
Clathrate Hydrates of Organic Solvents as Auxiliary Intermediates in Pharmaceutical Research and Development: Improving Dissolution Behaviour of a New Anti-Tuberculosis Drug, Perchlozon

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials & Samples Preparation
2.2. Methods
2.2.1. Freeze-Drying
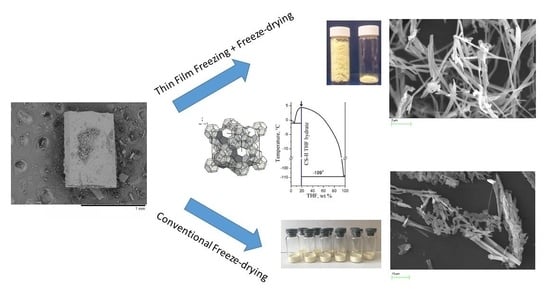
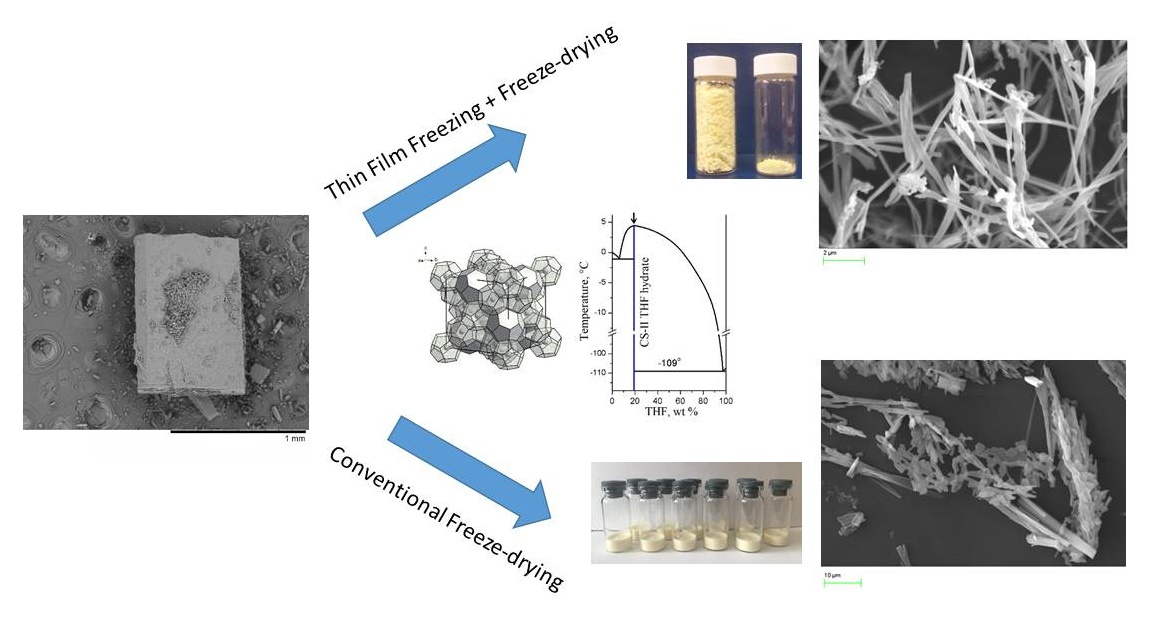
Ultrafine Pz Preparation Method (Thin Film Freezing (TFF) + Freeze-Drying (FD))
2.2.2. Pz Formulations Preparation Method
- (a)
- Preliminary experiments: 10 vials (1.00 mL aliquots; 5 mL vials, Sci/Spec, B69308);
- (b)
- Main experiments: 20 vials (2.00 mL aliquots; 15 mL injection vials made of colorless borosilicate glass tubing with 13 mm 2 leg freeze-drying stoppers (Jiangsu Runde Ltd., Jiangsu, China)) and frozen in air thermostat at −20 °C. Vials were placed on a shelf, which was pre-cooled to −10 °C, and freeze-dried at a chamber pressure of 100/400 mTorr.
2.2.3. Thermoanalytical (TA) Experiments
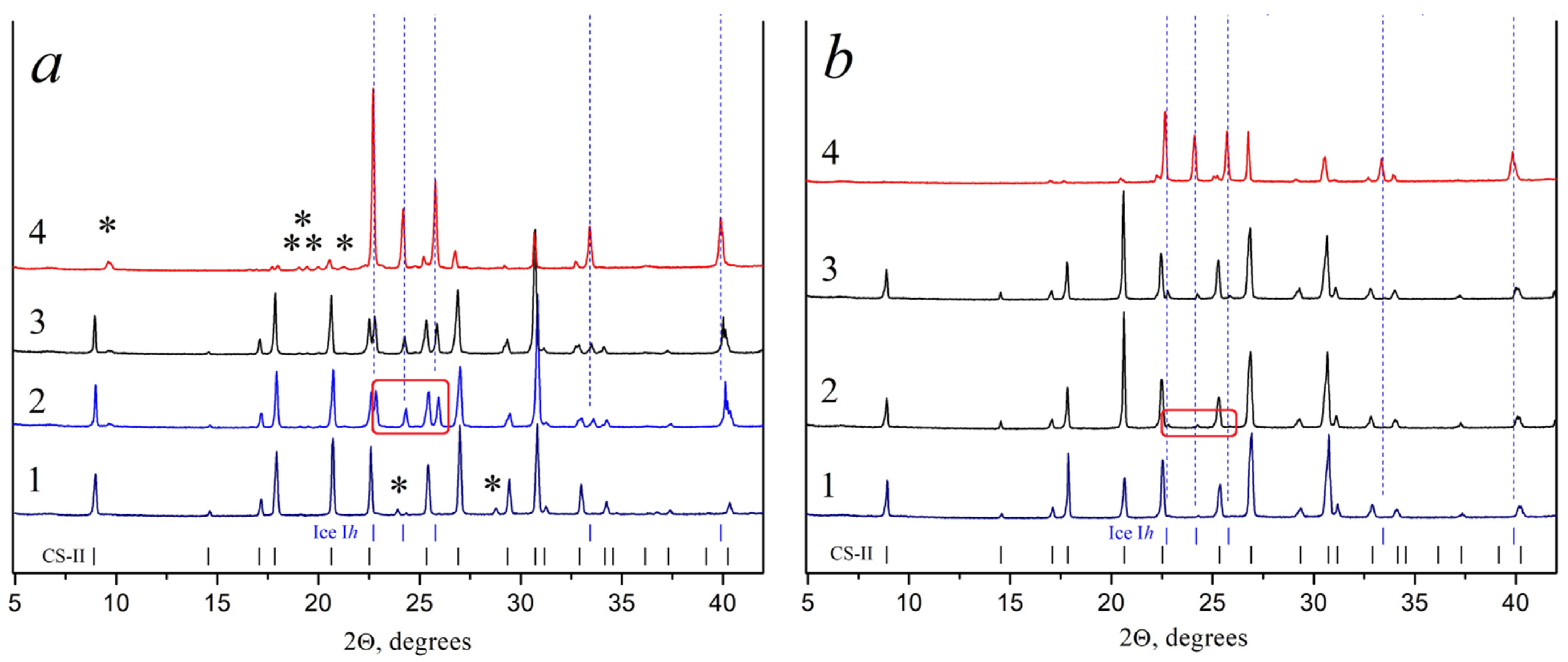
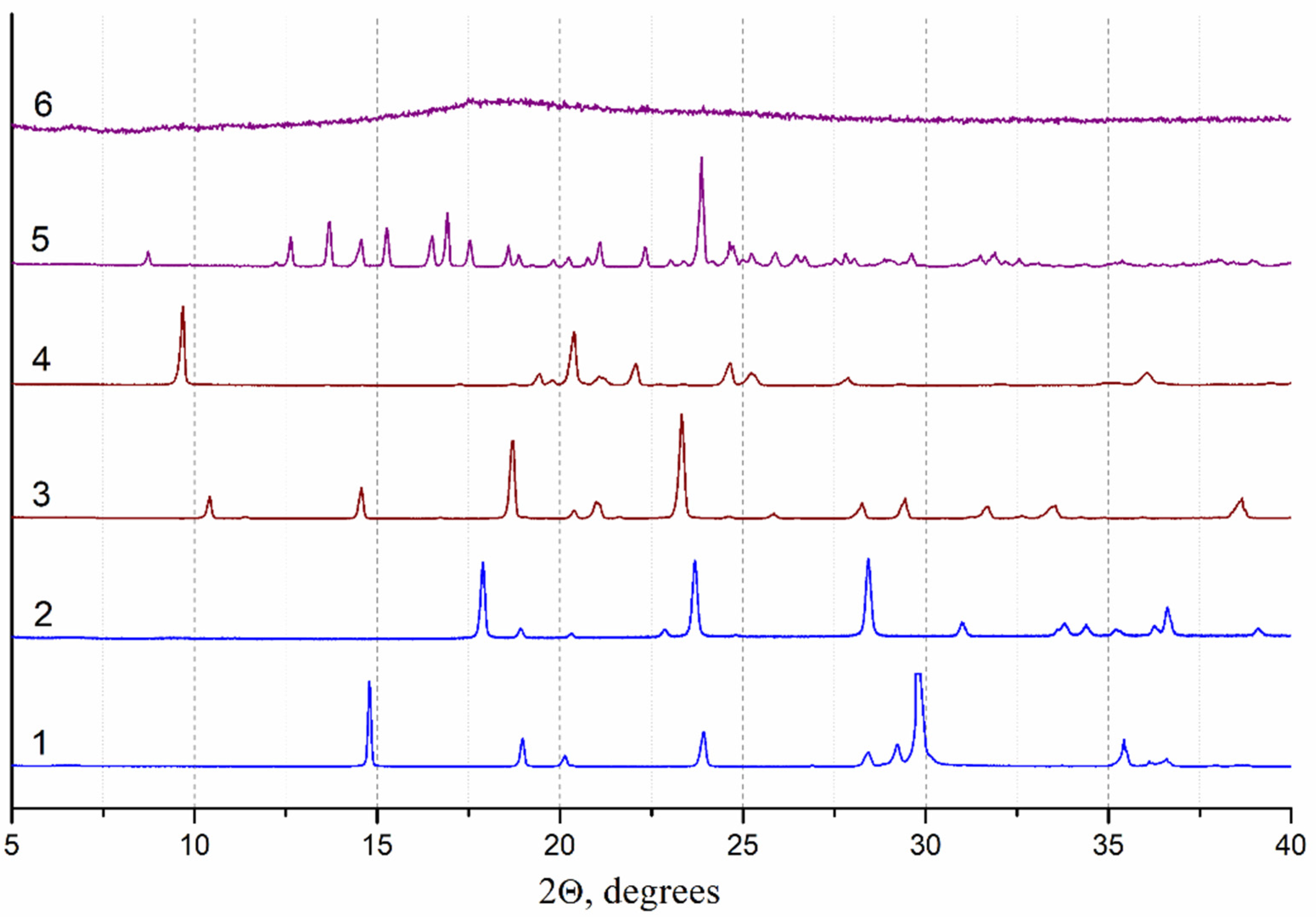
