
Hyaluronic Acid Nanoparticles for Immunogenic Chemotherapy of Leukemia and T-Cell Lymphoma

 , and
, and
Abstract
:1. Introduction
2. Materials
3. Methods
3.1. Synthesis of HA–DOX–CPT
3.2. Physical Characterization
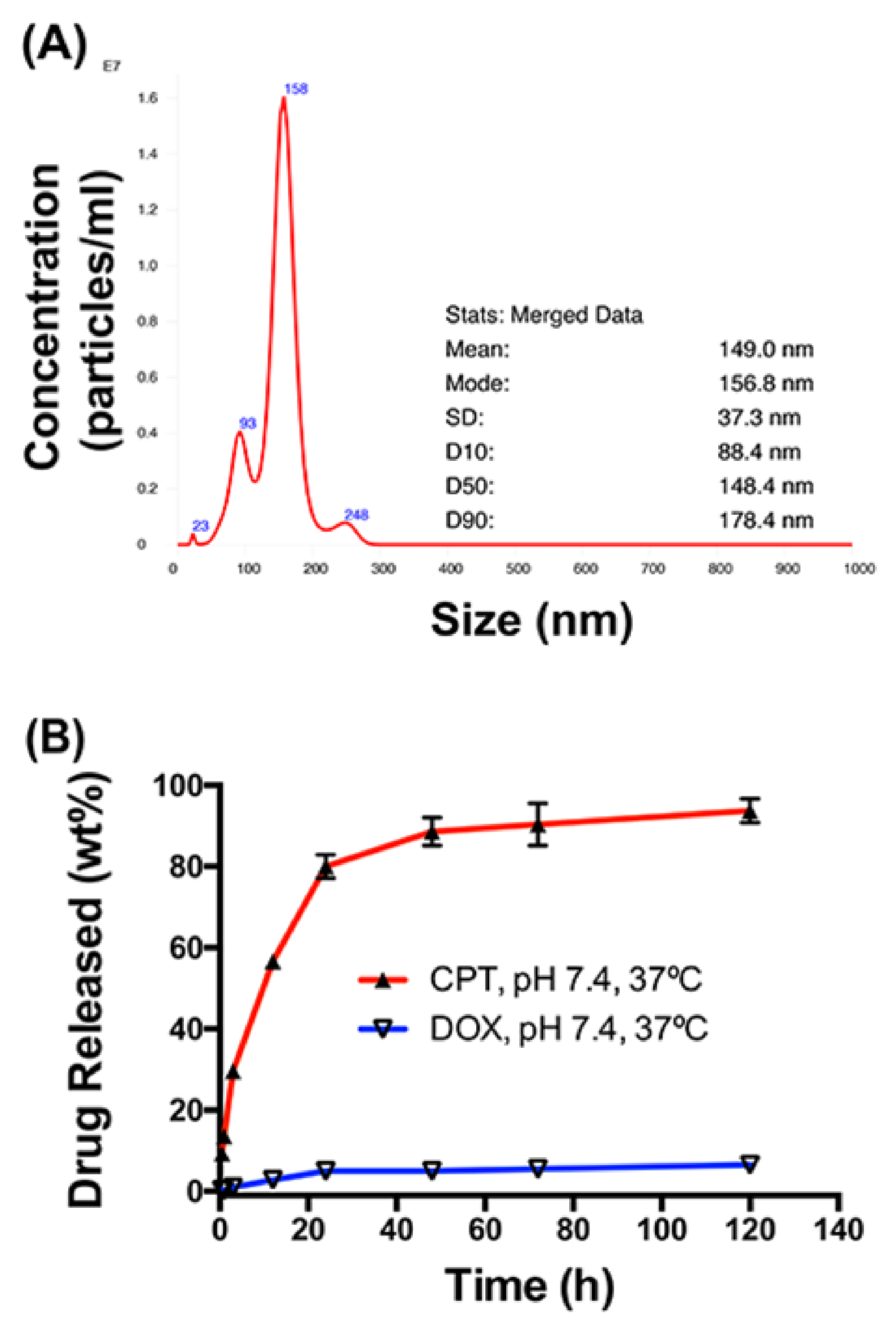
3.2.1. Size
3.2.2. In Vitro Release Studies
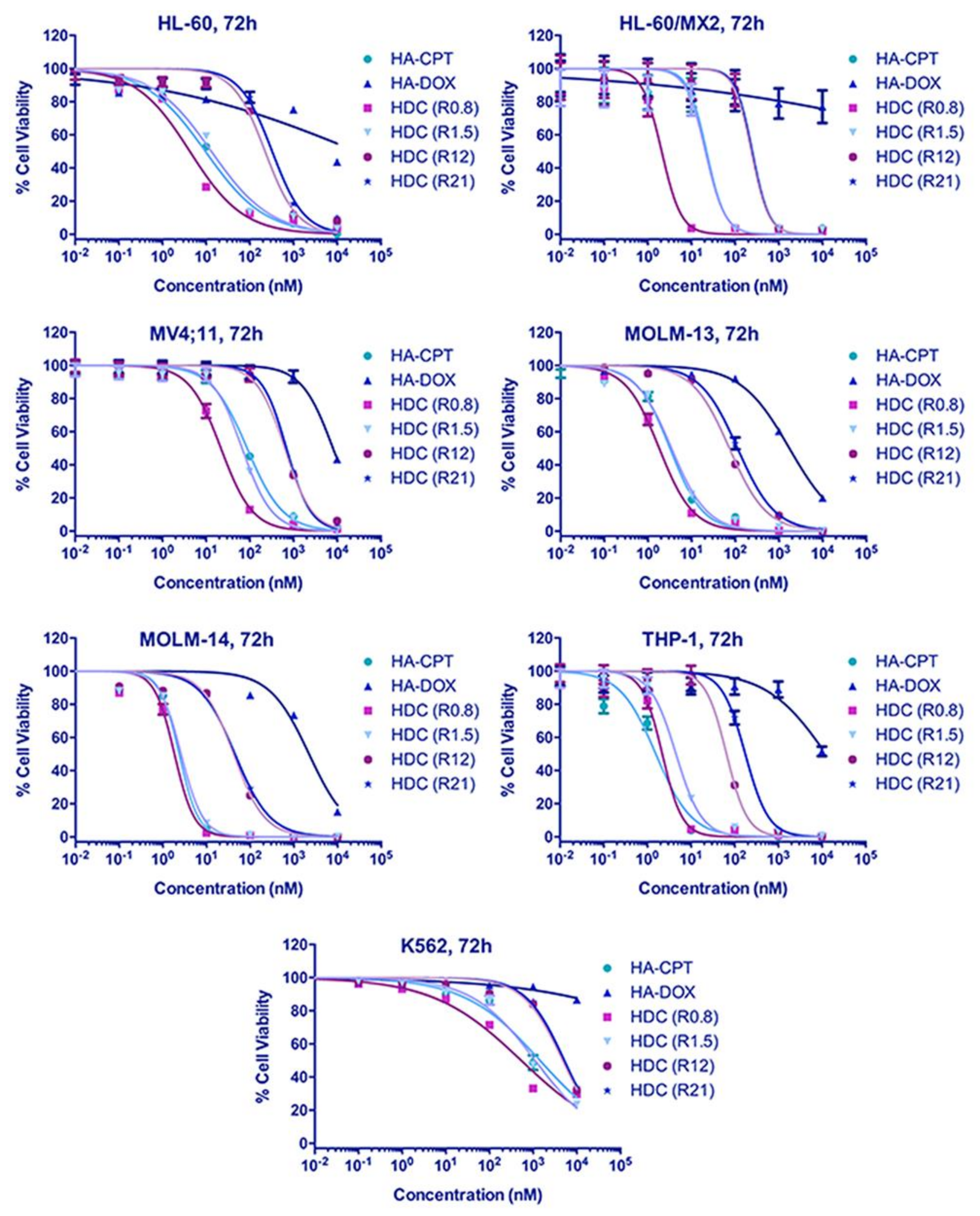
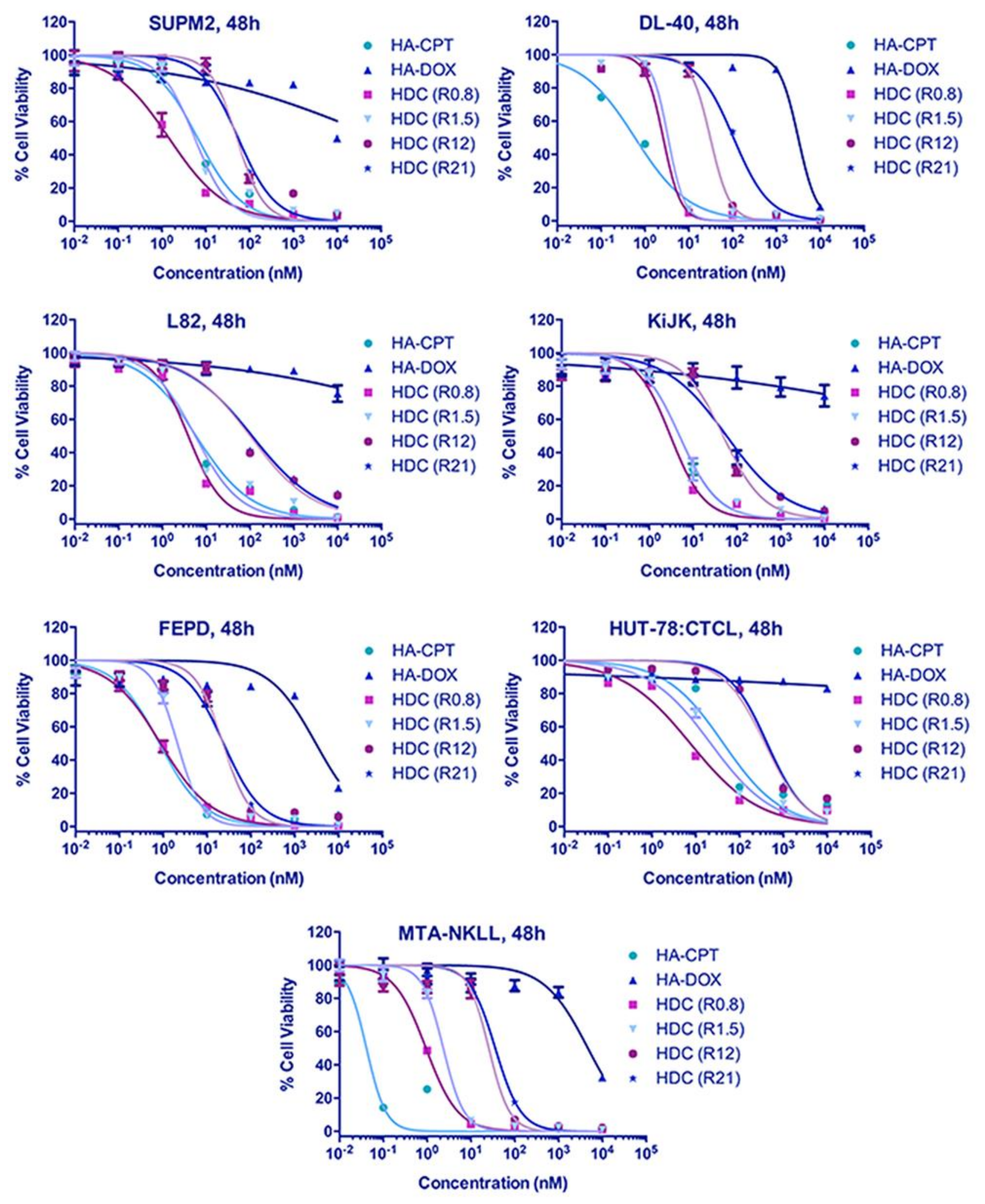
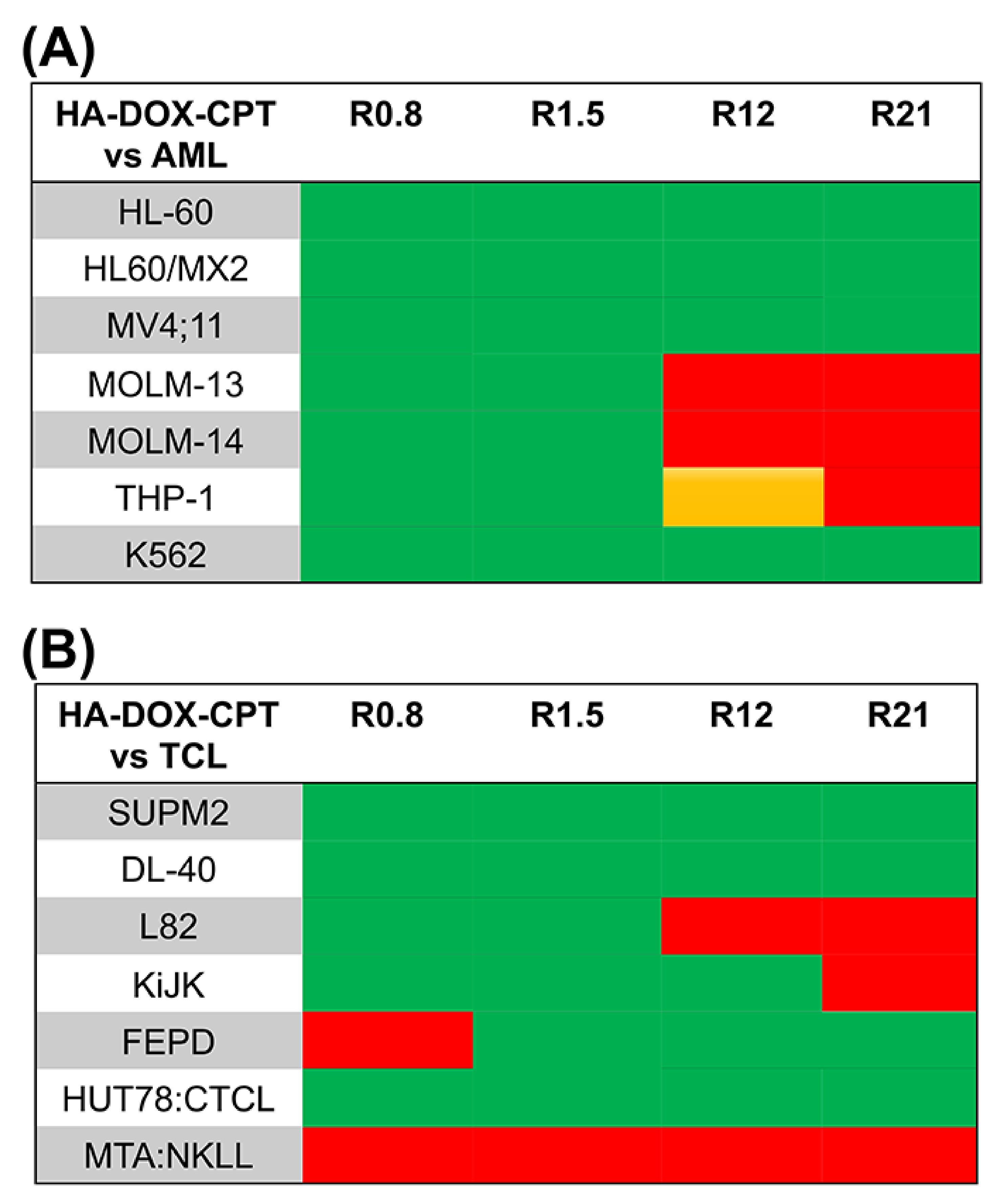
3.3. In Vitro Toxicity Analysis
3.4. In Vitro Anticancer Immunogenic and Drug-Resistant Marker Assay
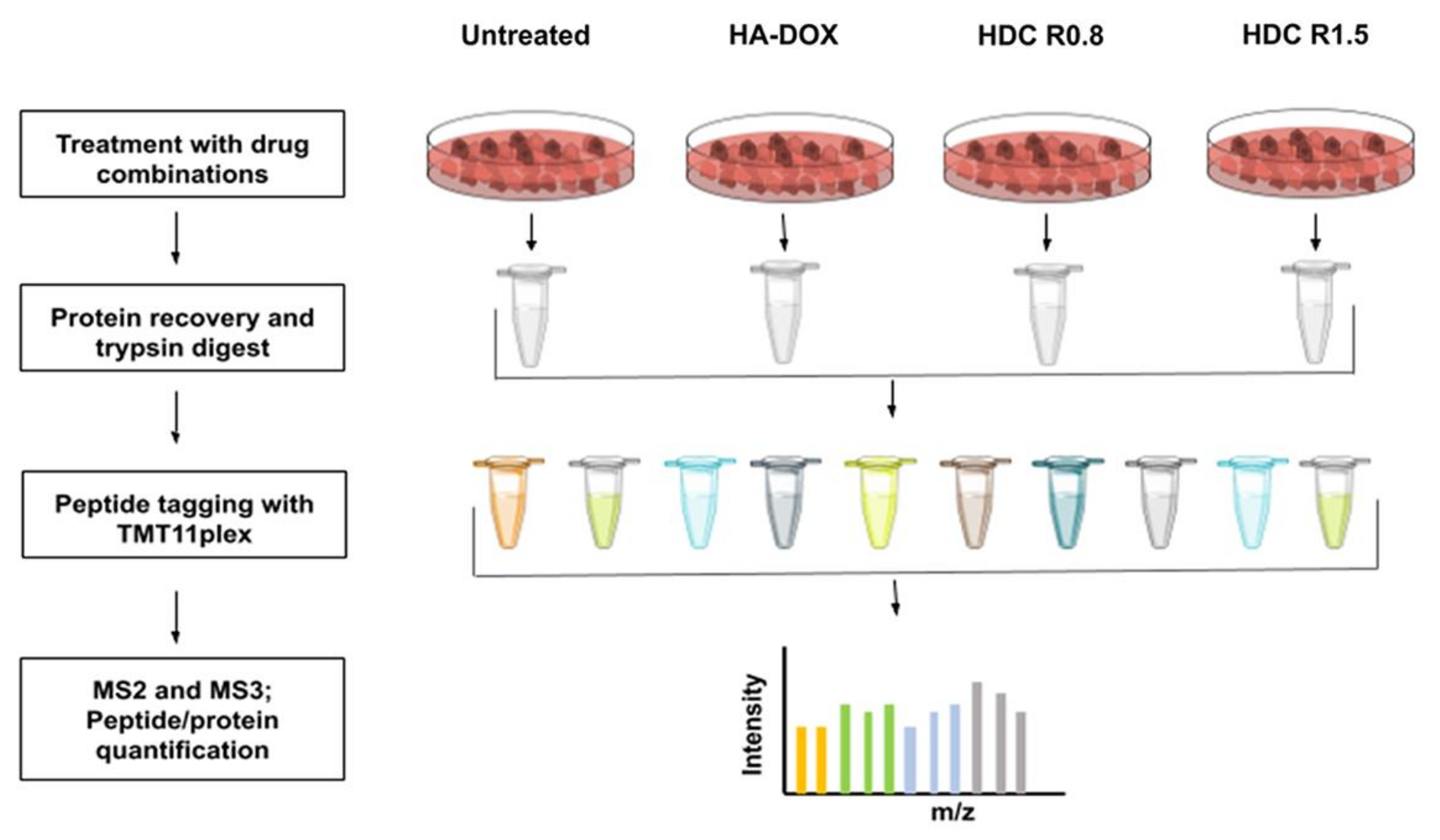
3.5. Protein Extraction from Cells
3.6. Hi pH Separation and Mass Spectrometry Analysis
3.7. Statistical Analysis
4. Results
4.1. Physical Characterization of HA–DOX–CPT
4.2. Increased Synergistic Potency of HA–DOX–CPT
4.3. Anticancer Immunogenicity of HA–DOX–CPT
4.4. Proteomic Profiling of DOX-Resistant Leukemia Cell Line (HL-60/MX2)
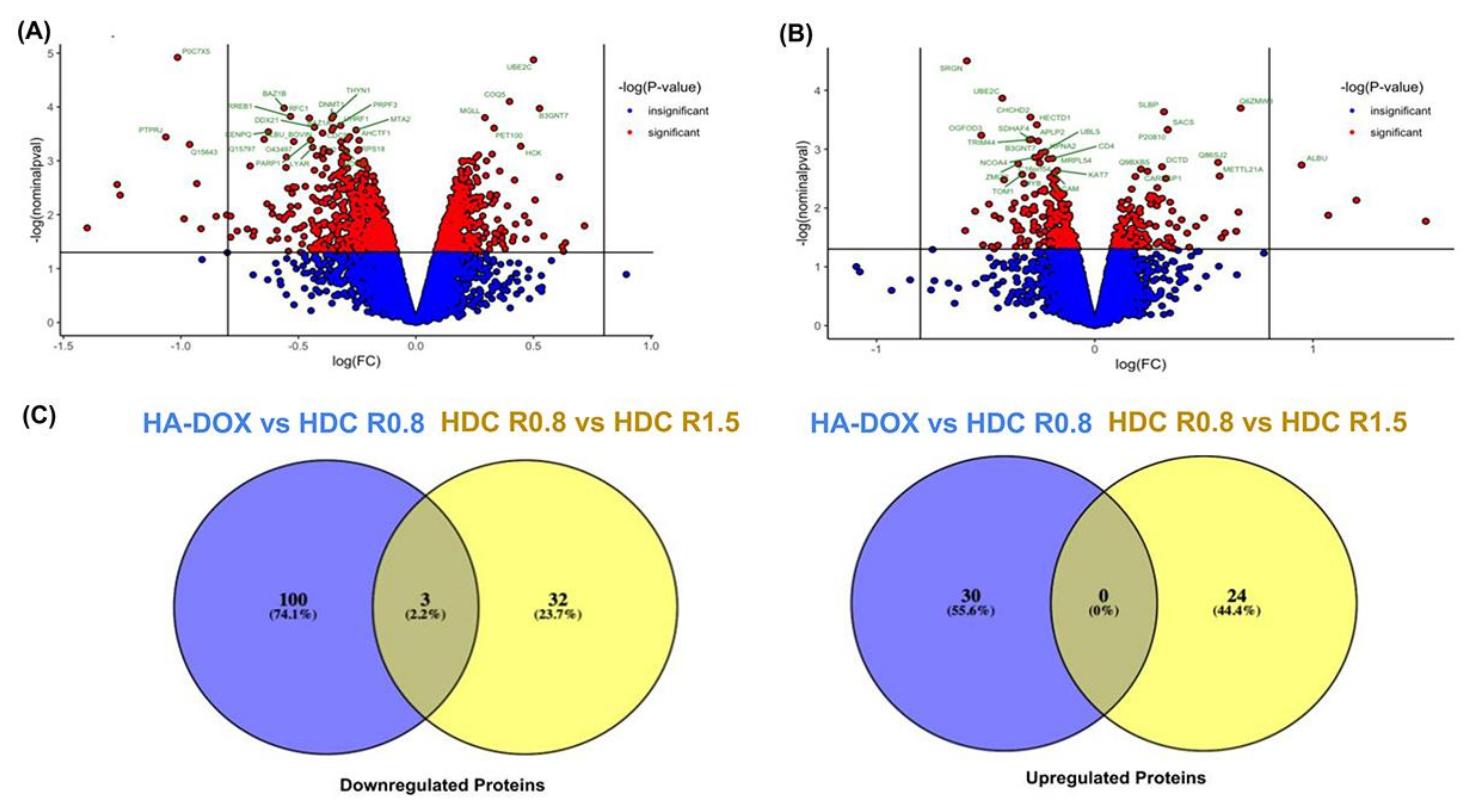
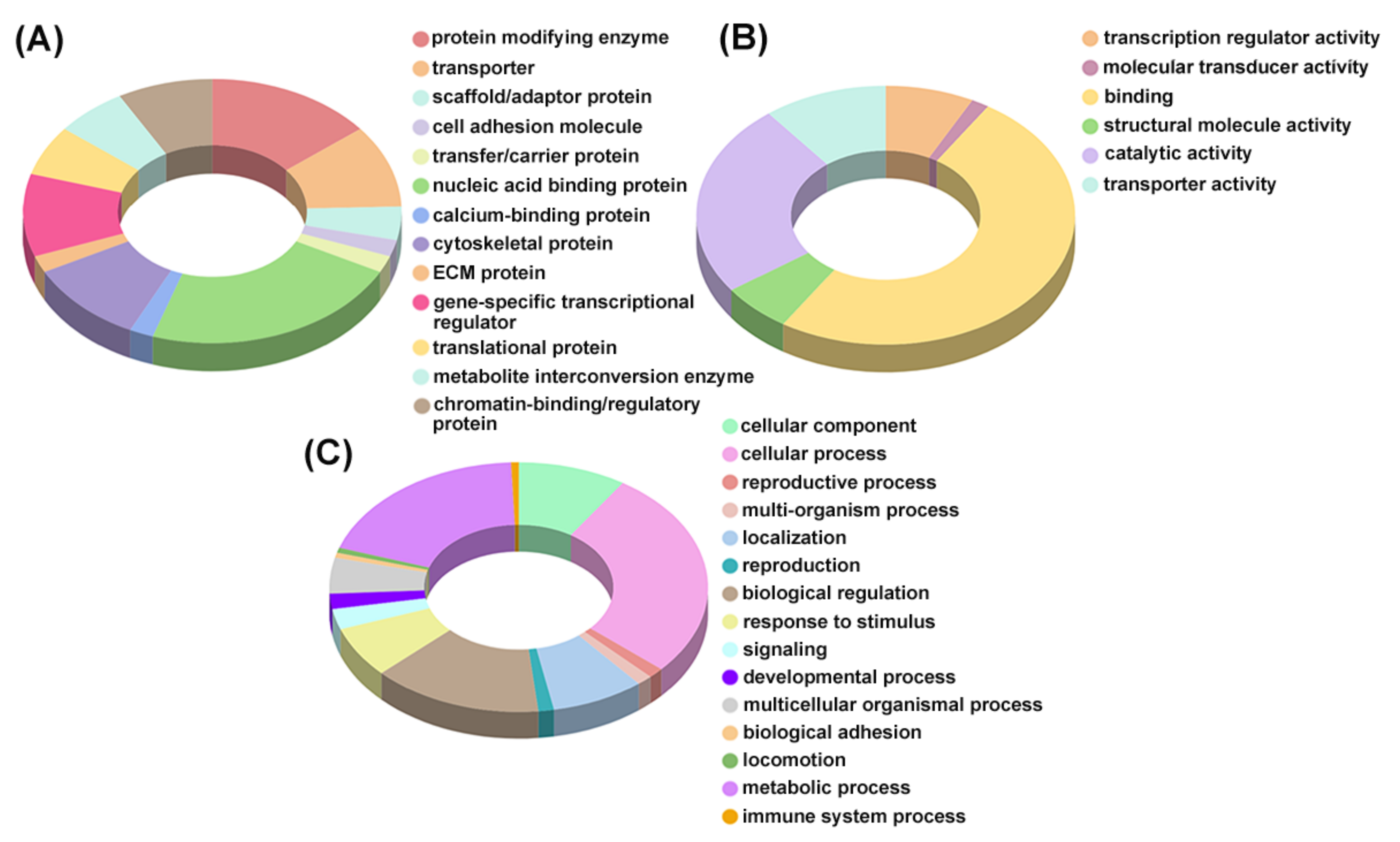
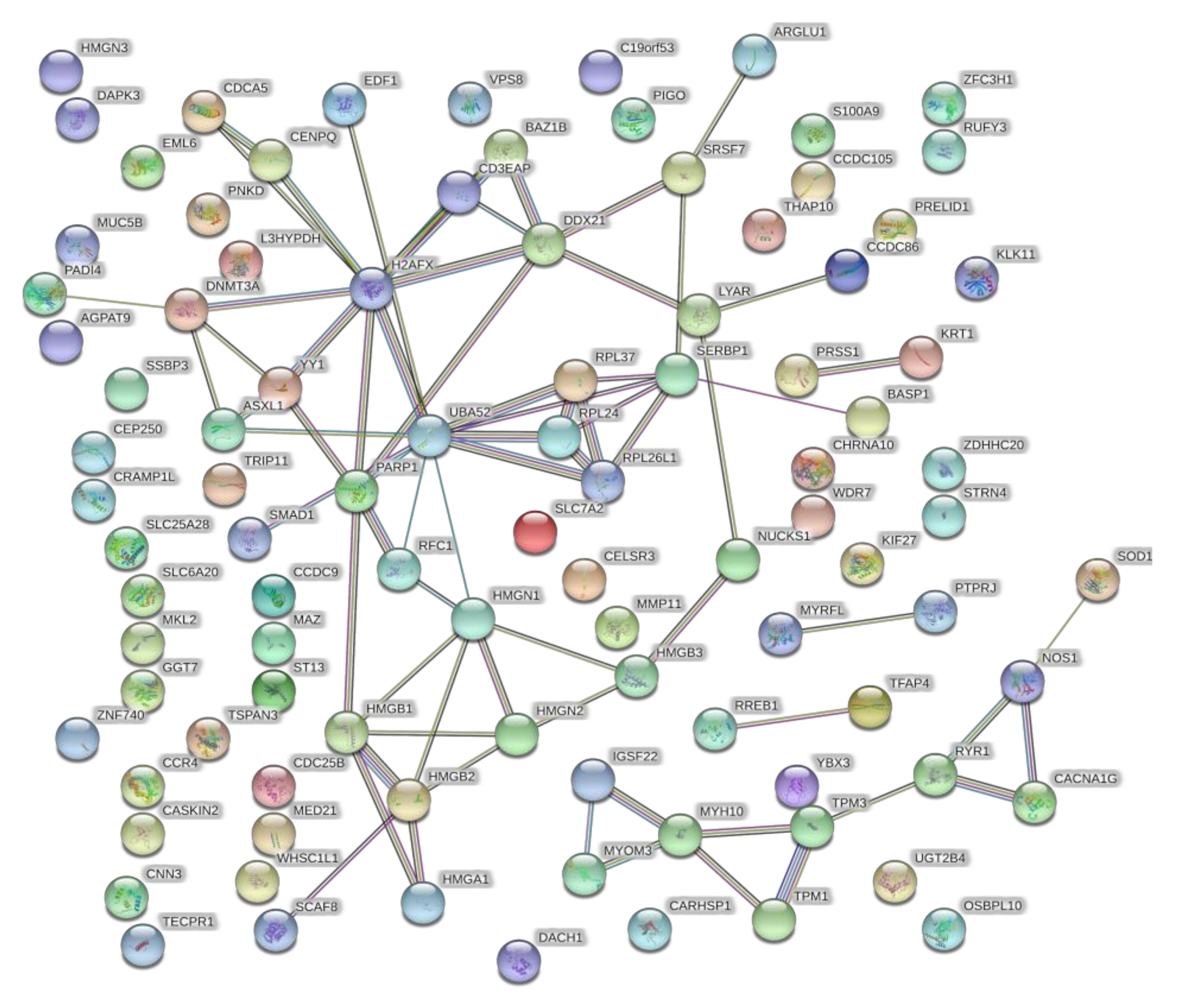
4.5. Bioinformatic Analysis—Identification and Enrichment of Differentially Expressed Proteins
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamaguchi, M.; Suzuki, R.; Kwong, Y.L.; Kim, W.S.; Hasegawa, Y.; Izutsu, K.; Suzumiya, J.; Okamura, T.; Nakamura, S.; Kawa, K.; et al. Phase I study of dexamethasone, methotrexate, ifosfamide, L-asparaginase, and etoposide (SMILE) chemotherapy for advanced-stage, relapsed or refractory extranodal natural killer (NK)/T-cell lymphoma and leukemia. Cancer Sci. 2008, 99, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Carrick, S.; Parker, S.; Thornton, C.E.; Ghersi, D.; Simes, J.; Wilcken, N. Single agent versus combination chemotherapy for metastatic breast cancer. Cochrane Database Syst. Rev. 2009, 2021, CD003372. [Google Scholar] [CrossRef]
- Yap, T.A.; Omlin, A.; de Bono, J.S. Development of therapeutic combinations targeting major cancer signaling pathways. J. Clin. Oncol. 2013, 31, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Chang, J.; Liu, L.G.; Hu, D.; Zhang, W.H.; Yan, Y.; Ma, L.Y.; Li, Z.C.; Ma, Y.J.; Hao, S.G.; et al. High-dose methotrexate, etoposide, dexamethasone and pegaspargase (MEDA) combination chemotherapy is effective for advanced and relapsed/refractory extranodal natural killer/T cell lymphoma: A retrospective study. Int. J. Hematol. 2015, 102, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Gao, G.X.; Chen, J.P.; Wang, J.S.; Wang, X.M.; Zeng, Y.; Bai, Q.X.; Zhang, T.; Yang, L.; Dong, B.X.; et al. A phase 2 study of methotrexate, etoposide, dexamethasone, and pegaspargase chemotherapy for newly diagnosed, relapsed, or refractory extranodal natural killer/T-cell lymphoma, nasal type: A multicenter trial in Northwest China. Hematol. Oncol. 2017, 35, 619–629. [Google Scholar] [CrossRef]
