
The Potential of Nanomedicine to Unlock the Limitless Applications of mRNA


Abstract
:1. Prospects and Limitations of mRNA
1.1. Structure and Chemical Modifications of mRNA
2. Delivery Vehicles for mRNA
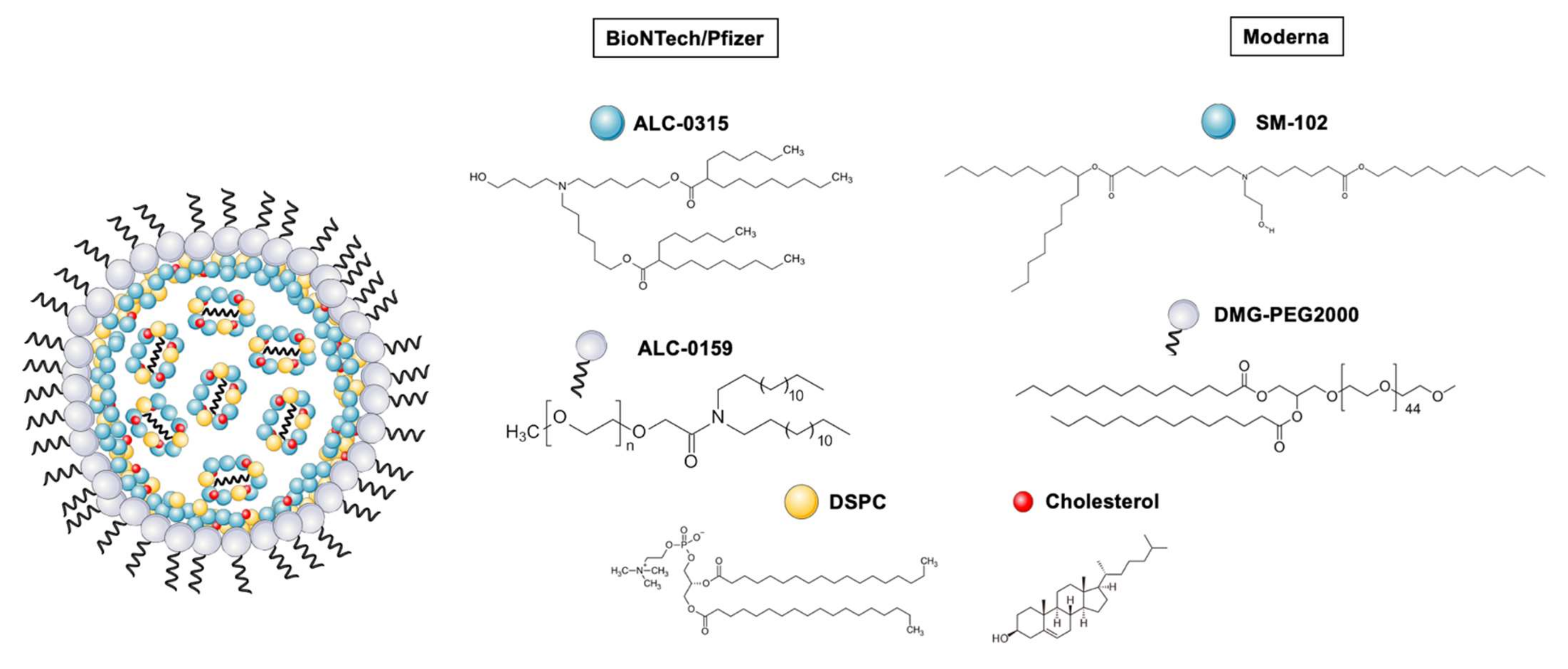
2.1. Lipid Nanoparticles (LNPs), the Delivery Vehicles behind COVID-19 mRNA Vaccines
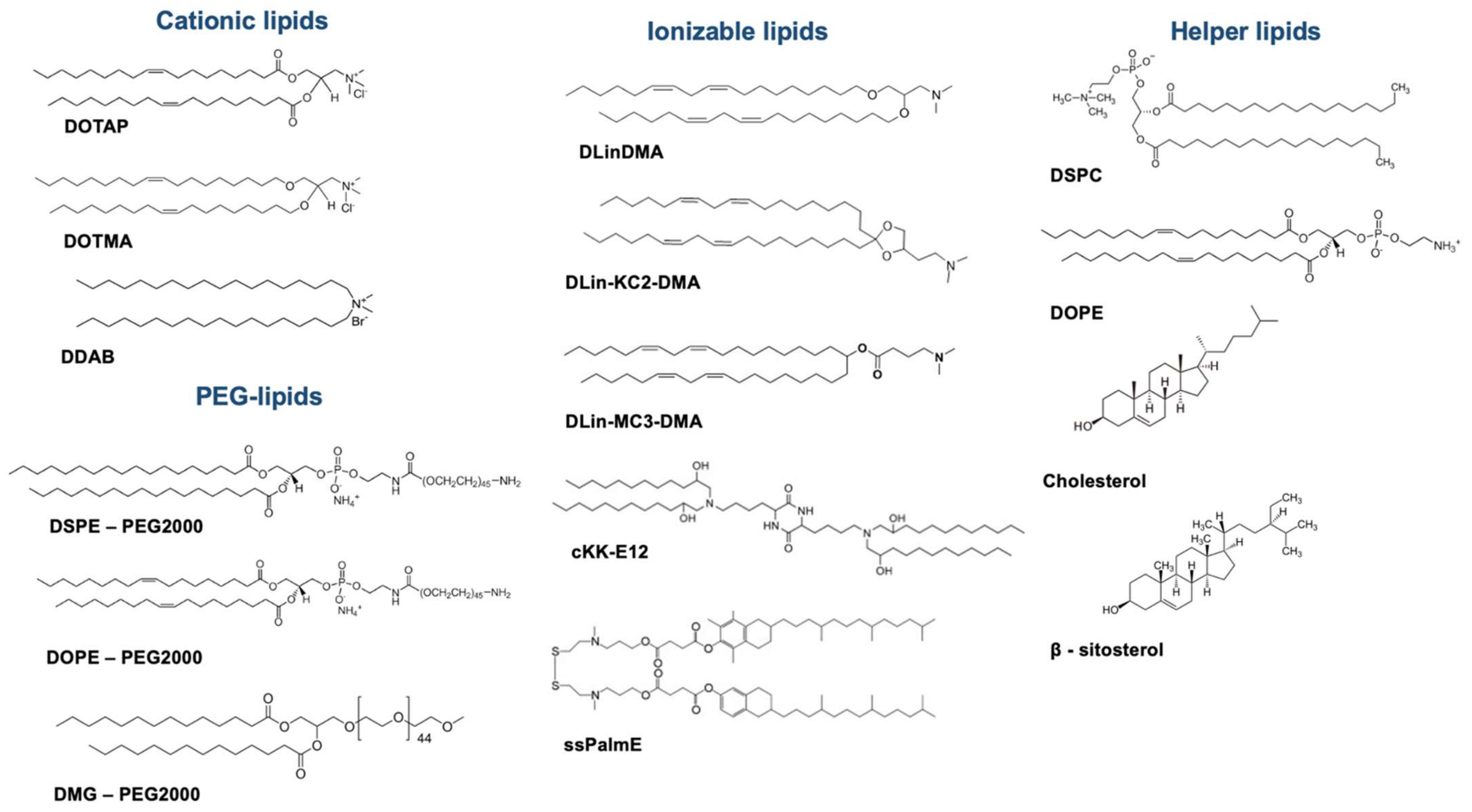
2.1.1. Unraveling LNP Composition
2.1.2. Manufacturing Processes for LNPs
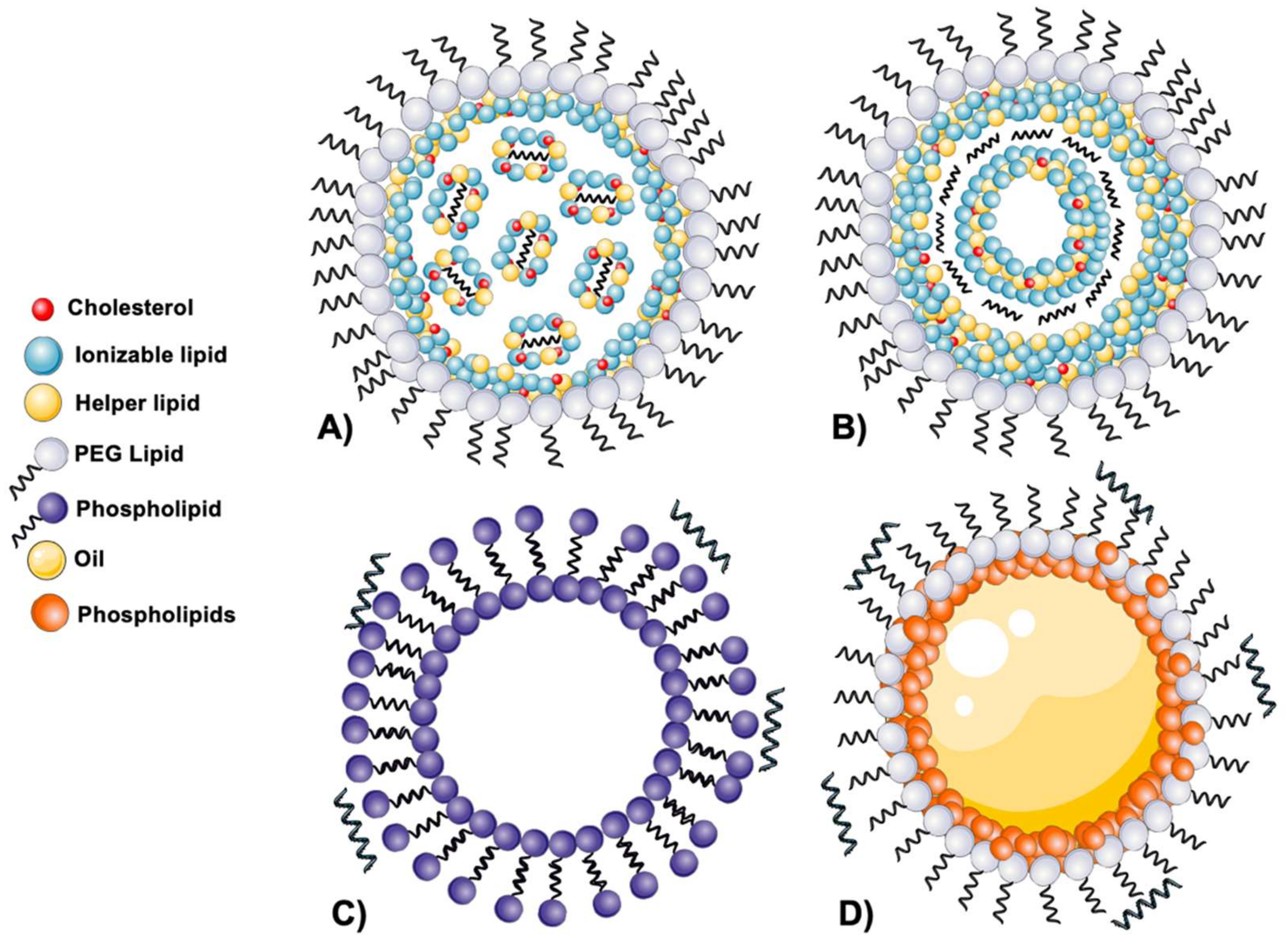
2.1.3. Disclosing the LNPs’ Structure
2.1.4. Stability Considerations
2.2. Additional Lipid-Based Delivery Vehicles for mRNA
2.2.1. Lipoplexes
2.2.2. Cationic Nanoemulsions
2.3. Additional COVID-19 Vaccines Based on mRNA and Delivery Vehicles
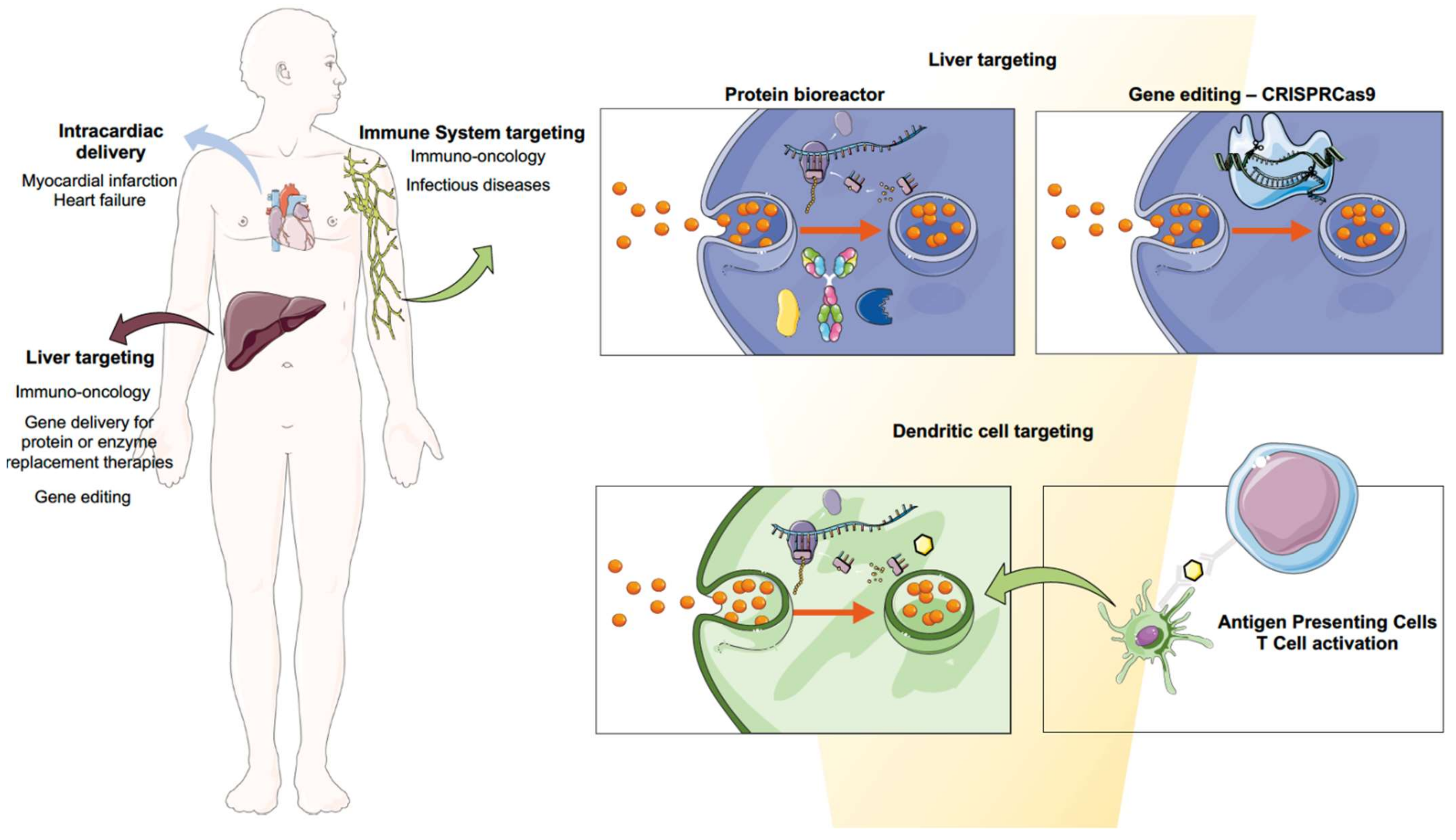
3. Biomedical Applications of mRNA Using Nanomedicine
3.1. Infectious Diseases
3.2. Immuno-Oncology
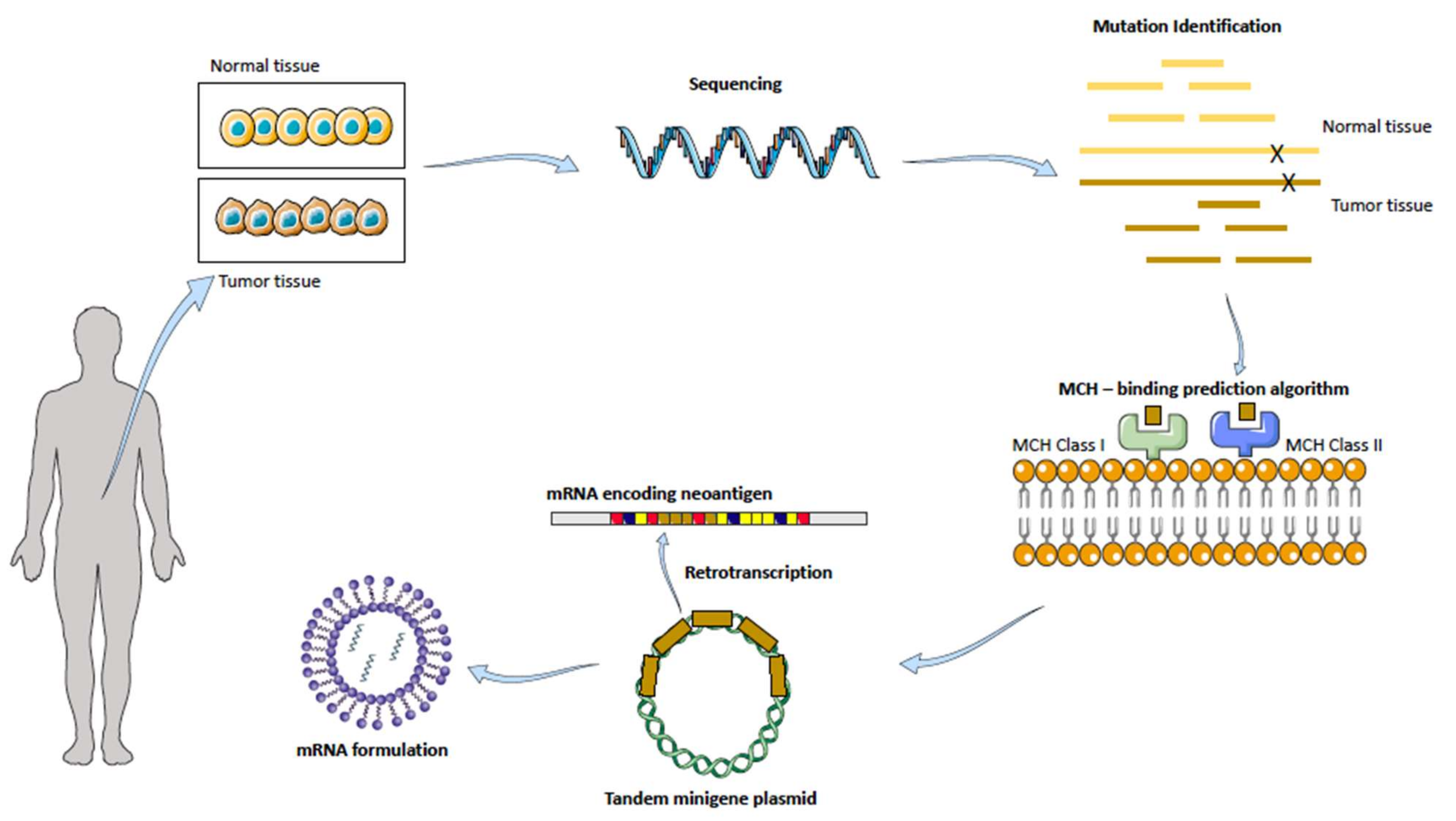
3.2.1. Cancer Vaccines: Targeting Dendritic Cells
3.2.2. The Liver as a Factory of Immunomodulatory Proteins
3.2.3. CAR-T Cells
3.3. RNA-Based Protein Replacement Therapies
3.3.1. Cystic Fibrosis
3.3.2. Rare Metabolic Diseases
Acute Intermittent Porphyria
Propionic Acidaemia (PA)
Fabry Disease
Methylmalonic Acidemia (MMA)
Ornithine Transcarbamylase Deficiency (OTD)
3.4. Gene Editing
3.5. Autoimmune Diseases
3.6. Cardiovascular Diseases
4. Highlights and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gros, F.; Hiatt, H.; Gilbert, W.; Kurland, C.G.; Risebrough, R.W.; Watson, J.D. Unstable Ribonucleic Acid Revealed by Pulse Labelling of Escherichia Coli. Nature 1961, 190, 581–585. [Google Scholar] [CrossRef]
- Brenner, S.; Jacob, F.; Meselson, M. An Unstable Intermediate Carrying Information from Genes to Ribosomes for Protein Synthesis. Nature 1961, 190, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Monod, J. Genetic Regulatory Mechanisms in the Synthesis of Proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. MRNA-Based Therapeutics—Developing a New Class of Drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.; Li, C.; Yang, T.; Hu, B.; Zhang, M.; Guo, S.; Xiao, H.; Liang, X.-J.; Huang, Y. The Challenge and Prospect of MRNA Therapeutics Landscape. Biotechnol. Adv. 2020, 40, 107534. [Google Scholar] [CrossRef]
- Hajj, K.A.; Whitehead, K.A. Tools for Translation: Non-Viral Materials for Therapeutic MRNA Delivery. Nat. Rev. Mater. 2017, 2, 17056. [Google Scholar] [CrossRef]
- Hornung, V.; Barchet, W.; Schlee, M.; Hartmann, G. RNA Recognition via TLR7 and TLR8. In Toll-Like Receptors (TLRs) and Innate Immunity; Stefan, B., Hartmann, G., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 71–86. ISBN 978-3-540-72167-3. [Google Scholar]
- Pogocki, D.; Schöneich, C. Chemical Stability of Nucleic Acid-Derived Drugs. J. Pharm. Sci. 2000, 89, 443–456. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. MRNA Vaccines—A New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic MRNA Delivery. J. Soc. Gene Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [Green Version]