2.2.4. X-ray Powder Diffraction Analysis
2.2.5. Residual THF Level Determination
2.2.6. Scanning Electron Microscopy (SEM)
2.2.7. Elemental Analysis
2.2.8. Nebulizer Test
2.2.9. Determination of Pz Content
3. Results and Discussion
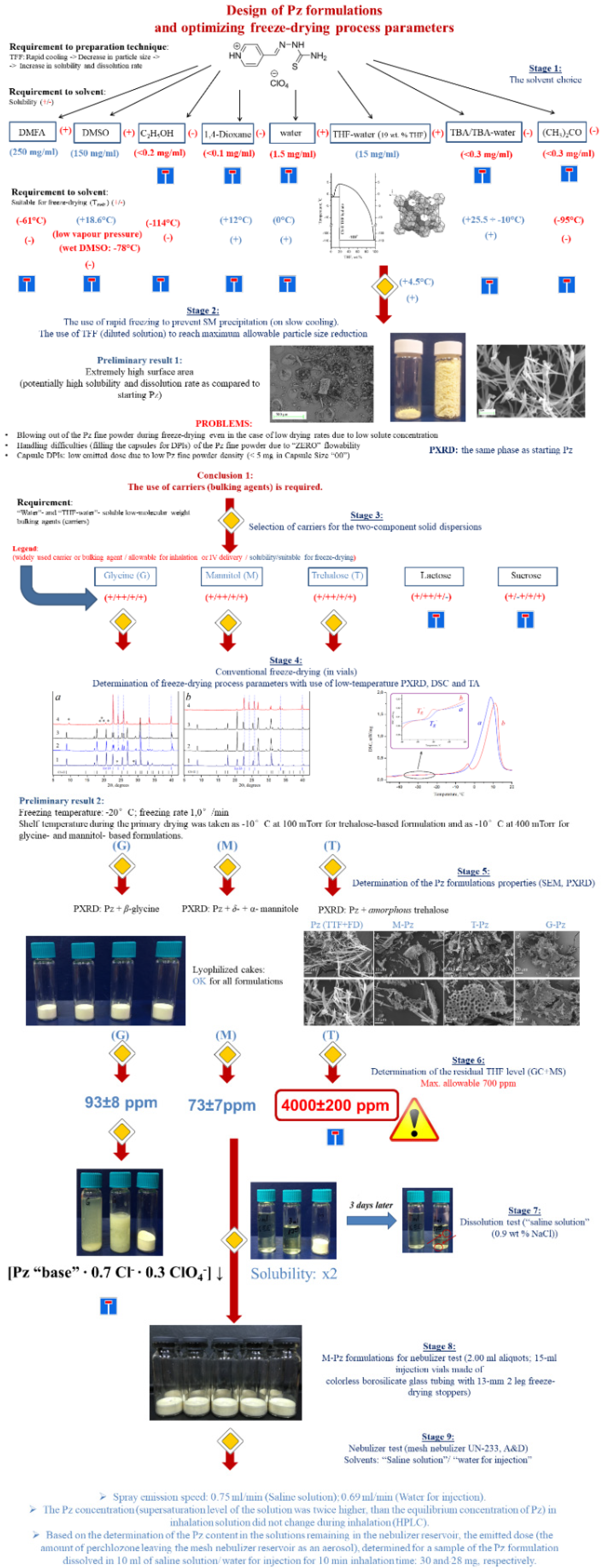
3.1. The Solvent Choice
3.2. Trial Experiments with Precipitation of Pure Pz from Frozen THF–Water Solutions
3.3. Selection of a Carrier of Pz for the Two-Component Solid Dispersions
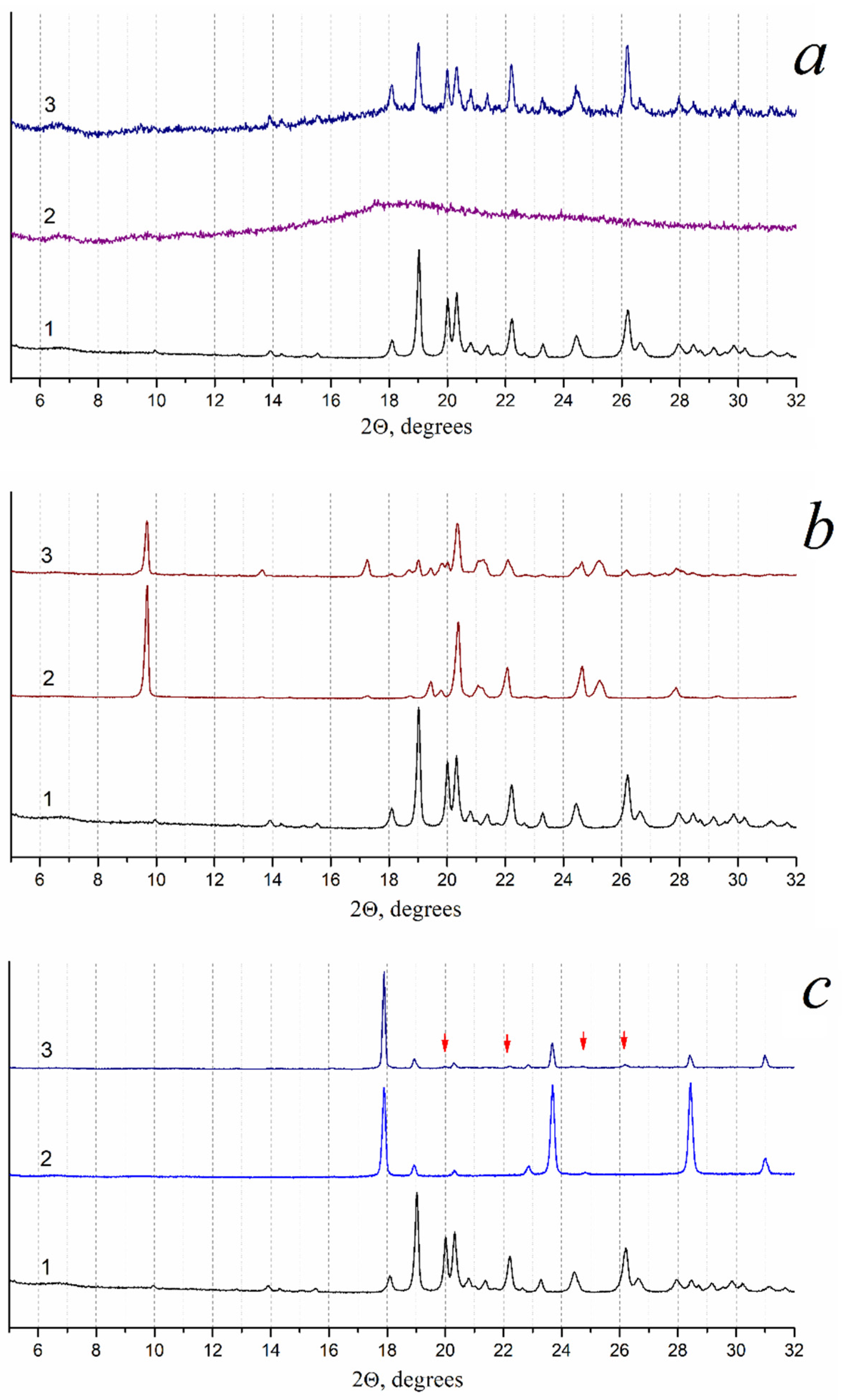
3.3.1. Optimizing the Freeze-Drying Protocol Based on the TA and XRD Experiments

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Crystalline Phases (PXRD) | Melting Temperature ± s.d., °C | Comments |
|---|---|---|---|
| THF–water (19.1 wt % THF) | THF hydrate | 4.2 ± 0.1 (4.2 ± 0.1 [49]; 4.5 °C [44]) | Congruent melting of the THF hydrate in binary system |
| Glycine–water (5 wt % glycine) | ice Ih + β-glycine | −3.5 ± 0.1 (−3.5 ± 0.1 [69]; −3.6 °C [70]) −0.8 ± 0.2 | Glycine–water eutectic ice Ih melting (liquidus line) |
