
NanoSIMS Imaging Reveals the Impact of Ligand-ASO Conjugate Stability on ASO Subcellular Distribution

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mammalian Cell Culture
2.2. Hepatic Spheroid Formation and Maintenance
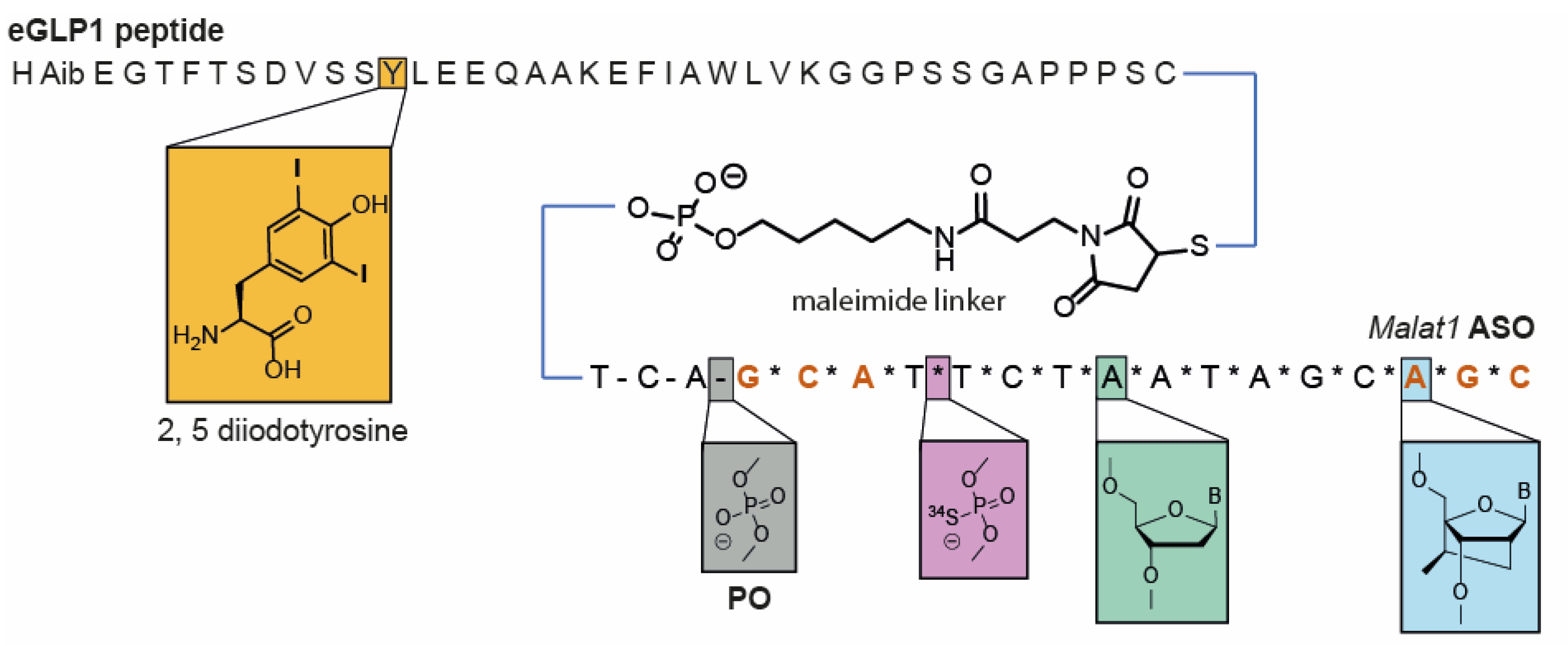
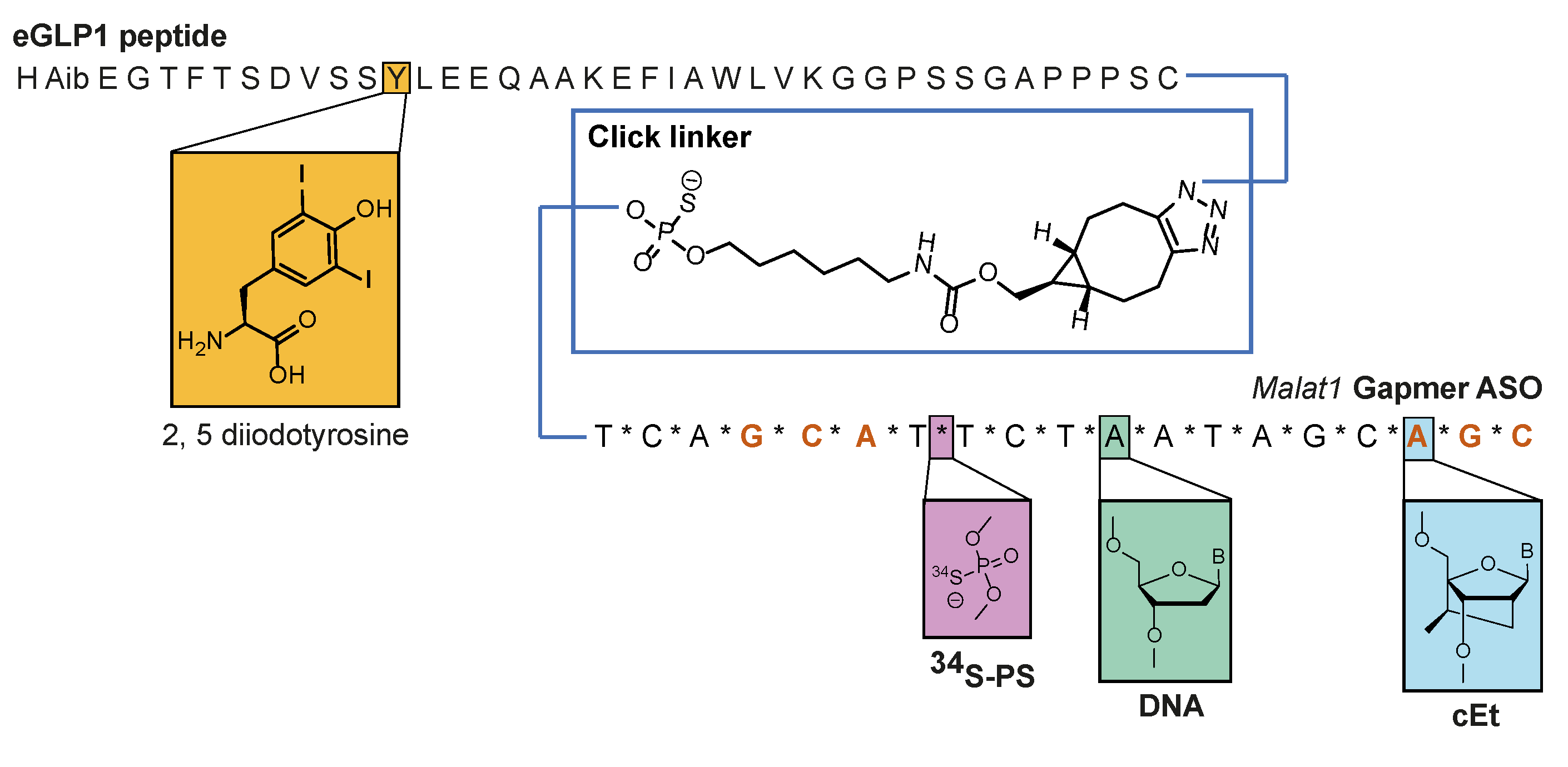
2.3. Synthesis of Peptides, PS-ASOs, and eGLP1-ASO Conjugates
2.3.1. Malat1 PS-ASO Synthesis
2.3.2. GalNAc Conjugate Synthesis
2.3.3. eGLP1 Peptide Synthesis
2.3.4. ASO eGLP1 Peptide Conjugations Using Maleimide
2.3.5. ASO eGLP1 Peptide Conjugation Using Strain-Promoted Azide Alkyne Click Reaction
2.4. GLP1R Internalization DiscoverX Assay
2.5. ASO Incubations
2.6. Immunofluorescence Microscopy
2.7. Quantitative RT-PCR
2.8. Electron Microscopy
2.9. NanoSIMS Imaging
2.10. Image Analysis
2.11. Data Analysis and Statistics
3. Results
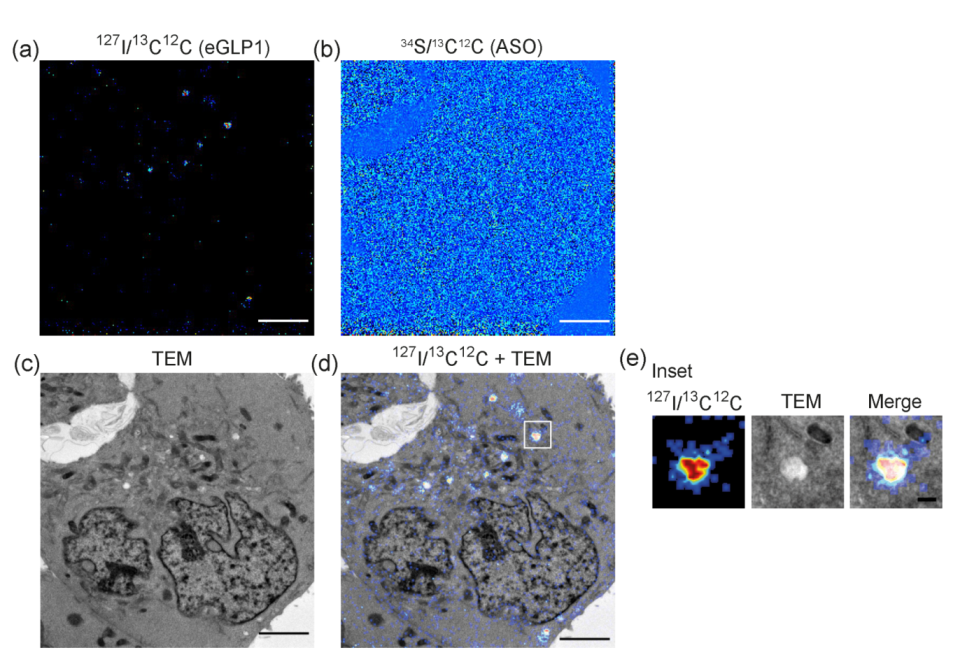
3.1. Detection of Isotopically Labeled ASOs Delivered by Ligand-Mediated Endocytosis
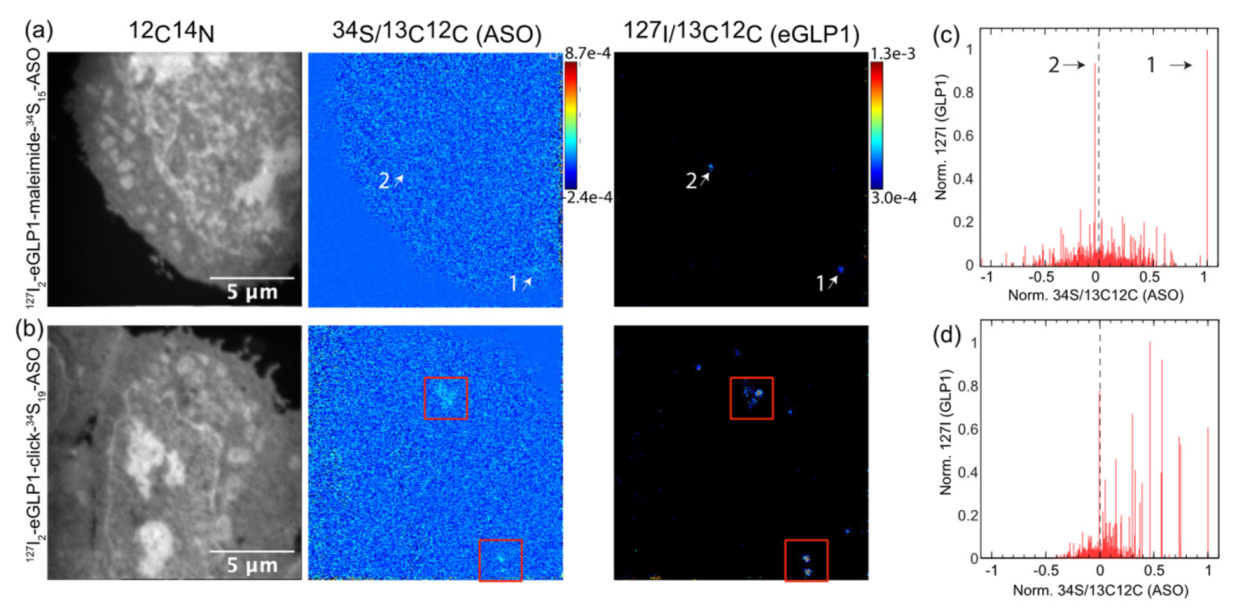
3.2. Dual Labelling to Track Both Targeting Ligand and PS-ASO
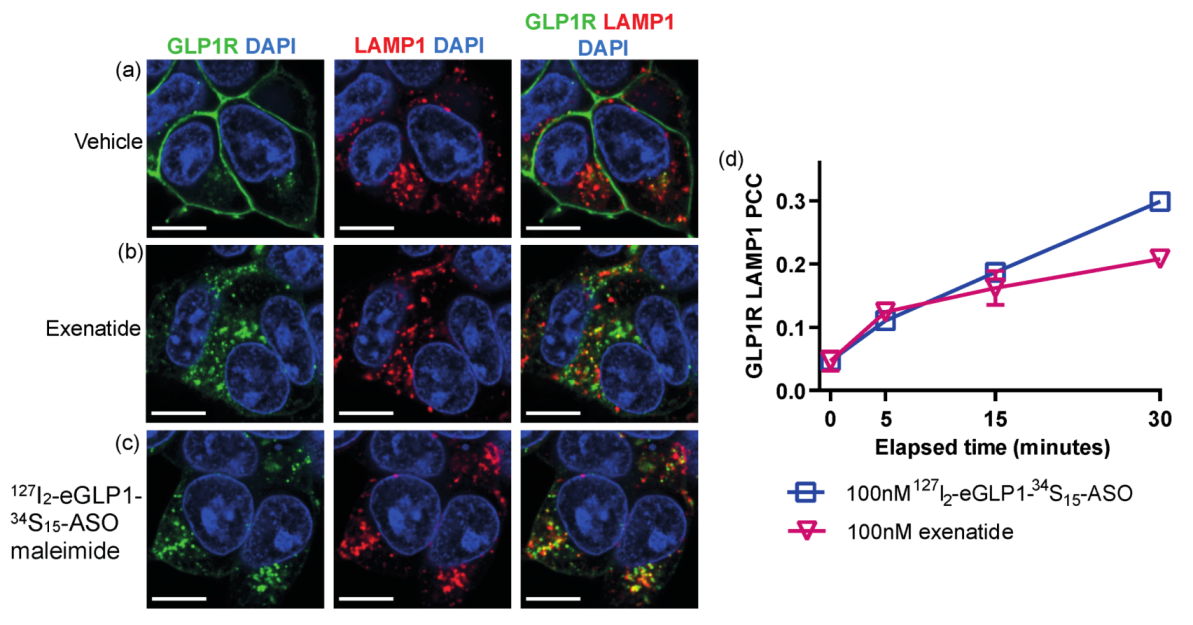
3.3. Identifying a Subcellular Compartment for eGLP1-ASO Localization
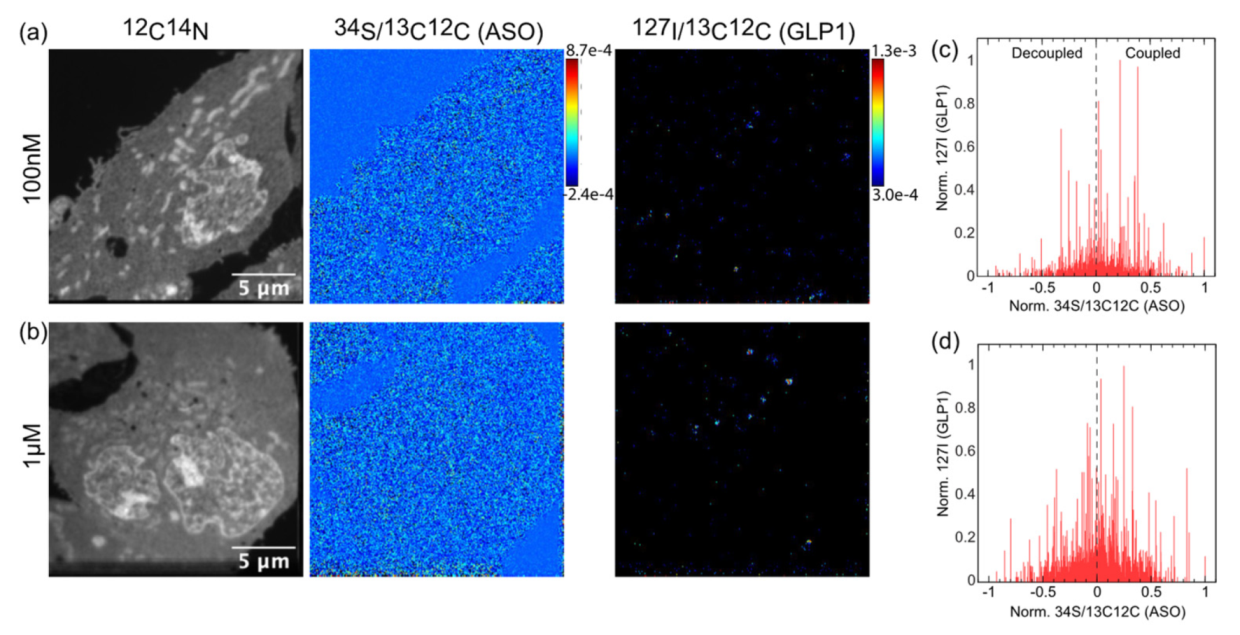
3.4. Quantification of Both Targeting Ligand and Drug Moiety
3.5. Increasing the Stability of the Linker between eGLP1 and ASO Increases Colocalization of Conjugate Components and Reduces RNA Knockdown Efficacy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valeur, E.; Gueret, S.M.; Adihou, H.; Gopalakrishnan, R.; Lemurell, M.; Waldmann, H.; Grossmann, T.N.; Plowright, A.T. New Modalities for Challenging Targets in Drug Discovery. Angew. Chem. Int. Ed. Engl. 2017, 56, 10294–10323. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Pallan, P.S.; Allerson, C.R.; Berdeja, A.; Seth, P.P.; Swayze, E.E.; Prakash, T.P.; Egli, M. Structure and nuclease resistance of 2′,4′-constrained 2′-O-methoxyethyl (cMOE) and 2′-O-ethyl (cEt) modified DNAs. Chem. Commun. 2012, 48, 8195–8197. [Google Scholar] [CrossRef]
- Koshkin, A.A.; Singh, S.K.; Nielsen, P.; Rajwanshi, V.K.; Kumar, R.; Meldgaard, M.; Olsen, C.E.; Wengel, J. LNA (Locked Nucleic Acids): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron 1998, 54, 3607–3630. [Google Scholar] [CrossRef]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Tanowitz, M.; Hettrick, L.; Revenko, A.; Kinberger, G.A.; Prakash, T.P.; Seth, P.P. Asialoglycoprotein receptor 1 mediates productive uptake of N-acetylgalactosamine-conjugated and unconjugated phosphorothioate antisense oligonucleotides into liver hepatocytes. Nucleic Acids Res. 2017, 45, 12388–12400. [Google Scholar] [CrossRef]
- Reed, J.; Bain, S.; Kanamarlapudi, V. Recent advances in understanding the role of glucagon-like peptide 1. F1000Research 2020, 9, (F1000 Faculty Rev):239. [Google Scholar] [CrossRef]
- Ammala, C.; Drury, W.J., 3rd; Knerr, L.; Ahlstedt, I.; Stillemark-Billton, P.; Wennberg-Huldt, C.; Andersson, E.M.; Valeur, E.; Jansson-Lofmark, R.; Janzen, D.; et al. Targeted delivery of antisense oligonucleotides to pancreatic beta-cells. Sci. Adv. 2018, 4, eaat3386. [Google Scholar] [CrossRef] [Green Version]