- Blakney, A.K.; Ip, S.; Geall, A.J. An Update on Self-Amplifying MRNA Vaccine Development. Vaccines 2021, 9, 97. [Google Scholar] [CrossRef]
- Ballesteros-Briones, M.C.; Silva-Pilipich, N.; Herrador-Cañete, G.; Vanrell, L.; Smerdou, C. A New Generation of Vaccines Based on Alphavirus Self-Amplifying RNA. Curr. Opin. Virol. 2020, 44, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kiledjian, M. Regulation of MRNA Decapping. WIREs RNA 2010, 1, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; Paoletti, E.; Moss, B. Purification of MRNA Guanylyltransferase and MRNA (Guanine-7-) Methyltransferase from Vaccinia Virions. J. Biol. Chem. 1975, 250, 9322–9329. [Google Scholar] [CrossRef]
- Strenkowska, M.; Grzela, R.; Majewski, M.; Wnek, K.; Kowalska, J.; Lukaszewicz, M.; Zuberek, J.; Darzynkiewicz, E.; Kuhn, A.N.; Sahin, U.; et al. Cap Analogs Modified with 1,2-Dithiodiphosphate Moiety Protect MRNA from Decapping and Enhance Its Translational Potential. Nucleic Acids Res. 2016, 44, 9578–9590. [Google Scholar] [CrossRef] [PubMed]
- Kormann, M.S.D.; Hasenpusch, G.; Aneja, M.K.; Nica, G.; Flemmer, A.W.; Herber-Jonat, S.; Huppmann, M.; Mays, L.E.; Illenyi, M.; Schams, A.; et al. Expression of Therapeutic Proteins after Delivery of Chemically Modified MRNA in Mice. Nat. Biotechnol. 2011, 29, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Mauger, D.M.; Cabral, B.J.; Presnyak, V.; Su, S.V.; Reid, D.W.; Goodman, B.; Link, K.; Khatwani, N.; Reynders, J.; Moore, M.J.; et al. MRNA Structure Regulates Protein Expression through Changes in Functional Half-Life. Proc. Natl. Acad. Sci. USA 2019, 116, 24075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine Into MRNA Yields Superior Nonimmunogenic Vector with Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the Optimal MRNA for Therapy: HPLC Purification Eliminates Immune Activation and Improves Translation of Nucleoside-Modified, Protein-Encoding MRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Muramatsu, H.; Weissman, D.; Karikó, K. In Vitro Transcription of Long RNA Containing Modified Nucleosides. In Synthetic Messenger RNA and Cell Metabolism Modulation; Humana Press: Totowa, NJ, USA, 2013. [Google Scholar]
- Kwon, H.; Kim, M.; Seo, Y.; Moon, Y.S.; Lee, H.J.; Lee, K.; Lee, H. Emergence of Synthetic MRNA: In Vitro Synthesis of MRNA and Its Applications in Regenerative Medicine. Biomaterials 2018, 156, 172–193. [Google Scholar] [CrossRef]
- EMA Spikevax: Summary of Products Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/spikevax-previously-covid-19-vaccine-moderna-epar-product-information_en.pdf (accessed on 16 October 2021).
- EMA Comirnaty: Summary of Products Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/comirnaty-epar-product-information_en.pdf (accessed on 16 October 2021).
