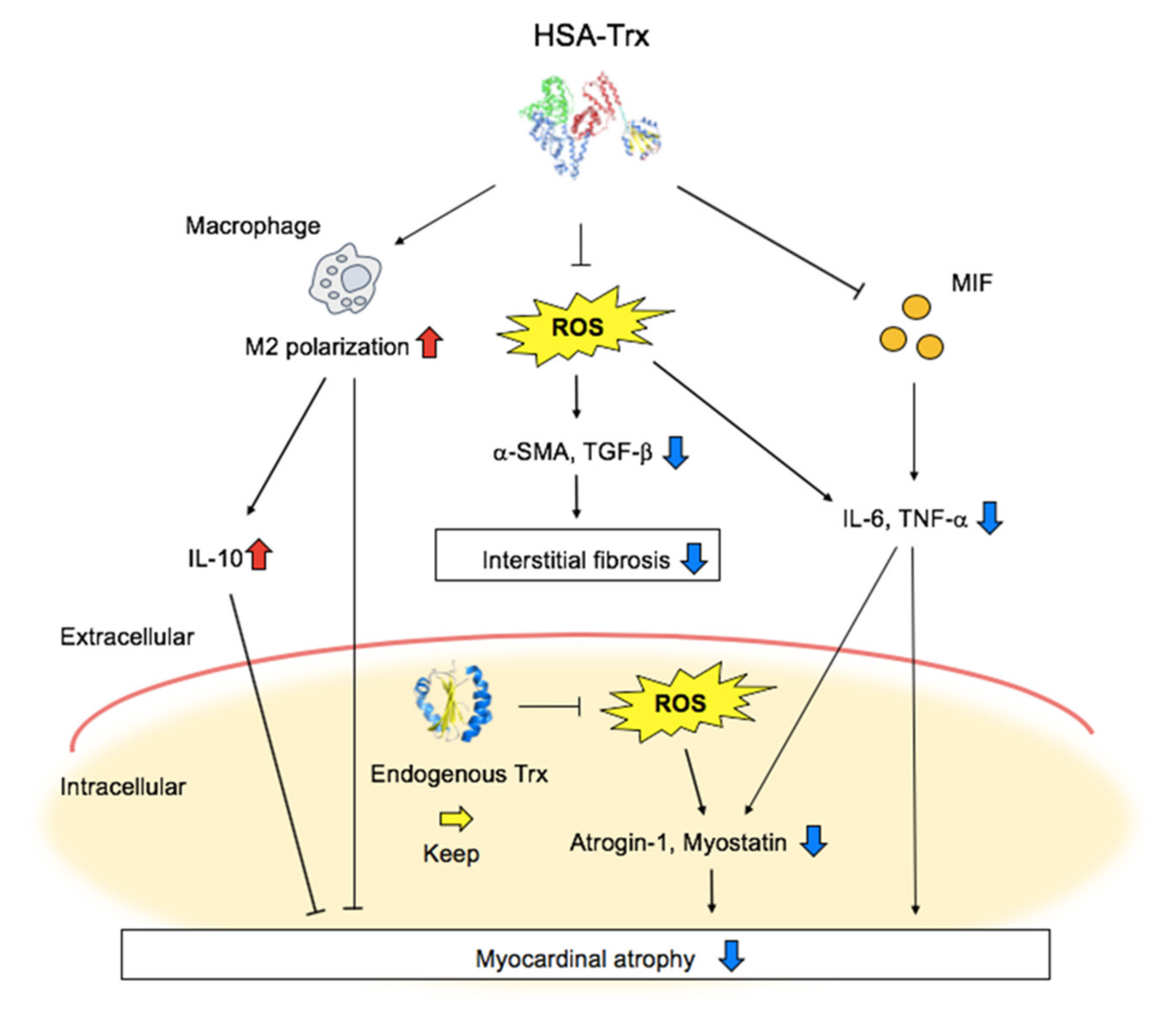
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kurian, G.A.; Rajagopal, R.; Vedantham, S.; Rajesh, M. The Role of Oxidative Stress in Myocardial Ischemia and Reperfusion Injury and Remodeling: Revisited. Oxid. Med. Cell Longev. 2016, 2016, 1656450. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Hoshino, Y.; Ito, T.; Nariai, T.; Mohri, T.; Obana, M.; Hayata, N.; Uozumi, Y.; Maeda, M.; Fujio, Y.; et al. Atrogin-1 ubiquitin ligase is upregulated by doxorubicin via p38-MAP kinase in cardiac myocytes. Cardiovasc. Res. 2008, 79, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, J.A.; Olson, E.N. Cardiac plasticity. N. Engl. J. Med. 2008, 358, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.K.; Tyagi, N.; Sen, U.; Joshua, I.G.; Tyagi, S.C. Synergism in hyperhomocysteinemia and diabetes: Role of PPAR gamma and tempol. Cardiovasc. Diabetol. 2010, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neidhardt, S.; Garbade, J.; Emrich, F.; Klaeske, K.; Borger, M.A.; Lehmann, S.; Jawad, K.; Dieterlen, M.T. Ischemic Cardiomyopathy Affects the Thioredoxin System in the Human Myocardium. J. Card. Fail. 2019, 25, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Huang, Y.; Que, B.; Chang, C.; Liu, W.; Hu, H.; Liu, L.; Shi, Y.; Wang, Y.; Wang, M.; et al. Interleukin-12p35 Knock Out Aggravates Doxorubicin-Induced Cardiac Injury and Dysfunction by Aggravating the Inflammatory Response, Oxidative Stress, Apoptosis and Autophagy in Mice. EBioMedicine 2018, 35, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Octavia, Y.; Kararigas, G.; de Boer, M.; Chrifi, I.; Kietadisorn, R.; Swinnen, M.; Duimel, H.; Verheyen, F.K.; Brandt, M.M.; Fliegner, D.; et al. Folic acid reduces doxorubicin-induced cardiomyopathy by modulating endothelial nitric oxide synthase. J. Cell Mol. Med. 2017, 21, 3277–3287. [Google Scholar] [CrossRef] [Green Version]
- Dong, Q.; Chen, L.; Lu, Q.; Sharma, S.; Li, L.; Morimoto, S.; Wang, G. Quercetin attenuates doxorubicin cardiotoxicity by modulating Bmi-1 expression. Br. J. Pharmacol. 2014, 171, 4440–4454. [Google Scholar] [CrossRef] [Green Version]
- Shimauchi, T.; Numaga-Tomita, T.; Ito, T.; Nishimura, A.; Matsukane, R.; Oda, S.; Hoka, S.; Ide, T.; Koitabashi, N.; Uchida, K.; et al. TRPC3-Nox2 complex mediates doxorubicin-induced myocardial atrophy. JCI Insight 2017, 2, e93358. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.M.; Liu, Y.; White, D.; Su, Y.; Drew, B.G.; Bruce, C.R.; Kiriazis, H.; Xu, Q.; Jennings, N.; Bobik, A.; et al. Deletion of macrophage migration inhibitory factor protects the heart from severe ischemia-reperfusion injury: A predominant role of anti-inflammation. J. Mol. Cell Cardiol. 2011, 50, 991–999. [Google Scholar] [CrossRef]