| TheIMannitol–water (5 wt % mannitol) | ice Ih + β-mannitol | −1.5 ± 0.1 (−1.5 °C [71,72]) −0.5 ± 0.2 | Mannitol–water eutectic ice Ih melting (liquidus line) |
| Trehalose–water (5 wt % trehalose) | ice Ih | N/A * −0.2 ± 0.1 | --- ice Ih melting (liquidus line) |
| Glycine (5 wt %)–THF–water (19.1 wt % THF) | THF hydrate + β-glycine | −3.9 °C (−3.9 °C [49]) | Ternary peritectic |
| +3.0 ± 0.1 | THF hydrate melting in ternary system | ||
| Mannitol (5 wt %)—THF–water (19.1 wt % THF) | THF hydrate + + β-mannitol | −2.4 ± 0.2 | Ternary peritectic |
| +3.2 ± 0.2 | THF hydrate melting in ternary system | ||
| Trehalose (5 wt %)—THF–water (19.1 wt % THF) | THF hydrate | N/A * | ---- |
| +3.5 ± 0.2 | THF hydrate melting in ternary system | ||
| Pz (1.5 wt %)– THF–water (19.1 wt % THF) | THF hydrate + Pz | −2.0 ± 0.2 | Ternary peritectic |
| +4.0 ± 0.2 | THF hydrate melting in ternary system |
3.3.2. Determination of the Pz Formulations’ Properties
3.3.3. Determination of the Residual THF Level of the Pz Formulations
3.3.4. Estimating the Dissolution of the Pz Formulations with Mannitol and Glycine
3.4. Nebulizer Test Using Formulation of Pz with Mannitol (1:4)
- (1)
- To test the stability of the prepared inhalation solution based on the formulation of Pz with mannitol (1:4) under an external action (vibrating membrane) during a time period typical for the total duration of the inhalation treatment (10–15 min);
- (2)
- To estimate the spraying rate of the solution (mL/min). This is important, since if the spraying speed of the solution is very low (less than 0.2 mL/min), the therapeutically needed dose of a drug will not be delivered to the lungs during a reasonable duration of an inhalation treatment;
- (3)
- To estimate the dose released during 10–15 min of an inhalation treatment.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Tuberculosis Report 2017. Available online: https://www.who.int/tb/publications/global_report/gtbr2017_main_text.pdf (accessed on 28 December 2021).
- Global Tuberculosis Report 2021. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports (accessed on 28 December 2021).