- Dolgin, E. CureVac COVID Vaccine Let-down Spotlights MRNA Design Challenges. Nature 2021, 594, 483. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 MRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Moreira, E.D.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Polack, F.P.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 MRNA Covid-19 Vaccine through 6 Months. N. Engl. J. Med. 2021, 385, 1761–1773. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; el Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the MRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Collier, A.Y.; Yu, J.; McMahan, K.; Liu, J.; Chandrashekar, A.; Maron, J.S.; Atyeo, C.; Martinez, D.R.; Ansel, J.L.; Aguayo, R.; et al. Differential Kinetics of Immune Responses Elicited by Covid-19 Vaccines. N. Engl. J. Med. 2021, 385, 2010–2012. [Google Scholar] [CrossRef]
- Walter, E.B.; Talaat, K.R.; Sabharwal, C.; Gurtman, A.; Lockhart, S.; Paulsen, G.C.; Barnett, E.D.; Muñoz, F.M.; Maldonado, Y.; Pahud, B.A.; et al. Evaluation of the BNT162b2 Covid-19 Vaccine in Children 5 to 11 Years of Age. N. Engl. J. Med. 2021, 386, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.; Berman, G.; Zhou, H.; Deng, W.; Faughnan, V.; Coronado-Voges, M.; Ding, B.; Dooley, J.; Girard, B.; Hillebrand, W.; et al. Evaluation of MRNA-1273 SARS-CoV-2 Vaccine in Adolescents. N. Engl. J. Med. 2021, 385, 2241–2251. [Google Scholar] [CrossRef]
- Frenck, R.W.; Klein, N.P.; Kitchin, N.; Gurtman, A.; Absalon, J.; Lockhart, S.; Perez, J.L.; Walter, E.B.; Senders, S.; Bailey, R.; et al. Safety, Immunogenicity, and Efficacy of the BNT162b2 Covid-19 Vaccine in Adolescents. N. Engl. J. Med. 2021, 385, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.J.W.; Jewell, N.P. Vaccine Effectiveness Studies in the Field. N. Engl. J. Med. 2021, 385, 650–651. [Google Scholar] [CrossRef]
- Wu, K.; Werner, A.P.; Koch, M.; Choi, A.; Narayanan, E.; Stewart-Jones, G.; Colpitts, T.; Bennett, H.; Boyoglu-Barnum, S.; Shi, W.; et al. Serum Neutralizing Activity Elicited by MRNA-1273 Vaccine. N. Engl. J. Med. 2021, 384, 1468–1470. [Google Scholar] [CrossRef]
- Abu-Raddad, L.; Chemaitelly, H.; Butt, A.A. Effectiveness of the BNT162b2 Covid-19 Vaccine against the B.1.1.7 and B.1.351 Variants. N. Engl. J. Med. 2021, 385, 187–189. [Google Scholar] [CrossRef]
- Gardner, B.J.; Kilpatrick, A.M. Estimates of Reduced Vaccine Effectiveness against Hospitalization, Infection, Transmission and Symptomatic Disease of a New SARS-CoV-2 Variant, Omicron (B.1.1.529), Using Neutralizing Antibody Titers. medRxiv 2021. [Google Scholar] [CrossRef]
- Rössler, A.; Riepler, L.; Bante, D.; von Laer, D.; Kimpel, J. SARS-CoV-2 Omicron Variant Neutralization in Serum from Vaccinated and Convalescent Persons. N. Engl. J. Med. 2022, 386, 698–700. [Google Scholar] [CrossRef]