- Xu, P.P.; Xiong, J.; Cheng, S.; Zhao, X.; Wang, C.F.; Cai, G.; Zhong, H.J.; Huang, H.Y.; Chen, J.Y.; Zhao, W.L. A Phase II Study of Methotrexate, Etoposide, Dexamethasone and Pegaspargase Sandwiched with Radiotherapy in the Treatment of Newly Diagnosed, Stage IE to IIE Extranodal Natural-Killer/T-Cell Lymphoma, Nasal-Type. EBioMedicine 2017, 25, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Levato, L.; Gianfelici, V.; Caglioti, F.; Russo, A.; Giugliano, E.; Mannella, A.; Molica, S. Sustained Complete Remission with Incomplete Hematologic Recovery (CRi) in a Patient with Relapsed AML and Concurrent BCR-ABL1 and CBFB Rearrangement Treated with a Combination of Venetoclax and 5-Azacytidine. Chemotherapy 2020, 65, 51–53. [Google Scholar] [CrossRef]
- Vitale, C.; Ferrajoli, A. The combination of venetoclax and rituximab for the treatment of patients with recurrent chronic lymphocytic leukemia. Expert Rev. Hematol. 2020, 13, 885–894. [Google Scholar] [CrossRef]
- Ganta, S.; Amiji, M. Coadministration of Paclitaxel and curcumin in nanoemulsion formulations to overcome multidrug resistance in tumor cells. Mol. Pharm. 2009, 6, 928–939. [Google Scholar] [CrossRef]
- Yardley, D.A. Drug resistance and the role of combination chemotherapy in improving patient outcomes. Int. J. Breast Cancer 2013, 2013, 137414. [Google Scholar] [CrossRef] [Green Version]
- Nikanjam, M.; Liu, S.; Kurzrock, R. Dosing targeted and cytotoxic two-drug combinations: Lessons learned from analysis of 24,326 patients reported 2010 through 2013. Int. J. Cancer 2016, 139, 2135–2141. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Seebacher, N.; Shi, H.; Kan, Q.; Duan, Z. Novel strategies to prevent the development of multidrug resistance (MDR) in cancer. Oncotarget 2017, 8, 84559–84571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, A.C.; Chidley, C.; Sorger, P.K. A curative combination cancer therapy achieves high fractional cell killing through low cross-resistance and drug additivity. Elife 2019, 8, e50036. [Google Scholar] [CrossRef] [PubMed]