- Zumla, P.S.A.; Nahid, P.; Cole, S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 2013, 12, 388–404. [Google Scholar] [CrossRef]
- Jüptner, A.; Scherließ, R. Spray Dried Formulations for Inhalation—Meaningful Characterisation of Powder Properties. Pharmaceutics 2019, 12, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nodilo, L.N.; Ugrina, I.; Špoljarić, D.; Klarić, D.A.; Brala, C.J.; Perkušić, M.; Pepić, I.; Lovrić, J.; Saršon, V.; Kučuk, M.S.; et al. A Dry Powder Platform for Nose-to-Brain Delivery of Dexamethasone: Formulation Development and Nasal Deposition Studies. Pharmaceutics 2021, 13, 795. [Google Scholar] [CrossRef] [PubMed]
- Pardeshi, S.; More, M.; Patil, P.; Pardeshi, C.; Deshmukh, P.; Mujumdar, A.; Naik, J. A meticulous overview on drying-based (spray-, freeze-, and spray-freeze) particle engineering approaches for pharmaceutical technologies. Dry. Technol. 2021, 39, 1447–1491. [Google Scholar] [CrossRef]
- Hertel, N.; Birk, G.; Scherließ, R. Performance tuning of particle engineered mannitol in dry powder inhalation formulations. Int. J. Pharm. 2020, 586, 119592. [Google Scholar] [CrossRef]
- Patil, T.S.; Deshpande, A.; Shende, P.K.; Deshpande, S.; Gaud, R. Evaluation of Nanocarrier-Based Dry Powder Formulations for Inhalation with Special Reference to Anti-Tuberculosis Drugs. Crit. Rev. Ther. Drug Carr. Syst. 2019, 36, 239–276. [Google Scholar] [CrossRef]
- Khadka, P.; Hill, P.C.; Zhang, B.; Katare, R.; Dummer, J.; Das, S.C. A study on polymorphic forms of rifampicin for inhaled high dose delivery in tuberculosis treatment. Int. J. Pharm. 2020, 587, 119602. [Google Scholar] [CrossRef]
- de Castro, R.R.; Todaro, V.; da Silva, L.C.R.P.; Simon, A.; Carmo, F.A.D.; de Sousa, V.P.; Rodrigues, C.R.; Sarmento, B.; Healy, A.M.; Cabral, L.M. Development of inhaled formulation of modified clofazimine as an alternative to treatment of tuberculosis. J. Drug Deliv. Sci. Technol. 2020, 58, 101805. [Google Scholar] [CrossRef]
- Mehta, P.; Bothiraja, C.; Kadam, S.; Pawar, A. Potential of dry powder inhalers for tuberculosis therapy: Facts, fidelity and future. Artif. Cells Nanomed. Biotechnol. 2018, 46, S791–S806. [Google Scholar] [CrossRef]
- Rossi, I.; Bettini, R.; Buttini, F. Resistant Tuberculosis: The Latest Advancements of Second-line Antibiotic Inhalation Products. Curr. Pharm. Des. 2021, 27, 1436–1452. [Google Scholar] [CrossRef]
- Patil, T.S.; Deshpande, A.S.; Deshpande, S.; Shende, P. Targeting pulmonary tuberculosis using nanocarrier-based dry powder inhalation: Current status and futuristic need. J. Drug Target. 2019, 27, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Makled, S.; Boraie, N.; Nafee, N. Nanoparticle-mediated macrophage targeting—a new inhalation therapy tackling tuberculosis. Drug Deliv. Transl. Res. 2021, 11, 1037–1055. [Google Scholar] [CrossRef] [PubMed]
- Chogale, M.M.; Dhoble, S.B.; Patravale, V.B. A triple combination ’nano’ dry powder inhaler for tuberculosis: In vitro and in vivo pulmonary characterization. Drug Deliv. Transl. Res. 2021, 11, 1520–1531. [Google Scholar] [CrossRef] [PubMed]
- Vidyadevi, B. Direct Lungs Targeting: An Alternative Treatment Approach for Pulmonary Tuberculosis. Asian J. Pharm. 2022, 15. [Google Scholar] [CrossRef]
- Joshi, M.; Prabhakar, B. Development of respirable rifampicin loaded bovine serum albumin formulation for the treatment of pulmonary tuberculosis. J. Drug Deliv. Sci. Technol. 2021, 61, 102197. [Google Scholar] [CrossRef]
- Kuzovlev, A.N.; Moroz, V.V.; Golubev, A.M. Inhaled Antibiotics in Treatment of Nosocomial Pneumonia. Anesteziol. I Reanimatol. 2015, 60, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Sievert, D.M.; Ricks, P.; Edwards, J.R.; Schneider, A.; Patel, J.; Srinivasan, A.; Kallen, A.; Limbago, B.; Fridkin, S.; National Healthcare Safety Network, T.; et al. Antimicrobial-resistant pathogens associated with healthcare-associated infections: Summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect. Control. Hosp. Epidemiol. 2013, 34, 1–14. [Google Scholar] [CrossRef]
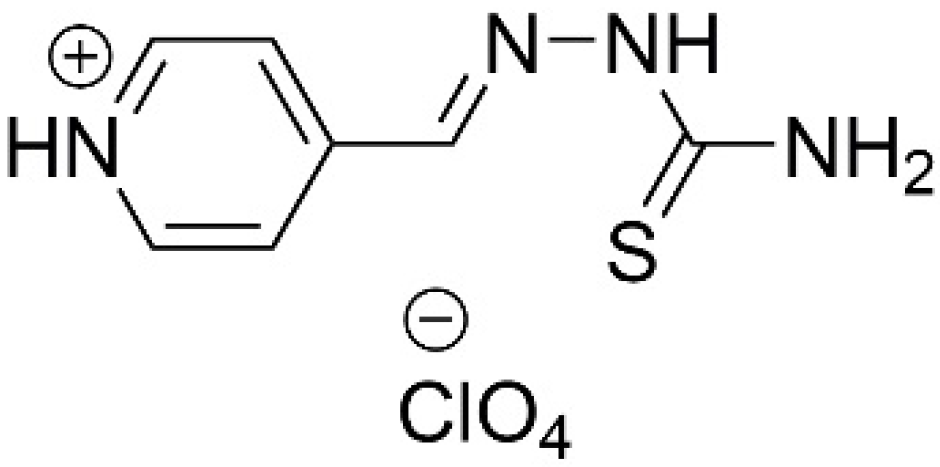
- Alexandrova, A.E.; Vinogradova, T.I.; Elokhina, V.N.; Kalihman, I.D.; Karnaukhova, R.V.; Nakhmanovich, A.S.; Shegoleva, R.A. 4-thioureidoiminomethylpyridinium Perchlorate with Tuberculostatic Activity. RF Patent No. 1621449, 25 November 1993. [Google Scholar]