- Girada, S.B.; Kuna, R.S.; Bele, S.; Zhu, Z.; Chakravarthi, N.R.; DiMarchi, R.D.; Mitra, P. Gαs regulates Glucagon-Like Peptide 1 Receptor-mediated cyclic AMP generation at Rab5 endosomal compartment. Mol. Metab. 2017, 6, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Kuna, R.S.; Girada, S.B.; Asalla, S.; Vallentyne, J.; Maddika, S.; Patterson, J.T.; Smiley, D.L.; DiMarchi, R.D.; Mitra, P. Glucagon-like peptide-1 receptor-mediated endosomal cAMP generation promotes glucose-stimulated insulin secretion in pancreatic β-cells. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E161–E170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roed, S.N.; Wismann, P.; Underwood, C.R.; Kulahin, N.; Iversen, H.; Cappelen, K.A.; Schaffer, L.; Lehtonen, J.; Hecksher-Soerensen, J.; Secher, A.; et al. Real-time trafficking and signaling of the glucagon-like peptide-1 receptor. Mol. Cell Endocrinol. 2014, 382, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.; Buenaventura, T.; Kanda, N.; Chabosseau, P.; Owen, B.M.; Scott, R.; Goldin, R.; Angkathunyakul, N.; Corrêa, I.R., Jr.; Bosco, D.; et al. Targeting GLP-1 receptor trafficking to improve agonist efficacy. Nat. Commun. 2018, 9, 1602. [Google Scholar] [CrossRef]
- Fang, Z.; Chen, S.; Manchanda, Y.; Bitsi, S.; Pickford, P.; David, A.; Shchepinova, M.M.; Corrêa, I.R., Jr.; Hodson, D.J.; Broichhagen, J.; et al. Ligand-Specific Factors Influencing GLP-1 Receptor Post-Endocytic Trafficking and Degradation in Pancreatic Beta Cells. Int. J. Mol. Sci. 2020, 21, 8404. [Google Scholar] [CrossRef]