- White, D.A.; Fang, L.; Chan, W.; Morand, E.F.; Kiriazis, H.; Duffy, S.J.; Taylor, A.J.; Dart, A.M.; Du, X.J.; Gao, X.M. Pro-inflammatory action of MIF in acute myocardial infarction via activation of peripheral blood mononuclear cells. PLoS ONE 2013, 8, e76206. [Google Scholar] [CrossRef]
- Spyrou, G.; Enmark, E.; Miranda-Vizuete, A.; Gustafsson, J. Cloning and expression of a novel mammalian thioredoxin. J. Biol. Chem. 1997, 272, 2936–2941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.A.; Kirnarsky, L.; Sherman, S.; Gladyshev, V.N. Selenoprotein oxidoreductase with specificity for thioredoxin and glutathione systems. Proc. Natl. Acad. Sci. USA 2001, 98, 3673–3678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, K.C.; Das, C.K. Thioredoxin, a singlet oxygen quencher and hydroxyl radical scavenger: Redox independent functions. Biochem. Biophys. Res. Commun. 2000, 277, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Kasuno, K.; Shirakawa, K.; Yoshida, H.; Mori, K.; Kimura, H.; Takahashi, N.; Nobukawa, Y.; Shigemi, K.; Tanabe, S.; Yamada, N.; et al. Renal redox dysregulation in AKI: Application for oxidative stress marker of AKI. Am. J. Physiol. Renal. Physiol. 2014, 307, F1342–F1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, H.; Herzenberg, L.A.; Bai, J.; Araya, S.; Kondo, N.; Nishinaka, Y.; Yodoi, J. Circulating thioredoxin suppresses lipopolysaccharide-induced neutrophil chemotaxis. Proc. Natl. Acad. Sci. USA 2001, 98, 15143–15148. [Google Scholar] [CrossRef] [Green Version]
- Kondo, N.; Ishii, Y.; Son, A.; Sakakura-Nishiyama, J.; Kwon, Y.W.; Tanito, M.; Nishinaka, Y.; Matsuo, Y.; Nakayama, T.; Taniguchi, M.; et al. Cysteine-dependent immune regulation by TRX and MIF/GIF family proteins. Immunol. Lett. 2004, 92, 143–147. [Google Scholar] [CrossRef]
- Nakamura, T.; Hoshino, Y.; Yamada, A.; Teratani, A.; Furukawa, S.; Okuyama, H.; Ueda, S.; Wada, H.; Yodoi, J.; Nakamura, H. Recombinant human thioredoxin-1 becomes oxidized in circulation and suppresses bleomycin-induced neutrophil recruitment in the rat airway. Free Radic. Res. 2007, 41, 1089–1098. [Google Scholar] [CrossRef]
- Hara, T.; Kondo, N.; Nakamura, H.; Okuyama, H.; Mitsui, A.; Hoshino, Y.; Yodoi, J. Cell-surface thioredoxin-1: Possible involvement in thiol-mediated leukocyte-endothelial cell interaction through lipid rafts. Antioxid. Redox. Signal 2007, 9, 1427–1437. [Google Scholar] [CrossRef] [Green Version]