- Yablonskiy, P.; Vinogradova, T.I.; Levashev, Y.N.; Pavlova, M.V.; Zilber, E.K.; Starshinova, A.A.; Sapozhnikova, N.V.; Chernokhaeva, I.V.; Archakova, L.I.; Zabolotnykh, N.V.; et al. Preclinical and clinical trials of the new tuberculosis drug perchlozon. Ter. arkhiv 2016, 88, 111–115. [Google Scholar] [CrossRef]
- Nakhmanovich, A.S.; Elokhina, V.N.; Dolgushin, G.V.; Gushin, A.S.; Polyakov, R.A.; Volkova, K.A.; Puniya, V.S. Method for Producing 4-thioureidoiminomethylpyridinium Perchlorate with Tuberculostatic Activity. RF Patent No. 2265014, 2004. [Google Scholar]
- Pharmasyntez JSC; Perchlozone®. New Opportunities in MDR TB Treatment; Working Group on New TB Drugs, JSC Pharmasyntez: Moscow, Russia, 2012. [Google Scholar]
- Trofimov, B.A.; Amosova, S.V.; Elokhina, V.N.; Yaroshenko, T.I.; Potapov, V.A. Method for Production Pharmacopeanic 4-thioureidoiminomethylpyridinium Perchlorate with High Tuberculostatic Activity. RF Patent No. 2476426, 2013. [Google Scholar]
- Malík, I.; Čižmárik, J.; Pecháčová, M. Focus on perchlozone, an anti-tuberculosis drug from the Russian Federation. Čes. slov. Farm. 2020, 69, 203–210. [Google Scholar]
- Safonova, S.G.; Peretokina, I.V.; Makarova, M.V.; Krylova, L.Y.; Mikhailova, Y.D.; Grigorash, D.V. Minimal inhibiting concentration of an anti-tuberculosis drug Perchloson® with respect to mycobacteria with different resistivity. Tuberc. Soc. Important Dis. 2020, 1, 26–33. Available online: https://elibrary.ru/item.asp?id=43808436 (accessed on 28 December 2021). (In Russian).
- Bogomolova, E.S.; Vokina, V.A. Estimation of Genotoxicity and Embriotoxicity of an Anti-Tuberculosis Drug Perchloson®. Health and Quality of Life 2018, Proceed. III Nation. Confer. with Foreign Participation, Irkutsk. pp. 25–31. Available online: https://elibrary.ru/item.asp?id=36787805 (accessed on 28 December 2021). (In Russian).
- Bogomolova, E.S. Antimicrobial Properties of a New Anti-Tuberculosis Drug, Perchloson®. In: Prospects of Development of Biomedical Technologies in Baikal Region 2019, Proceed. Intern. Confer., Irkutsk. pp. 9–10. Available online: https://elibrary.ru/item.asp?id=38334159 (accessed on 28 December 2021). (In Russian).
- Mokrousov, I.; Vyazovaya, A.; Akhmedova, G.; Solovieva, N.; Turkin, E.; Zhuravlev, V. Genetic Variation Putatively Associated with Mycobacterium tuberculosis Resistance to Perchlozone, a New Thiosemicarbazone: Clues from Whole Genome Sequencing and Implications for Treatment of Multidrug-Resistant Tuberculosis. Antibiotics 2020, 9, 669. [Google Scholar] [CrossRef] [PubMed]
- Churilov, L.; Korzhikov-Vlakh, V.; Sinitsyna, E.; Polyakov, D.; Darashkevich, O.; Poida, M.; Platonova, G.; Vinogradova, T.; Utekhin, V.; Zabolotnykh, N.; et al. Enhanced Delivery of 4-Thioureidoiminomethylpyridinium Perchlorate in Tuberculosis Models with IgG Functionalized Poly(Lactic Acid)-Based Particles. Pharmaceutics 2018, 11, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antosova, Z.; Mackova, M.; Král, V.; Macek, T. Therapeutic application of peptides and proteins: Parenteral forever? Trends Biotechnol. 2009, 27, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Merisko-Liversidge, E.; Liversidge, G.G.; Cooper, E.R. Nanosizing: A formulation approach for poorly-water-soluble compounds. Eur. J. Pharm. Sci. 2003, 18, 113–120. [Google Scholar] [CrossRef]
- Nestor, J.J., Jr. The Medicinal Chemistry of Peptides. Curr. Med. Chem. 2009, 16, 4399–4418. [Google Scholar] [CrossRef]
- Chow, A.H.L.; Tong, H.H.Y.; Chattopadhyay, T.P.; Shekunov, B.Y. Particle Engineering for Pulmonary Drug Delivery. Pharm. Res. 2007, 24, 411–437. [Google Scholar] [CrossRef]
- Weers, J.G.; Bell, J.; Chan, H.-K.; Cipolla, D.; Dunbar, C.; Hickey, A.J.; Smith, I.J. Pulmonary Formulations: What Remains to be Done? J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, S-5. [Google Scholar] [CrossRef]
- Boldyreva, E. Glycine: The Gift that Keeps on Giving. Isr. J. Chem. 2021, 61, 828–850. [Google Scholar] [CrossRef]
- Teagarden, D.L.; Baker, D.S. Practical aspects of lyophilization using non-aqueous co-solvent systems. Eur. J. Pharm. Sci. 2002, 15, 115–133. [Google Scholar] [CrossRef]
- Guidance for Industry. Q3C Impurities: Residual Solvents. Available online: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073395.pdf (accessed on 28 December 2021).