- Finan, B.; Yang, B.; Ottaway, N.; Stemmer, K.; Müller, T.D.; Yi, C.-X.; Habegger, K.; Schriever, S.C.; García-Cáceres, C.; Kabra, D.G.; et al. Targeted estrogen delivery reverses the metabolic syndrome. Nat. Med. 2012, 18, 1847–1856. [Google Scholar] [CrossRef] [Green Version]
- Knerr, L.; Prakash, T.P.; Lee, R.; Drury, W.J., III; Nikan, M.; Fu, W.; Pirie, E.; Maria, L.D.; Valeur, E.; Hayen, A.; et al. Glucagon Like Peptide 1 Receptor Agonists for Targeted Delivery of Antisense Oligonucleotides to Pancreatic Beta Cell. J. Am. Chem. Soc. 2021, 143, 3416–3429. [Google Scholar] [CrossRef]
- Juliano, R.L. Intracellular Trafficking and Endosomal Release of Oligonucleotides: What We Know and What We Don’t. Nucleic Acid Ther. 2018, 28, 166–177. [Google Scholar] [CrossRef]
- Deprey, K.; Batistatou, N.; Kritzer, J.A. A critical analysis of methods used to investigate the cellular uptake and subcellular localization of RNA therapeutics. Nucleic Acids Res. 2020, 48, 7623–7639. [Google Scholar] [CrossRef]
- Pendergraff, H.; Schmidt, S.; Vikesa, J.; Weile, C.; Overup, C.; Lindholm, M.W.; Koch, T. Nuclear and Cytoplasmatic Quantification of Unconjugated, Label-Free Locked Nucleic Acid Oligonucleotides. Nucleic Acid Ther. 2020, 30, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Thomen, A.; Najafinobar, N.; Penen, F.; Kay, E.; Upadhyay, P.P.; Li, X.; Phan, N.T.N.; Malmberg, P.; Klarqvist, M.; Andersson, S.; et al. Subcellular Mass Spectrometry Imaging and Absolute Quantitative Analysis across Organelles. ACS Nano 2020, 14, 