- Guerrera, G.; Picozza, M.; D’Orso, S.; Placido, R.; Pirronello, M.; Verdiani, A.; Termine, A.; Fabrizio, C.; Giannessi, F.; Sambucci, M.; et al. BNT162b2 Vaccination Induces Durable SARS-CoV-2–Specific T Cells with a Stem Cell Memory Phenotype. Sci. Immunol. 2021, 6, eabl5344. [Google Scholar] [CrossRef]
- Martins, J.P.; das Neves, J.; de la Fuente, M.; Celia, C.; Florindo, H.; Günday-Türeli, N.; Popat, A.; Santos, J.L.; Sousa, F.; Schmid, R.; et al. The Solid Progress of Nanomedicine. Drug Deliv. Transl. Res. 2020, 10, 726–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the Clinic: An Update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paliwal, S.R.; Kenwat, R.; Maiti, S.; Paliwal, R. Nanotheranostics for Cancer Therapy and Detection: State of the Art. Curr. Pharm. Des. 2020, 26, 5503–5517. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The Current Landscape of Nucleic Acid Therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of Univalent Ions across the Lamellae of Swollen Phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Gregoriadis, G. Drug Entrapment in Liposomes. FEBS Lett. 1973, 36, 292–296. [Google Scholar] [CrossRef] [Green Version]
- Gregoriadis, G. The Carrier Potential of Liposomes in Biology and Medicine (Second of Two Parts). N. Engl. J. Med. 1976, 295, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Gregoriadis, G.; Ryman, B.E. Liposomes as Carriers of Enzymes or Drugs: A New Approach to the Treatment of Storage Diseases. Biochem. J. 1971, 124, 58P. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, T.M.; Cullis, P.R. Liposomal Drug Delivery Systems: From Concept to Clinical Applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J.A. MRNA-Lipid Nanoparticle COVID-19 Vaccines: Structure and Stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of Cationic Lipids and Cationic Polymers in Gene Delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid Nanoparticles for Nucleic Acid Delivery: Current Perspectives. Adv. Drug Deliv. Rev. 2020, 154–155, 37–63. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef] [Green Version]
- Heyes, J.; Palmer, L.; Bremner, K.; MacLachlan, I. Cationic Lipid Saturation Influences Intracellular Delivery of Encapsulated Nucleic Acids. J. Control. Release Off. J. Control. Release Soc. 2005, 107, 276–287. [Google Scholar] [CrossRef]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational Design of Cationic Lipids for SiRNA Delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- Zimmermann, T.S.; Lee, A.C.H.; Akinc, A.; Bramlage, B.; Bumcrot, D.; Fedoruk, M.N.; Harborth, J.; Heyes, J.A.; Jeffs, L.B.; John, M.; et al. RNAi-Mediated Gene Silencing in Non-Human Primates. Nature 2006, 441, 111–114. [Google Scholar] [CrossRef]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the Potency of SiRNA Lipid Nanoparticles for Hepatic Gene Silencing in Vivo. Angew. Chem. 2012, 51, 8529–8533. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro Story and the Clinical Translation of Nanomedicines Containing Nucleic Acid-Based Drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Francia, V.; Schiffelers, R.M.; Cullis, P.R.; Witzigmann, D. The Biomolecular Corona of Lipid Nanoparticles for Gene Therapy. Bioconjug. Chem. 2020, 31, 2046–2059. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted Delivery of RNAi Therapeutics With Endogenous and Exogenous Ligand-Based Mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R.; et al. Efficiency of SiRNA Delivery by Lipid Nanoparticles Is Limited by Endocytic Recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastiani, F.; Arteta, M.Y.; Lerche, M.; Porcar, L.; Lang, C.; Bragg, R.A.; Elmore, C.S.; Krishnamurthy, V.R.; Russell, R.A.; Darwish, T.; et al. Apolipoprotein E Binding Drives Structural and Compositional Rearrangement of MRNA-Containing Lipid Nanoparticles. ACS Nano 2021, 15, 6709–6722. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.J.; Peel, S.E.; Schantz, A.; England, R.M.; Beano, M.; Bates, S.M.; Desai, A.S.; Puri, S.; Ashford, M.B.; Jones, A.T. Endocytic Profiling of Cancer Cell Models Reveals Critical Factors Influencing LNP-Mediated MRNA Delivery and Protein Expression. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 1950–1962. [Google Scholar] [CrossRef]
- Maugeri, M.; Nawaz, M.; Papadimitriou, A.; Angerfors, A.; Camponeschi, A.; Na, M.; Hölttä, M.; Skantze, P.; Johansson, S.; Sundqvist, M.; et al. Linkage between Endosomal Escape of LNP-MRNA and Loading into EVs for Transport to Other Cells. Nat. Commun. 2019, 10, 4333. [Google Scholar] [CrossRef] [Green Version]
- Akinc, A.; Zumbuehl, A.; Goldberg, M.; Leshchiner, E.S.; Busini, V.; Hossain, N.; Bacallado, S.A.; Nguyen, D.N.; Fuller, J.; Alvarez, R.; et al. A Combinatorial Library of Lipid-like Materials for Delivery of RNAi Therapeutics. Nat. Biotechnol. 2008, 26, 561–569. [Google Scholar] [CrossRef]
- Dong, Y.; Love, K.T.; Dorkin, J.R.; Sirirungruang, S.; Zhang, Y.; Chen, D.; Bogorad, R.L.; Yin, H.; Chen, Y.; Vegas, A.J.; et al. Lipopeptide Nanoparticles for Potent and Selective SiRNA Delivery in Rodents and Nonhuman Primates. Proc. Natl. Acad. Sci. USA 2014, 111, 3955–3960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, M.A.; Jayaraman, M.; Matsuda, S.; Liu, J.; Barros, S.; Querbes, W.; Tam, Y.K.; Ansell, S.M.; Kumar, V.; Qin, J.; et al. Biodegradable Lipids Enabling Rapidly Eliminated Lipid Nanoparticles for Systemic Delivery of RNAi Therapeutics. Mol. Ther. 2013, 21, 1570–1578. [Google Scholar] [CrossRef] [Green Version]
- Shirazi, R.S.; Ewert, K.K.; Leal, C.; Majzoub, R.N.; Bouxsein, N.F.; Safinya, C.R. Synthesis and Characterization of Degradable Multivalent Cationic Lipids with Disulfide-Bond Spacers for Gene Delivery. Biochim. Biophys. Acta (BBA) Biomembr. 2011, 1808, 2156–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akita, H.; Noguchi, Y.; Hatakeyama, H.; Sato, Y.; Tange, K.; Nakai, Y.; Harashima, H. Molecular Tuning of a Vitamin E-Scaffold PH-Sensitive and Reductive Cleavable Lipid-like Material for Accelerated in Vivo Hepatic SiRNA Delivery. ACS Biomater. Sci. Eng. 2015, 1, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Leung, J.; Tam, Y.Y.C.; Cullis, P.R. On the Role of Helper Lipids in Lipid Nanoparticle Formulations of SiRNA. Nanoscale 2019, 11, 21733–21739. [Google Scholar] [CrossRef]