- Scharovsky, O.G.; Mainetti, L.E.; Rozados, V.R. Metronomic chemotherapy: Changing the paradigm that more is better. Curr. Oncol. 2009, 16, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Li, S.D.; Huang, L. Pharmacokinetics and biodistribution of nanoparticles. Mol. Pharm. 2008, 5, 496–504. [Google Scholar] [CrossRef]
- Xu, X.; Ho, W.; Zhang, X.; Bertrand, N.; Farokhzad, O. Cancer nanomedicine: From targeted delivery to combination therapy. Trends Mol. Med. 2015, 21, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Yhee, J.Y.; Son, S.; Lee, H.; Kim, K. Nanoparticle-Based Combination Therapy for Cancer Treatment. Curr. Pharm. Des. 2015, 21, 3158–3166. [Google Scholar] [CrossRef]
- Li, L.; He, H.; Jiang, S.; Qi, J.; Lu, Y.; Ding, N.; Lin, H.S.; Wu, W.; Xiang, X. Simulation of the In Vivo Fate of Polymeric Nanoparticles Traced by Environment-Responsive Near-Infrared Dye: A Physiologically Based Pharmacokinetic Modelling Approach. Molecules 2021, 26, 1271. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, Z.; Xiang, H.; Xu, X.; Zou, J.; Lu, C. Biocompatible Superparamagnetic Europium-Doped Iron Oxide Nanoparticle Clusters as Multifunctional Nanoprobes for Multimodal In Vivo Imaging. ACS Appl. Mater. Interfaces 2021, 13, 33850–33861. [Google Scholar] [CrossRef]
- Fatima, I.; Rahdar, A.; Sargazi, S.; Barani, M.; Hassanisaadi, M.; Thakur, V.K. Quantum Dots: Synthesis, Antibody Conjugation, and HER2-Receptor Targeting for Breast Cancer Therapy. J. Funct. Biomater. 2021, 12, 75. [Google Scholar] [CrossRef]
- Camacho, K.M.; Kumar, S.; Menegatti, S.; Vogus, D.R.; Anselmo, A.C.; Mitragotri, S. Synergistic antitumor activity of camptothecin-doxorubicin combinations and their conjugates with hyaluronic acid. J. Control Release 2015, 210, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Vogus, D.R.; Evans, M.A.; Pusuluri, A.; Barajas, A.; Zhang, M.; Krishnan, V.; Nowak, M.; Menegatti, S.; Helgeson, M.E.; Squires, T.M.; et al. A hyaluronic acid conjugate engineered to synergistically and sequentially deliver gemcitabine and doxorubicin to treat triple negative breast cancer. J. Control Release 2017, 267, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Peng, K.; Sarode, A.; Prakash, S.; Zhao, Z.; Filippov, S.K.; Todorova, K.; Sell, B.R.; Lujano, O.; Bakre, S.; et al. Hyaluronic acid conjugates for topical treatment of skin cancer lesions. Sci. Adv. 2021, 7, eabe6627. [Google Scholar] [CrossRef] [PubMed]