4316–4325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Migawa, M.T.; Chen, K.; Weston, T.A.; Tanowitz, M.; Song, W.; Guagliardo, P.; Iyer, K.S.; Bennett, C.F.; Fong, L.G.; et al. High-resolution visualization and quantification of nucleic acid-based therapeutics in cells and tissues using Nanoscale secondary ion mass spectrometry (NanoSIMS). Nucleic Acids Res. 2021, 49, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Stulz, R.; Meuller, J.; Bazdarevic, D.; Wennberg Huldt, C.; Stromberg, R.; Andersson, S.; Dahlen, A. A Versatile and Convenient Synthesis of 34S-Labeled Phosphorothioate Oligonucleotides. ChemBioChem 2018, 19, 2114–2119. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.B.; Pyke, C.; Rasch, M.G.; Dahl, A.B.; Knudsen, L.B.; Secher, A. Characterization of the Glucagonlike Peptide-1 Receptor in Male Mouse Brain Using a Novel Antibody and In Situ Hybridization. Endocrinology 2018, 159, 665–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogovic, J.A.; Hanslovsky, P.; Wong, A.; Saalfeld, S. Robust registration of calcium images by learned contrast synthesis. In Proceedings of the 2016 IEEE 13th International Symposium on Biomedical Imaging (ISBI), Prague, Czech Republic, 13–16 April 2016; pp. 1123–1126. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Debacker, A.J.; Voutila, J.; Catley, M.; Blakey, D.; Habib, N. Delivery of Oligonucleotides to the Liver with GalNAc: From Research to Registered Therapeutic Drug. Mol. Ther. 2020, 28, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Miki, K.; Kubota, K.; Inoue, Y.; Vera, D.R.; Makuuchi, M. Receptor measurements via Tc-GSA kinetic modeling are proportional to functional hepatocellular mass. J. Nucl. Med. 2001, 42, 733–737. [Google Scholar]