- Tamaki, H.; Nakamura, H.; Nishio, A.; Nakase, H.; Ueno, S.; Uza, N.; Kido, M.; Inoue, S.; Mikami, S.; Asada, M.; et al. Human thioredoxin-1 ameliorates experimental murine colitis in association with suppressed macrophage inhibitory factor production. Gastroenterology 2006, 131, 1110–1121. [Google Scholar] [CrossRef] [Green Version]
- El Hadri, K.; Mahmood, D.F.; Couchie, D.; Jguirim-Souissi, I.; Genze, F.; Diderot, V.; Syrovets, T.; Lunov, O.; Simmet, T.; Rouis, M. Thioredoxin-1 promotes anti-inflammatory macrophages of the M2 phenotype and antagonizes atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1445–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shioji, K.; Kishimoto, C.; Nakamura, H.; Masutani, H.; Yuan, Z.; Oka, S.; Yodoi, J. Overexpression of thioredoxin-1 in transgenic mice attenuates adriamycin-induced cardiotoxicity. Circulation 2002, 106, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Adluri, R.S.; Thirunavukkarasu, M.; Zhan, L.; Akita, Y.; Samuel, S.M.; Otani, H.; Ho, Y.S.; Maulik, G.; Maulik, N. Thioredoxin 1 enhances neovascularization and reduces ventricular remodeling during chronic myocardial infarction: A study using thioredoxin 1 transgenic mice. J. Mol. Cell Cardiol. 2011, 50, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Ago, T.; Zhai, P.; Abdellatif, M.; Sadoshima, J. Thioredoxin 1 negatively regulates angiotensin II-induced cardiac hypertrophy through upregulation of miR-98/let-7. Circ. Res. 2011, 108, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Ikuta, S.; Chuang, V.T.; Ishima, Y.; Nakajou, K.; Furukawa, M.; Watanabe, H.; Maruyama, T.; Otagiri, M. Albumin fusion of thioredoxin--the production and evaluation of its biological activity for potential therapeutic applications. J. Control. Release 2010, 147, 17–23. [Google Scholar] [CrossRef]
- Furukawa, M.; Tanaka, R.; Chuang, V.T.; Ishima, Y.; Taguchi, K.; Watanabe, H.; Maruyama, T.; Otagiri, M. Human serum albumin-thioredoxin fusion protein with long blood retention property is effective in suppressing lung injury. J. Control Release 2011, 154, 189–195. [Google Scholar] [CrossRef]
- Tanaka, R.; Watanabe, H.; Kodama, A.; Chuang, V.T.; Ishima, Y.; Hamasaki, K.; Tanaka, K.; Mizushima, T.; Otagiri, M.; Maruyama, T. Long-acting human serum albumin-thioredoxin fusion protein suppresses bleomycin-induced pulmonary fibrosis progression. J. Pharmacol. Exp. Ther. 2013, 345, 271–283. [Google Scholar] [CrossRef] [Green Version]