- Tzankov, A.; Pehrs, A.C.; Zimpfer, A.; Ascani, S.; Lugli, A.; Pileri, S.; Dirnhofer, S. Prognostic significance of CD44 expression in diffuse large B cell lymphoma of activated and germinal centre B cell-like types: A tissue microarray analysis of 90 cases. J. Clin. Pathol. 2003, 56, 747–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quere, R.; Andradottir, S.; Brun, A.C.; Zubarev, R.A.; Karlsson, G.; Olsson, K.; Magnusson, M.; Cammenga, J.; Karlsson, S. High levels of the adhesion molecule CD44 on leukemic cells generate acute myeloid leukemia relapse after withdrawal of the initial transforming event. Leukemia 2011, 25, 515–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, V.M.; Szoka, F.C., Jr. Anticancer therapeutics: Targeting macromolecules and nanocarriers to hyaluronan or CD44, a hyaluronan receptor. Mol. Pharm. 2008, 5, 474–486. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, V.; Paul, M.K.; Mukhopadhyay, A.K. 6-mercaptopurine and daunorubicin double drug liposomes-preparation, drug-drug interaction and characterization. J. Liposome Res. 2005, 15, 141–155. [Google Scholar] [CrossRef]
- Harasym, T.O.; Tardi, P.G.; Harasym, N.L.; Harvie, P.; Johnstone, S.A.; Mayer, L.D. Increased preclinical efficacy of irinotecan and floxuridine coencapsulated inside liposomes is associated with tumor delivery of synergistic drug ratios. Oncol. Res. 2007, 16, 361–374. [Google Scholar] [CrossRef]
- Zhang, L.; Radovic-Moreno, A.F.; Alexis, F.; Gu, F.X.; Basto, P.A.; Bagalkot, V.; Jon, S.; Langer, R.S.; Farokhzad, O.C. Co-delivery of hydrophobic and hydrophilic drugs from nanoparticle-aptamer bioconjugates. ChemMedChem 2007, 2, 1268–1271. [Google Scholar] [CrossRef]
- Lammers, T.; Subr, V.; Ulbrich, K.; Peschke, P.; Huber, P.E.; Hennink, W.E.; Storm, G. Simultaneous delivery of doxorubicin and gemcitabine to tumors in vivo using prototypic polymeric drug carriers. Biomaterials 2009, 30, 3466–3475. [Google Scholar] [CrossRef]
- Tekade, R.K.; Dutta, T.; Tyagi, A.; Bharti, A.C.; Das, B.C.; Jain, N.K. Surface-engineered dendrimers for dual drug delivery: A receptor up-regulation and enhanced cancer targeting strategy. J. Drug Target 2008, 16, 758–772. [Google Scholar] [CrossRef] [PubMed]
- Green, J.E.; Hudson, T. The promise of genetically engineered mice for cancer prevention studies. Nat. Rev. Cancer 2005, 5, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Spisek, R.; Charalambous, A.; Mazumder, A.; Vesole, D.H.; Jagannath, S.; Dhodapkar, M.V. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: Therapeutic implications. Blood 2007, 109, 4839–4845. [Google Scholar] [CrossRef] [PubMed]