- Mateus, A.; Matsson, P.; Artursson, P. Rapid measurement of intracellular unbound drug concentrations. Mol. Pharm. 2013, 10, 2467–2478. [Google Scholar] [CrossRef]
- Jensen, E.P.; Poulsen, S.S.; Kissow, H.; Holstein-Rathlou, N.H.; Deacon, C.F.; Jensen, B.L.; Holst, J.J.; Sorensen, C.M. Activation of GLP-1 receptors on vascular smooth muscle cells reduces the autoregulatory response in afferent arterioles and increases renal blood flow. Am. J. Physiol. Renal Physiol. 2015, 308, F867–F877. [Google Scholar] [CrossRef] [Green Version]
- Pickford, P.; Lucey, M.; Fang, Z.; Bitsi, S.; de la Serna, J.B.; Broichhagen, J.; Hodson, D.J.; Minnion, J.; Rutter, G.A.; Bloom, S.R.; et al. Signalling, trafficking and glucoregulatory properties of glucagon-like peptide-1 receptor agonists exendin-4 and lixisenatide. Br. J. Pharmacol. 2020, 177, 3905–3923. [Google Scholar] [CrossRef]
- Koller, E.; Vincent, T.M.; Chappell, A.; De, S.; Manoharan, M.; Bennett, C.F. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011, 39, 4795–4807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, M.R.; Ming, X.; Dixit, V.; Fisher, M.; Chen, X.; Juliano, R.L.J.O. The biological effect of an antisense oligonucleotide depends on its route of endocytosis and trafficking. Oligonucleotides 2010, 20, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnane, E.; Davey, P.; Zhang, P.; Puri, S.; Edbrooke, M.; Chiarparin, E.; Revenko, A.S.; Macleod, A.R.; Norman, J.C.; Ross, S.J. Differential