- Kodama, A.; Watanabe, H.; Tanaka, R.; Tanaka, H.; Chuang, V.T.; Miyamoto, Y.; Wu, Q.; Endo, M.; Hamasaki, K.; Ishima, Y.; et al. A human serum albumin-thioredoxin fusion protein prevents experimental contrast-induced nephropathy. Kidney Int. 2013, 83, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Kodama, A.; Watanabe, H.; Tanaka, R.; Kondo, M.; Chuang, V.T.; Wu, Q.; Endo, M.; Ishima, Y.; Fukagawa, M.; Otagiri, M.; et al. Albumin fusion renders thioredoxin an effective anti-oxidative and anti-inflammatory agent for preventing cisplatin-induced nephrotoxicity. Biochim. Biophys. Acta 2014, 1840, 1152–1162. [Google Scholar] [CrossRef]
- Tanaka, R.; Ishima, Y.; Maeda, H.; Kodama, A.; Nagao, S.; Watanabe, H.; Chuang, V.T.; Otagiri, M.; Maruyama, T. Albumin fusion prolongs the antioxidant and anti-inflammatory activities of thioredoxin in mice with acetaminophen-induced hepatitis. Mol. Pharm. 2014, 11, 1228–1238. [Google Scholar] [CrossRef]
- Tanaka, R.; Ishima, Y.; Enoki, Y.; Kimachi, K.; Shirai, T.; Watanabe, H.; Chuang, V.T.; Maruyama, T.; Otagiri, M. Therapeutic impact of human serum albumin-thioredoxin fusion protein on influenza virus-induced lung injury mice. Front. Immunol. 2014, 5, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, K.; Watanabe, H.; Ogaki, S.; Kodama, A.; Tanaka, R.; Imafuku, T.; Ishima, Y.; Chuang, V.T.; Toyoda, M.; Kondoh, M.; et al. Renoprotective effect of long acting thioredoxin by modulating oxidative stress and macrophage migration inhibitory factor against rhabdomyolysis-associated acute kidney injury. Sci. Rep. 2015, 5, 14471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.I.; Shimoda, M.; Chuang, V.T.G.; Nishida, K.; Kawahara, M.; Ishida, T.; Otagiri, M.; Maruyama, T.; Ishima, Y. Thioredoxin-albumin fusion protein prevents copper enhanced zinc-induced neurotoxicity via its antioxidative activity. Int. J. Pharm. 2018, 535, 140–147. [Google Scholar] [CrossRef]
- Herman, E.H.; Ferrans, V.J. Preclinical animal models of cardiac protection from anthracycline-induced cardiotoxicity. Semin. Oncol. 1998, 25, 15–21. [Google Scholar] [PubMed]
- Nishida, K.; Watanabe, H.; Murata, R.; Tokumaru, K.; Fujimura, R.; Oshiro, S.; Nagasaki, T.; Miyahisa, M.; Hiramoto, Y.; Nosaki, H.; et al. Recombinant Long-Acting Thioredoxin Ameliorates AKI to CKD Transition via Modulating Renal Oxidative Stress and Inflammation. Int. J. Mol. Sci. 2021, 22, 5600. [Google Scholar] [CrossRef]
- Tokarska-Schlattner, M.; Zaugg, M.; Zuppinger, C.; Wallimann, T.; Schlattner, U. New insights into doxorubicin-induced cardiotoxicity: The critical role of cellular energetics. J. Mol. Cell Cardiol. 2006, 41, 389–405. [Google Scholar] [CrossRef]