- Kauffman, K.J.; Dorkin, J.R.; Yang, J.H.; Heartlein, M.W.; DeRosa, F.; Mir, F.F.; Fenton, O.S.; Anderson, D.G. Optimization of Lipid Nanoparticle Formulations for MRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. [Google Scholar] [CrossRef]
- Zhang, R.; El-Mayta, R.; Murdoch, T.J.; Warzecha, C.C.; Billingsley, M.M.; Shepherd, S.J.; Gong, N.; Wang, L.; Wilson, J.M.; Lee, D.; et al. Helper Lipid Structure Influences Protein Adsorption and Delivery of Lipid Nanoparticles to Spleen and Liver. Biomater. Sci. 2021, 9, 1449–1463. [Google Scholar] [CrossRef]
- Pozzi, D.; Marchini, C.; Cardarelli, F.; Amenitsch, H.; Garulli, C.; Bifone, A.; Caracciolo, G. Transfection Efficiency Boost of Cholesterol-Containing Lipoplexes. Biochim. Biophys. Acta 2012, 1818, 2335–2343. [Google Scholar] [CrossRef] [Green Version]
- Tenchov, B.G.; MacDonald, R.C.; Siegel, D.P. Cubic Phases in Phosphatidylcholine-Cholesterol Mixtures: Cholesterol as Membrane “Fusogen”. Biophys. J. 2006, 91, 2508–2516. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Ashwanikumar, N.; Robinson, E.; Xia, Y.; Mihai, C.; Griffith, J.P.; Hou, S.; Esposito, A.A.; Ketova, T.; Welsher, K.; et al. Naturally-Occurring Cholesterol Analogues in Lipid Nanoparticles Induce Polymorphic Shape and Enhance Intracellular Delivery of MRNA. Nat. Commun. 2020, 11, 983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambegia, E.; Ansell, S.; Cullis, P.; Heyes, J.; Palmer, L.; MacLachlan, I. Stabilized Plasmid-Lipid Particles Containing PEG-Diacylglycerols Exhibit Extended Circulation Lifetimes and Tumor Selective Gene Expression. Biochim. Biophys. Acta 2005, 1669, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Tam, P.; Monck, M.; Lee, D.; Ludkovski, O.; Leng, E.C.; Clow, K.; Stark, H.; Scherrer, P.; Graham, R.W.; Cullis, P.R. Stabilized Plasmid-Lipid Particles for Systemic Gene Therapy. Gene Ther. 2000, 7, 1867–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.Y.; Ahkong, Q.F.; Rong, Q.; Wang, Z.; Ansell, S.; Hope, M.J.; Mui, B. Characterization of the Inhibitory Effect of PEG-Lipid Conjugates on the Intracellular Delivery of Plasmid and Antisense DNA Mediated by Cationic Lipid Liposomes. Biochim. Biophys. Acta 2002, 1558, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Mui, B.L.; Tam, Y.K.; Jayaraman, M.; Ansell, S.M.; Du, X.; Tam, Y.Y.; Lin, P.J.; Chen, S.; Narayanannair, J.K.; Rajeev, K.G.; et al. Influence of Polyethylene Glycol Lipid Desorption Rates on Pharmacokinetics and Pharmacodynamics of SiRNA Lipid Nanoparticles. Mol. Ther. Nucleic Acids 2013, 2, e139. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective Organ Targeting (SORT) Nanoparticles for Tissue-Specific MRNA Delivery and CRISPR–Cas Gene Editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef]