uptake, kinetics and mechanisms of intracellular trafficking of next-generation antisense oligonucleotides across human cancer cell lines. Nucleic Acids Res. 2019, 47, 4375–4392. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.; Alam, M.R.; Fisher, M.; Yan, Y.; Chen, X.; Juliano, R.L. Intracellular delivery of an antisense oligonucleotide via endocytosis of a G protein-coupled receptor. Nucleic Acids Res. 2010, 38, 6567–6576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Sun, H.; Tanowitz, M.; Liang, X.-h.; Crooke, S.T. Intra-endosomal trafficking mediated by lysobisphosphatidic acid contributes to intracellular release of phosphorothioate-modified antisense oligonucleotides. Nucleic Acids Res. 2017, 45, 5309–5322. [Google Scholar] [CrossRef]
- Castanotto, D.; Lin, M.; Kowolik, C.; Koch, T.; Hansen, B.R.; Oerum, H.; Stein, C.A. Protein kinase C-α is a critical protein for antisense oligonucleotide-mediated silencing in mammalian cells. Mol. Ther. 2016, 24, 1117–1125. [Google Scholar] [CrossRef] [Green Version]
- Castanotto, D.; Lin, M.; Kowolik, C.; Wang, L.; Ren, X.-Q.; Soifer, H.S.; Koch, T.; Hansen, B.R.; Oerum, H.; Armstrong, B.; et al. A cytoplasmic pathway for gapmer antisense oligonucleotide-mediated gene silencing in mammalian cells. Nucleic Acids Res. 2015, 43, 9350–9361. [Google Scholar] [CrossRef]
- Wang, S.; Allen, N.; Vickers, T.A.; Revenko, A.S.; Sun, H.; Liang, X.-h.; Crooke, S.T. Cellular uptake mediated by epidermal growth factor receptor facilitates the intracellular activity of phosphorothioate-modified antisense oligonucleotides. Nucleic Acids Res. 2018, 46, 3579–3594. [Google Scholar] [CrossRef]