- Watanabe, H.; Bi, J.; Murata, R.; Fujimura, R.; Nishida, K.; Imafuku, T.; Nakamura, Y.; Maeda, H.; Mukunoki, A.; Takeo, T.; et al. A synthetic retinoic acid receptor agonist Am80 ameliorates renal fibrosis via inducing the production of alpha-1-acid glycoprotein. Sci. Rep. 2020, 10, 11424. [Google Scholar] [CrossRef]
- Sudi, S.B.; Tanaka, T.; Oda, S.; Nishiyama, K.; Nishimura, A.; Sunggip, C.; Mangmool, S.; Numaga-Tomita, T.; Nishida, M. TRPC3-Nox2 axis mediates nutritional deficiency-induced cardiomyocyte atrophy. Sci. Rep. 2019, 9, 9785. [Google Scholar] [CrossRef]
- Son, A.; Kato, N.; Horibe, T.; Matsuo, Y.; Mochizuki, M.; Mitsui, A.; Kawakami, K.; Nakamura, H.; Yodoi, J. Direct association of thioredoxin-1 (TRX) with macrophage migration inhibitory factor (MIF): Regulatory role of TRX on MIF internalization and signaling. Antioxid. Redox. Signal 2009, 11, 2595–2605. [Google Scholar] [CrossRef] [Green Version]
- Ago, T.; Yeh, I.; Yamamoto, M.; Schinke-Braun, M.; Brown, J.A.; Tian, B.; Sadoshima, J. Thioredoxin1 upregulates mitochondrial proteins related to oxidative phosphorylation and TCA cycle in the heart. Antioxid. Redox. Signal 2006, 8, 1635–1650. [Google Scholar] [CrossRef]
- Heineke, J.; Auger-Messier, M.; Xu, J.; Sargent, M.; York, A.; Welle, S.; Molkentin, J.D. Genetic deletion of myostatin from the heart prevents skeletal muscle atrophy in heart failure. Circulation 2010, 121, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Biesemann, N.; Mendler, L.; Kostin, S.; Wietelmann, A.; Borchardt, T.; Braun, T. Myostatin induces interstitial fibrosis in the heart via TAK1 and p38. Cell Tissue Res. 2015, 361, 779–787. [Google Scholar] [CrossRef]
- Hulmi, J.J.; Nissinen, T.A.; Räsänen, M.; Degerman, J.; Lautaoja, J.H.; Hemanthakumar, K.A.; Backman, J.T.; Ritvos, O.; Silvennoinen, M.; Kivelä, R. Prevention of chemotherapy-induced cachexia by ACVR2B ligand blocking has different effects on heart and skeletal muscle. J. Cachexia Sarcopenia Muscle 2018, 9, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Keizer, H.G.; Pinedo, H.M.; Schuurhuis, G.J.; Joenje, H. Doxorubicin (adriamycin): A critical review of free radical-dependent mechanisms of cytotoxicity. Pharmacol. Ther. 1990, 47, 219–231. [Google Scholar] [CrossRef]
- Nakamura, H.; Hoshino, Y.; Okuyama, H.; Matsuo, Y.; Yodoi, J. Thioredoxin 1 delivery as new therapeutics. Adv. Drug Deliv. Rev. 2009, 61, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Garner, L.B.; Willis, M.S.; Carlson, D.L.; DiMaio, J.M.; White, M.D.; White, D.J.; Adams, G.A.; Horton, J.W.; Giroir, B.P. Macrophage migration inhibitory factor is a cardiac-derived myocardial depressant factor. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2500–H2509. [Google Scholar] [CrossRef] [Green Version]