- Goswami, R.; Chatzikleanthous, D.; Lou, G.; Giusti, F.; Bonci, A.; Taccone, M.; Brazzoli, M.; Gallorini, S.; Ferlenghi, I.; Berti, F.; et al. Mannosylation of LNP Results in Improved Potency for Self-Amplifying RNA (SAM) Vaccines. ACS Infect. Dis. 2019, 5, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; O’Hagan, D.T.; Adamo, R.; Baudner, B.C. Conjugation of Mannans to Enhance the Potency of Liposome Nanoparticles for the Delivery of RNA Vaccines. Pharmaceutics 2021, 13, 240. [Google Scholar] [CrossRef]
- Belliveau, N.M.; Huft, J.; Lin, P.J.; Chen, S.; Leung, A.K.; Leaver, T.J.; Wild, A.W.; Lee, J.B.; Taylor, R.J.; Tam, Y.K.; et al. Microfluidic Synthesis of Highly Potent Limit-Size Lipid Nanoparticles for In Vivo Delivery of SiRNA. Mol. Ther. Nucleic Acids 2012, 1, e37. [Google Scholar] [CrossRef]
- Leung, A.K.K.; Hafez, I.M.; Baoukina, S.; Belliveau, N.M.; Zhigaltsev, I.V.; Afshinmanesh, E.; Tieleman, D.P.; Hansen, C.L.; Hope, M.J.; Cullis, P.R. Lipid Nanoparticles Containing SiRNA Synthesized by Microfluidic Mixing Exhibit an Electron-Dense Nanostructured Core. J. Phys. Chem. C 2012, 116. [Google Scholar] [CrossRef]
- Roces, C.B.; Lou, G.; Jain, N.; Abraham, S.; Thomas, A.; Halbert, G.W.; Perrie, Y. Manufacturing Considerations for the Development of Lipid Nanoparticles Using Microfluidics. Pharmaceutics 2020, 12, 1095. [Google Scholar] [CrossRef]
- Terada, T.; Kulkarni, J.A.; Huynh, A.; Chen, S.; van der Meel, R.; Tam, Y.Y.C.; Cullis, P.R. Characterization of Lipid Nanoparticles Containing Ionizable Cationic Lipids Using Design-of-Experiments Approach. Langmuir 2021, 37, 1120–1128. [Google Scholar] [CrossRef]
- Viger-Gravel, J.; Schantz, A.; Pinon, A.C.; Rossini, A.J.; Schantz, S.; Emsley, L. Structure of Lipid Nanoparticles Containing SiRNA or MRNA by Dynamic Nuclear Polarization-Enhanced NMR Spectroscopy. J. Phys. Chem. B 2018, 122, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Darjuan, M.M.; Mercer, J.E.; Chen, S.; van der Meel, R.; Thewalt, J.L.; Tam, Y.Y.C.; Cullis, P.R. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and SiRNA. ACS Nano 2018, 12, 4787–4795. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, J.A.; Witzigmann, D.; Leung, J.; van der Meel, R.; Zaifman, J.; Darjuan, M.M.; Grisch-Chan, H.M.; Thöny, B.; Tam, Y.Y.C.; Cullis, P.R. Fusion-Dependent Formation of Lipid Nanoparticles Containing Macromolecular Payloads. Nanoscale 2019, 11, 9023–9031. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Thomson, S.B.; Zaifman, J.; Leung, J.; Wagner, P.K.; Hill, A.; Tam, Y.Y.C.; Cullis, P.R.; Petkau, T.L.; Leavitt, B.R. Spontaneous, Solvent-Free Entrapment of SiRNA within Lipid Nanoparticles. Nanoscale 2020, 12, 23959–23966. [Google Scholar] [CrossRef] [PubMed]