- Sukkurwala, A.Q.; Adjemian, S.; Senovilla, L.; Michaud, M.; Spaggiari, S.; Vacchelli, E.; Baracco, E.E.; Galluzzi, L.; Zitvogel, L.; Kepp, O.; et al. Screening of novel immunogenic cell death inducers within the NCI Mechanistic Diversity Set. Oncoimmunology 2014, 3, e28473. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Cui, L.; Li, T.; Chen, S.; Song, J.; Li, D. Oxaliplatin induces immunogenic cells death and enhances therapeutic efficacy of checkpoint inhibitor in a model of murine lung carcinoma. J. Recept. Signal Transduct. Res. 2019, 39, 208–214. [Google Scholar] [CrossRef]
- Du, B.; Waxman, D.J. Medium dose intermittent cyclophosphamide induces immunogenic cell death and cancer cell autonomous type I interferon production in glioma models. Cancer Lett. 2020, 470, 170–180. [Google Scholar] [CrossRef]
- Petricoin, E.F., 3rd; Bichsel, V.E.; Calvert, V.S.; Espina, V.; Winters, M.; Young, L.; Belluco, C.; Trock, B.J.; Lippman, M.; Fishman, D.A.; et al. Mapping molecular networks using proteomics: A vision for patient-tailored combination therapy. J. Clin. Oncol. 2005, 23, 3614–3621. [Google Scholar] [CrossRef]
- Ali, M.; Khan, S.A.; Wennerberg, K.; Aittokallio, T. Global proteomics profiling improves drug sensitivity prediction: Results from a multi-omics, pan-cancer modeling approach. Bioinformatics 2018, 34, 1353–1362. [Google Scholar] [CrossRef] [Green Version]
- Frantzi, M.; Latosinska, A.; Mischak, H. Proteomics in Drug Development: The Dawn of a New Era? Proteomics Clin. Appl. 2019, 13, e1800087. [Google Scholar] [CrossRef]
- Li, F.; Jiang, T.; Li, Q.; Ling, X. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: Did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am. J. Cancer Res. 2017, 7, 2350–2394. [Google Scholar] [PubMed]
- Li, J.; Hu, Z.E.; Yang, X.L.; Wu, W.X.; Xing, X.; Gu, B.; Liu, Y.H.; Wang, N.; Yu, X.Q. GSH/pH dual-responsive biodegradable camptothecin polymeric prodrugs combined with doxorubicin for synergistic anticancer efficiency. Biomater. Sci. 2019, 7, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- Pusuluri, A.; Krishnan, V.; Lensch, V.; Sarode, A.; Bunyan, E.; Vogus, D.R.; Menegatti, S.; Soh, H.T.; Mitragotri, S. Treating Tumors at Low Drug Doses Using an Aptamer-Peptide Synergistic Drug Conjugate. Angew. Chem. Int. Ed. Engl. 2019, 58, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J.; Cheshire, P.J.; Myers, L.; Stewart, C.F.; Synold, T.W.; Houghton, J.A. Evaluation of 9-dimethylaminomethyl-10-hydroxycamptothecin against xenografts derived from adult and childhood solid tumors. Cancer Chemother. Pharmacol. 