- Meijles, D.N.; Cull, J.J.; Markou, T.; Cooper, S.T.E.; Haines, Z.H.R.; Fuller, S.J.; O’Gara, P.; Sheppard, M.N.; Harding, S.E.; Sugden, P.H.; et al. Redox Regulation of Cardiac ASK1 (Apoptosis Signal-Regulating Kinase 1) Controls p38-MAPK (Mitogen-Activated Protein Kinase) and Orchestrates Cardiac Remodeling to Hypertension. Hypertension 2020, 76, 1208–1218. [Google Scholar] [CrossRef]
- Nagai, H.; Noguchi, T.; Takeda, K.; Ichijo, H. Pathophysiological roles of ASK1-MAP kinase signaling pathways. J. Biochem. Mol. Biol. 2007, 40, 1–6. [Google Scholar] [CrossRef]
- Yao, Y.; Chen, R.; Ying, C.; Zhang, G.; Rui, T.; Tao, A. Interleukin-33 attenuates doxorubicin-induced cardiomyocyte apoptosis through suppression of ASK1/JNK signaling pathway. Biochem. Biophys. Res. Commun. 2017, 493, 1288–1295. [Google Scholar] [CrossRef]
- Yamaguchi, O.; Higuchi, Y.; Hirotani, S.; Kashiwase, K.; Nakayama, H.; Hikoso, S.; Takeda, T.; Watanabe, T.; Asahi, M.; Taniike, M.; et al. Targeted deletion of apoptosis signal-regulating kinase 1 attenuates left ventricular remodeling. Proc. Natl. Acad. Sci. USA 2003, 100, 15883–15888. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Zhou, H.J.; Zhang, H.; Huang, Y.; Hinojosa-Kirschenbaum, F.; Fan, P.; Yao, L.; Belardinelli, L.; Tellides, G.; Giordano, F.J.; et al. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation 2015, 131, 1082–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murata, R.; Watanabe, H.; Nosaki, H.; Nishida, K.; Maeda, H.; Nishida, M.; Maruyama, T. Long-Acting Thioredoxin Ameliorates Doxorubicin-Induced Cardiomyopathy via Its Anti-Oxidative and Anti-Inflammatory Action. Pharmaceutics 2022, 14, 562. https://doi.org/10.3390/pharmaceutics14030562
Murata R, Watanabe H, Nosaki H, Nishida K, Maeda H, Nishida M, Maruyama T. Long-Acting Thioredoxin Ameliorates Doxorubicin-Induced Cardiomyopathy via Its Anti-Oxidative and Anti-Inflammatory Action. Pharmaceutics. 2022; 14(3):562. https://doi.org/10.3390/pharmaceutics14030562
Chicago/Turabian StyleMurata, Ryota, Hiroshi Watanabe, Hiroto Nosaki, Kento Nishida, Hitoshi Maeda, Motohiro Nishida, and Toru Maruyama. 2022. "Long-Acting Thioredoxin Ameliorates Doxorubicin-Induced Cardiomyopathy via Its Anti-Oxidative and Anti-Inflammatory Action" Pharmaceutics 14, no. 3: 562. https://doi.org/10.3390/pharmaceutics14030562