- Teagarden, D.L.; Wang, W.; Baker, D.S. Practical aspects of freeze-drying of pharmaceutical and biological products using nonaqueous cosolvent systems. In Freeze Drying/Lyophilization of Pharmaceutical and Biological Products, 3rd ed.; Rey, L., May, J.C., Eds.; Informa Healthcare: London, UK, 2010; pp. 254–287. [Google Scholar]
- Franks, F. Freeze-drying of bioproducts: Putting principles into practice. Eur. J. Pharm. Biopharm. 1998, 45, 221–229. [Google Scholar] [CrossRef]
- Tang, X.C.; Pikal, M.J. Design of Freeze-Drying Processes for Pharmaceuticals: Practical Advice. Pharm. Res. 2004, 21, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. Physical Characterization of Pharmaceutical Formulations in Frozen and Freeze-Dried Solid States: Techniques and Applications in Freeze-Drying Development. Pharm. Dev. Technol. 2006, 11, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.F.; Pikal, M.J.; Chang, B.S.; Randolph, T.W. Rational design of stable lyophilized protein formulations: Some practical advice. Pharm. Res. 1997, 14, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Dyadin, Y.A.; Bondaryuk, I.V.; Zhurko, F.V. Clathrate Hydrates at High Pressures; Atwood, J.L., Davies, J.E.D., MacNicol, D.D., Eds.; Inclusion compounds; Oxford University Press: Oxford, UK, 1991; Volume 5, pp. 214–275. [Google Scholar]
- Sloan, E.D.; Koh, C.A. Clathrate Hydrates of Natural Gases, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Manakov, A.; Kosyakov, V.; Solodovnikov, S. Structural Chemistry of Clathrate Hydrates and Related Compounds. In Comprehensive Supramolecular Chemistry II; Elsevier BV: Amsterdam, The Netherlands, 2017; Volume 7, pp. 161–206. [Google Scholar]
- Ogienko, A.G.; Boldyreva, E.V.; Manakov, A.Y.; Boldyrev, V.V.; Yunoshev, A.S.; Ogienko, A.; Myz, S.A.; Ancharov, A.I.; Achkasov, A.F.; Drebushchak, T.N. A New Method of Producing Monoclinic Paracetamol Suitable for Direct Compression. Pharm. Res. 2011, 28, 3116–3127. [Google Scholar] [CrossRef]
- Ogienko, A.G.; Drebushchak, V.A.; Bogdanova, E.G.; Yunoshev, A.S.; Boldyreva, E.V.; Manakov, A.Y. Thermodynamic aspects of freeze-drying. J. Therm. Anal. 2017, 127, 1593–1604. [Google Scholar] [CrossRef]
- Ogienko, A.; Bogdanova, E.; Trofimov, N.; Myz, S.; Kolesov, B.; Yunoshev, A.; Zubikov, N.; Manakov, A.; Boldyrev, V.; Boldyreva, E. Large porous particles for respiratory drug delivery. Glycine-based formulations. Eur. J. Pharm. Sci. 2017, 110, 148–156. [Google Scholar] [CrossRef]
- Ogienko, A.G.; Bogdanova, E.G.; Stoporev, A.; Ogienko, A.; Shinkorenko, M.P.; Yunoshev, A.S.; Manakov, A.Y. Preparation of fine powders by clathrate-forming freeze-drying: A case study of ammonium nitrate. Mendeleev Commun. 2018, 28, 211–213. [Google Scholar] [CrossRef]
- Smolentsev, A.I.; Lavrenova, L.G.; Elokhina, V.N.; Nakhmanovich, A.S.; Larina, L. Crystal structures of pyridine-4-aldehyde thiosemicarbazone perchlorate and trifluoromethane sulfonate. J. Struct. Chem. 2009, 50, 500–504. [Google Scholar] [CrossRef]
- Taga, T.; Senma, M.; Osaki, K. The crystal and molecular structure of trehalose dihydrate. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1972, 28, 3258–3263. [Google Scholar] [CrossRef]
- Fronczek, F.R.; Kamel, H.N.; Slattery, M. Three polymorphs (α, β, and δ) of D -mannitol at 100 K. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2003, 59, 567–570. [Google Scholar] [CrossRef]
- Boldyreva, E.V.; Drebushchak, V.A.; Drebushchak, T.N.; Paukov, I.E.; Kovalevskaya, Y.A.; Shutova, E.S. Polymorphism of glycine, Part I. J. Therm. Anal. 2003, 73, 409–418. [Google Scholar] [CrossRef]
- Rasmussen, D.H.; MacKenzie, A.P. Phase Diagram for the System Water–Dimethylsulphoxide. Nature 1968, 220, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Parks, M. What is the True Solubility Advantage for Amorphous Pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef]
- Serajuddin, A.T. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef]
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization and stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef]
- Rams-Baron, M.; Jachowicz, R.; Boldyreva, E.; Zhou, D.; Jamróz, W.; Paluch, M. Amorphous Drug Preparation Methods; Springer Science and Business Media LLC: Berlin, Germany, 2018; pp. 69–106. [Google Scholar]
- Zidan, A.S.; Rahman, Z.; Sayeed, V.; Raw, A.; Yu, L.; Khan, M. Crystallinity evaluation of tacrolimus solid dispersions by chemometric analysis. Int. J. Pharm. 2012, 423, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Chemburkar, S.R.; Bauer, J.; Deming, K.; Spiwek, H.; Patel, K.; Morris, J.; Henry, R.; Spanton, S.; Dziki, W.; Porter, W.; et al. Dealing with the Impact of Ritonavir Polymorphs on the Late Stages of Bulk Drug Process Development. Org. Process Res. Dev. 2000, 4, 413–417. [Google Scholar] [CrossRef]