- Wittrup, A.; Ai, A.; Liu, X.; Hamar, P.; Trifonova, R.; Charisse, K.; Manoharan, M.; Kirchhausen, T.; Lieberman, J. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotechnol. 2015, 33, 870–876. [Google Scholar] [CrossRef] [Green Version]
- Du Rietz, H.; Hedlund, H.; Wilhelmson, S.; Nordenfelt, P.; Wittrup, A. Imaging small molecule-induced endosomal escape of siRNA. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kay, E.; Stulz, R.; Becquart, C.; Lovric, J.; Tängemo, C.; Thomen, A.; Baždarević, D.; Najafinobar, N.; Dahlén, A.; Pielach, A.; et al. NanoSIMS Imaging Reveals the Impact of Ligand-ASO Conjugate Stability on ASO Subcellular Distribution. Pharmaceutics 2022, 14, 463. https://doi.org/10.3390/pharmaceutics14020463
Kay E, Stulz R, Becquart C, Lovric J, Tängemo C, Thomen A, Baždarević D, Najafinobar N, Dahlén A, Pielach A, et al. NanoSIMS Imaging Reveals the Impact of Ligand-ASO Conjugate Stability on ASO Subcellular Distribution. Pharmaceutics. 2022; 14(2):463. https://doi.org/10.3390/pharmaceutics14020463
Chicago/Turabian StyleKay, Emma, Rouven Stulz, Cécile Becquart, Jelena Lovric, Carolina Tängemo, Aurélien Thomen, Dženita Baždarević, Neda Najafinobar, Anders Dahlén, Anna Pielach, and et al. 2022. "NanoSIMS Imaging Reveals the Impact of Ligand-ASO Conjugate Stability on ASO Subcellular Distribution" Pharmaceutics 14, no. 2: 463. https://doi.org/10.3390/pharmaceutics14020463