1992, 31, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Husain, I.; Mohler, J.L.; Seigler, H.F.; Besterman, J.M. Elevation of topoisomerase I messenger RNA, protein, and catalytic activity in human tumors: Demonstration of tumor-type specificity and implications for cancer chemotherapy. Cancer Res. 1994, 54, 539–546. [Google Scholar]
- Bronstein, I.B.; Vorobyev, S.; Timofeev, A.; Jolles, C.J.; Alder, S.L.; Holden, J.A. Elevations of DNA topoisomerase I catalytic activity and immunoprotein in human malignancies. Oncol. Res. 1996, 8, 17–25. [Google Scholar]
- Ryan, C.W.; Fleming, G.F.; Janisch, L.; Ratain, M.J. A phase I study of liposomal doxorubicin (Doxil) with topotecan. Am. J. Clin. Oncol. 2000, 23, 297–300. [Google Scholar] [CrossRef]
- Garcia, A.A.; Roman, L.; Muderspach, L.; O’Meara, A.; Facio, G.; Edwards, S.; Burnett, A. Phase I clinical trial of topotecan and pegylated liposomal doxorubicin. Cancer Invest. 2005, 23, 665–670. [Google Scholar] [CrossRef]
- Mirchandani, D.; Hochster, H.; Hamilton, A.; Liebes, L.; Yee, H.; Curtin, J.P.; Lee, S.; Sorich, J.; Dellenbaugh, C.; Muggia, F.M. Phase I study of combined pegylated liposomal doxorubicin with protracted daily topotecan for ovarian cancer. Clin. Cancer Res. 2005, 11, 5912–5919. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, S.; Tsuda, H.; Hashiguchi, Y.; Kokawa, K.; Nishimura, R.; Ishiko, O.; Kamiura, S.; Hasegawa, K.; Umesaki, N. Phase II study of irinotecan plus doxorubicin for early recurrent or platinum-refractory ovarian cancer: Interim analysis. Int. J. Gynecol. Cancer 2007, 17, 159–163. [Google Scholar] [CrossRef]
- Goff, L.W.; Rothenberg, M.L.; Lockhart, A.C.; Roth, B.J.; VerMeulen, W.L.; Chan, E.; Berlin, J.D. A phase I trial of irinotecan alternating with epirubicin in patients with advanced malignancies. Am. J. Clin. Oncol. 