- Bauer, J.; Spanton, S.; Henry, R.; Quick, J.; Dziki, W.; Porter, W.; Morris, J. Ritonavir: An Extraordinary Example of Conformational Polymorphism. Pharm. Res. 2001, 18, 859–866. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef]
- Ogienko, A.G.; Stoporev, A.S.; Mel’gunov, M.S.; Adamova, T.P.; Yunoshev, A.S.; Manakov, A.Y.; Boldyreva, E.V. Discrepancy between thermodynamic and kinetic stabilities of thetert-butanol hydrates and its implication for obtaining pharmaceutical powders by freeze-drying. Chem. Commun. 2019, 55, 4262–4265. [Google Scholar] [CrossRef]
- Chang, B.S.; Fischer, N.L. Development of an Efficient Single-Step Freeze-Drying Cycle for Protein Formulations. Pharm. Res. 1995, 12, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Iitaka, Y. Crystal Structure of β-Glycine. Nature 1959, 183, 390–391. [Google Scholar] [CrossRef]
- Drebushchak, T.N.; Boldyreva, E.V.; Shutova, E.S. β-Glycine. Acta Crystallogr. Sect. E Struct. Rep. Online 2002, 58, 634–636. [Google Scholar] [CrossRef]
- Botez, C.E.; Stephens, P.W.; Nunes, C.; Suryanarayanan, R. Crystal structure of anhydrous δ-D-mannitol. Powder Diffr. 2003, 18, 214–218. [Google Scholar] [CrossRef] [Green Version]
- Kanev, A.N.; Kosyakov, V.I.; Malakhov, D.V.; Shalaev, E.Y. Determination of the eutectic composition in water–glycine system. Izv. Sib. Otd. Akad. Nauk SSSR Ser. Khim. Nauk. 1989, 3, 35–38. [Google Scholar]
- Drebushchak, V.A.; Ogienko, A.G.; Boldyreva, E.V. Polymorphic effects and the eutectic melting in the H2O-glycine system. J. Therm. Anal. Calorim. 2013, 111, 2187–2194. [Google Scholar] [CrossRef]
- Kim, A.I.; Akers, M.J.; Nail, S.L. The Physical State of Mannitol after Freeze-Drying: Effects of Mannitol Concentration, Freezing Rate, and a Noncrystallizing Cosolute. J. Pharm. Sci. 1998, 87, 931–935. [Google Scholar] [CrossRef]
- Liao, X.; Krishnamurthy, R.; Suryanarayanan, R. Influence of the Active Pharmaceutical Ingredient Concentration on the Physical State of Mannitol—Implications in Freeze-Drying. Pharm. Res. 2005, 22, 1978–1985. [Google Scholar] [CrossRef]
- Patel, S.M.; Nail, S.L.; Pikal, M.J.; Geidobler, R.; Winter, G.; Hawe, A.; Davagnino, J.; Gupta, S.R. Lyophilized Drug Product Cake Appearance: What Is Acceptable? J. Pharm. Sci. 2017, 106, 1706–1721. [Google Scholar] [CrossRef]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Vessot, S.; Andrieu, J. A review on freeze drying of drugs with tert-butanol (TBA) + water systems: Characteristics, advantages, drawbacks. Dry. Technol. 2012, 30, 377–385. [Google Scholar] [CrossRef]
- Choi, W.S.; Kim, K. Separation of acetic acid from acetic acid-water mixture by crystallization. J. Sep. Sci. Technol. 2013, 48, 1056–1061. [Google Scholar] [CrossRef]
- Zelenin, Y.M. Effect of Pressure on Clathrate Formation in a Water–Ethanol System. J. Struct. Chem. 2003, 44, 130–136. [Google Scholar] [CrossRef]
- Manakov, A.Y.; Aladko, L.S.; Ogienko, A.G.; Ancharov, A.I. Hydrate formation in the system n-propanol–water. J. Therm. Anal. Calorim. 2013, 111, 885–890. [Google Scholar] [CrossRef]
- Aladko, L.S.; Manakov, A.Y.; Ogienko, A.G.; Ancharov, A.I. New data on phase diagram and clathrate formation in the system water–isopropyl alcohol. J. Incl. Phenom. Macrocycl. Chem. 2009, 63, 151–157. [Google Scholar] [CrossRef]
- Kuznetsova, M.N.; Bergmann, A.G. Physicochemical analysis of the reaction of amines with acids. III. Thermal analysis of the ternary system urea-formic ac-id-water. J. General Chem. USSR 1956, 26, 1497–1504. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogienko, A.G.; Myz, S.A.; Nefedov, A.A.; Ogienko, A.A.; Adamova, T.P.; Voronkova, O.M.; Amosova, S.V.; Trofimov, B.A.; Boldyrev, V.V.; Boldyreva, E.V. Clathrate Hydrates of Organic Solvents as Auxiliary Intermediates in Pharmaceutical Research and Development: Improving Dissolution Behaviour of a New Anti-Tuberculosis Drug, Perchlozon. Pharmaceutics 2022, 14, 495. https://doi.org/10.3390/pharmaceutics14030495
Ogienko AG, Myz SA, Nefedov AA, Ogienko AA, Adamova TP, Voronkova OM, Amosova SV, Trofimov BA, Boldyrev VV, Boldyreva EV. Clathrate Hydrates of Organic Solvents as Auxiliary Intermediates in Pharmaceutical Research and Development: Improving Dissolution Behaviour of a New Anti-Tuberculosis Drug, Perchlozon. Pharmaceutics. 2022; 14(3):495. https://doi.org/10.3390/pharmaceutics14030495
Chicago/Turabian StyleOgienko, Andrey G., Svetlana A. Myz, Andrey A. Nefedov, Anna A. Ogienko, Tatyana P. Adamova, Olga M. Voronkova, Svetlana V. Amosova, Boris A. Trofimov, Vladimir V. Boldyrev, and Elena V. Boldyreva. 2022. "Clathrate Hydrates of Organic Solvents as Auxiliary Intermediates in Pharmaceutical Research and Development: Improving Dissolution Behaviour of a New Anti-Tuberculosis Drug, Perchlozon" Pharmaceutics 14, no. 3: 495. https://doi.org/10.3390/pharmaceutics14030495