2008, 31, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Morgensztern, D.; Baggstrom, M.Q.; Pillot, G.; Tan, B.; Fracasso, P.; Suresh, R.; Wildi, J.; Govindan, R. A phase I study of pegylated liposomal doxorubicin and irinotecan in patients with solid tumors. Chemotherapy 2009, 55, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Takatori, E.; Kaido, Y.; Omi, H.; Yokoyama, Y.; Mizunuma, H.; Kaiho, M.; Otsuki, T.; Takano, T.; Yaegashi, N.; et al. A phase I study of irinotecan and pegylated liposomal doxorubicin in recurrent ovarian cancer (Tohoku Gynecologic Cancer Unit 104 study). Cancer Chemother. Pharmacol. 2014, 73, 895–901. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Kall, L.; Storey, J.D.; Noble, W.S. Non-parametric estimation of posterior error probabilities associated with peptides identified by tandem mass spectrometry. Bioinformatics 2008, 24, i42–i48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandya, U.M.; Egbuta, C.; Abdullah Norman, T.M.; Chiang, C.E.; Wiersma, V.R.; Panchal, R.G.; Bremer, E.; Eggleton, P.; Gold, L.I. The Biophysical Interaction of the Danger-Associated Molecular Pattern (DAMP) Calreticulin with the Pattern-Associated Molecular Pattern (PAMP) Lipopolysaccharide. Int. J. Mol. Sci. 2019, 20, 408. [Google Scholar] [CrossRef] [Green Version]
- Solari, J.I.G.; Filippi-Chiela, E.; Pilar, E.S.; Nunes, V.; Gonzalez, E.A.; Figueiro, F.; Andrade, C.F.; Klamt, F. Damage-associated molecular patterns (DAMPs) related to immunogenic cell death are differentially triggered by clinically relevant chemotherapeutics in lung adenocarcinoma cells. BMC Cancer 2020, 20, 474. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Fucikova, J.; Kralikova, P.; Fialova, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Spisek, R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef] [Green Version]
- Kawano, M.; Tanaka, K.; Itonaga, I.; Iwasaki, T.; Miyazaki, M.; Ikeda, S.; Tsumura, H. Dendritic cells combined with doxorubicin induces immunogenic cell death and exhibits antitumor effects for osteosarcoma. Oncol. Lett. 2016, 11, 2169–2175. [Google Scholar] [CrossRef] [Green Version]
- Aoto, K.; Mimura, K.; Okayama, H.; Saito, M.; Chida, S.; Noda, M.; Nakajima, T.; Saito, K.; Abe, N.; Ohki, S.; et al. Immunogenic tumor cell death induced by chemotherapy in patients with breast cancer and esophageal squamous cell carcinoma. Oncol. Rep. 2018, 39, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlseder, J.; Wissing, D.; Holzer, G.; Orel, L.; Sliutz, G.; Auer, H.; Jaattela, M.; Simon, M.M. HSP70 overexpression mediates the escape of a doxorubicin-induced G2 cell cycle arrest. Biochem. Biophys. Res. Commun. 1996, 220, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Asling, J.; Morrison, J.; Mutsaers, A.J. Targeting HSP70 and GRP78 in canine osteosarcoma cells in combination with doxorubicin chemotherapy. Cell Stress Chaperones 2016, 21, 1065–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerikiotis, S.; Angelidis, C.; Dhima, I.; Naka, K.K.; Kasioumi, P.; Kalfakakou, V.; Peschos, D.; Vezyraki, P. The increased expression of the inducible Hsp70 (HSP70A1A) in serum of patients with heart failure and its protective effect against the cardiotoxic agent doxorubicin. Mol. Cell. Biochem. 2019, 455, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Dastsooz, H.; Cereda, M.; Donna, D.; Oliviero, S. A Comprehensive Bioinformatics Analysis of UBE2C in Cancers. Int. J. Mol. Sci. 2019, 20, 2228. [Google Scholar] [CrossRef] [Green Version]
- Vargas-Rondon, N.; Villegas, V.E.; Rondon-Lagos, M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers 2017, 10, 4. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
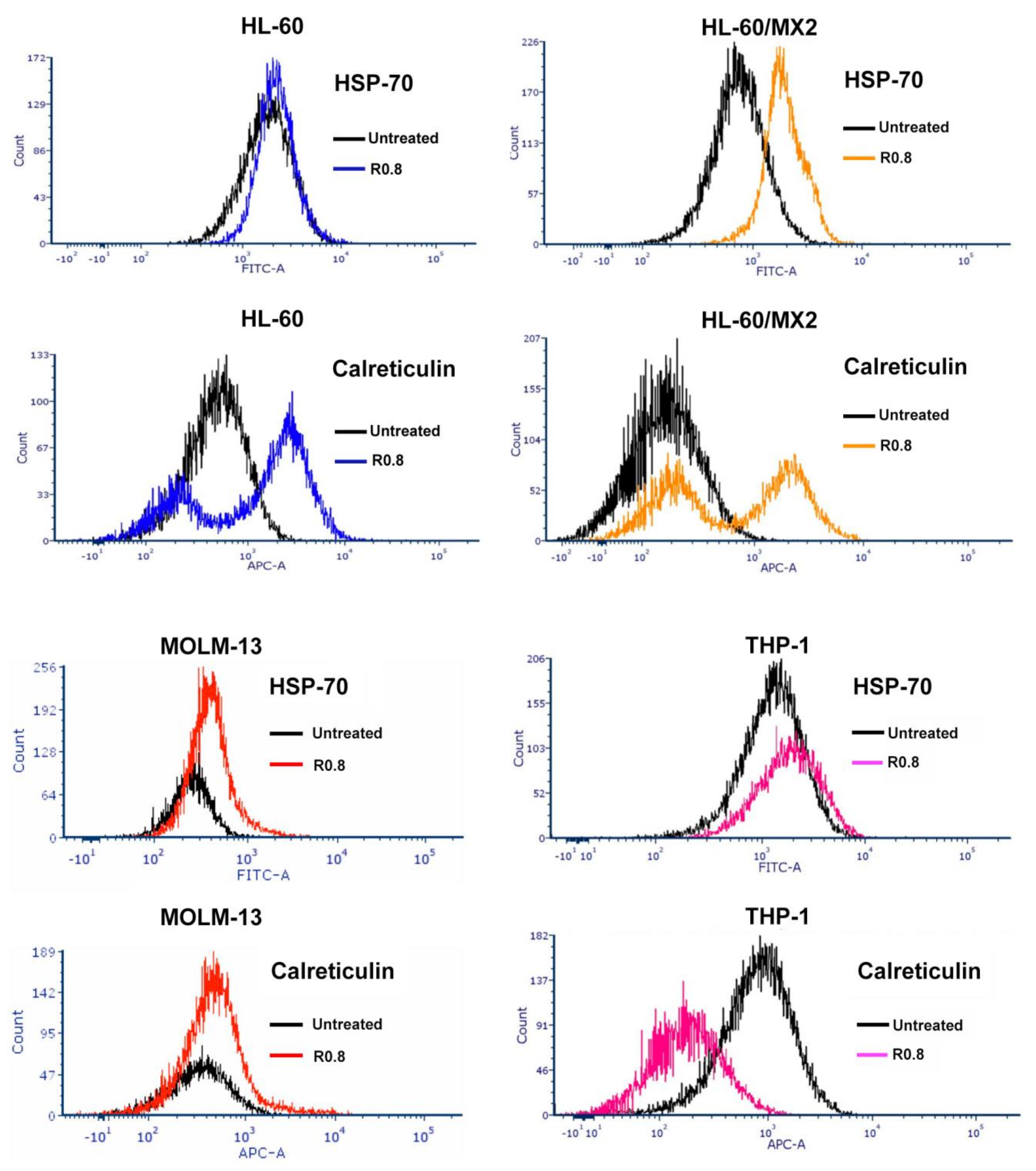
| HA–DOX–CPT R0.8, 48 h | HSP-70 Expression | Calreticulin Expression | ˜CI |
|---|---|---|---|
| HL-60 | increase | increase | 0.3 (synergistic) |
| HL-60/MX2 | increase | increase | 0.09 (synergistic) |
| MOLM-13 | no change | no change | 0.5 (synergistic) |
| THP-1 | increase | decrease | 0.1 (synergistic) |
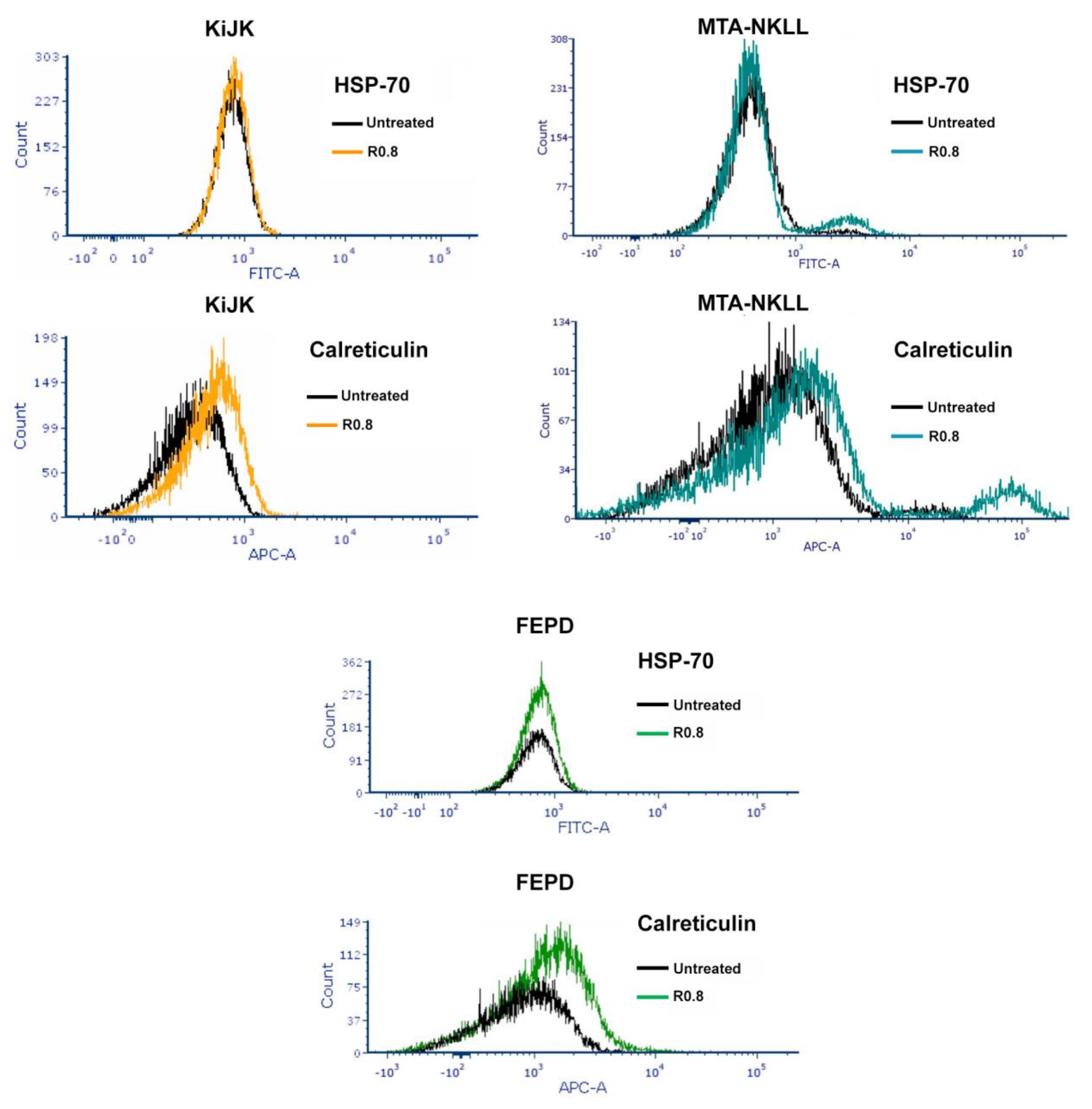
| HA–DOX–CPT R0.8, 48 h | HSP-70 Expression | Calreticulin Expression | ˜CI |
|---|---|---|---|
| KiJK | no change | increase | 0.4 (synergistic) |
| MTA-NKLL | no change | increase | >>>1 (highly antagonistic) |
| FEPD | no change | increase | 1.2 (antagonistic) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishnan, V.; Dharamdasani, V.; Bakre, S.; Dhole, V.; Wu, D.; Budnik, B.; Mitragotri, S. Hyaluronic Acid Nanoparticles for Immunogenic Chemotherapy of Leukemia and T-Cell Lymphoma. Pharmaceutics 2022, 14, 466. https://doi.org/10.3390/pharmaceutics14020466
Krishnan V, Dharamdasani V, Bakre S, Dhole V, Wu D, Budnik B, Mitragotri S. Hyaluronic Acid Nanoparticles for Immunogenic Chemotherapy of Leukemia and T-Cell Lymphoma. Pharmaceutics. 2022; 14(2):466. https://doi.org/10.3390/pharmaceutics14020466
Chicago/Turabian StyleKrishnan, Vinu, Vimisha Dharamdasani, Shirin Bakre, Ved Dhole, Debra Wu, Bogdan Budnik, and Samir Mitragotri. 2022. "Hyaluronic Acid Nanoparticles for Immunogenic Chemotherapy of Leukemia and T-Cell Lymphoma" Pharmaceutics 14, no. 2: 466. https://doi.org/10.3390/pharmaceutics14020466