- Arteta, M.Y.; Kjellman, T.; Bartesaghi, S.; Wallin, S.; Wu, X.; Kvist, A.J.; Dabkowska, A.; Székely, N.; Radulescu, A.; Bergenholtz, J.; et al. Successful Reprogramming of Cellular Protein Production through MRNA Delivered by Functionalized Lipid Nanoparticles. Proc. Natl. Acad. Sci. USA 2018, 115, E3351. [Google Scholar] [CrossRef] [Green Version]
- Eygeris, Y.; Patel, S.; Jozic, A.; Sahay, G. Deconvoluting Lipid Nanoparticle Structure for Messenger RNA Delivery. Nano Lett. 2020, 20, 4543–4549. [Google Scholar] [CrossRef]
- Blakney, A.K.; McKay, P.F.; Yus, B.I.; Aldon, Y.; Shattock, R.J. Inside out: Optimization of Lipid Nanoparticle Formulations for Exterior Complexation and in Vivo Delivery of SaRNA. Gene Ther. 2019, 26, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Ball, R.; Bajaj, P.; Whitehead, K. Achieving Long-Term Stability of Lipid Nanoparticles: Examining the Effect of PH, Temperature, and Lyophilization. Int. J. Nanomed. 2016, 12, 305. [Google Scholar] [CrossRef] [Green Version]
- Packer, M.; Gyawali, D.; Yerabolu, R.; Schariter, J.; White, P. A Novel Mechanism for the Loss of MRNA Activity in Lipid Nanoparticle Delivery Systems. Nat. Commun. 2021, 12, 6777. [Google Scholar] [CrossRef] [PubMed]
- EMA Onpattro: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/onpattro-epar-product-information_en.pdf (accessed on 16 October 2021).
- Ulkoski, D.; Bak, A.; Wilson, J.T.; Krishnamurthy, V.R. Recent Advances in Polymeric Materials for the Delivery of RNA Therapeutics. Expert. Opin. Drug Deliv. 2019, 16, 1149–1167. [Google Scholar] [CrossRef]
- Mendes, L.P.; Pan, J.; Torchilin, V.P. Dendrimers as Nanocarriers for Nucleic Acid and Drug Delivery in Cancer Therapy. Molecules 2017, 22, 1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DIMITRIADIS, G.J. Translation of Rabbit Globin MRNA Introduced by Liposomes into Mouse Lymphocytes. Nature 1978, 274, 923–924. [Google Scholar] [CrossRef] [PubMed]
- Ostro, M.J.; Giacomoni, D.; Lavelle, D.; Paxton, W.; Dray, S. Evidence for Translation of Rabbit Globin MRNA after Liposomemediated Insertion into a Human Cell Line. Nature 1978, 274, 921–923. [Google Scholar] [CrossRef]
- Tateshita, N.; Miura, N.; Tanaka, H.; Masuda, T.; Ohtsuki, S.; Tange, K.; Nakai, Y.; Yoshioka, H.; Akita, H. Development of a Lipoplex-Type MRNA Carrier Composed of an Ionizable Lipid with a Vitamin E Scaffold and the KALA Peptide for Use as an Ex Vivo Dendritic Cell-Based Cancer Vaccine. J. Control. Release 2019, 310, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Zhang, X.; Men, K.; Gao, Y.; Yang, X.; Wu, S.; Duan, X.; Wei, Y.; Tong, R. Efficient Colorectal Cancer Gene Therapy with IL-15 MRNA Nanoformulation. Mol. Pharm. 2020, 17, 3378–3391. [Google Scholar] [CrossRef]
- Zhang, X.; Men, K.; Zhang, Y.; Zhang, R.; Yang, L.; Duan, X. Local and Systemic Delivery of MRNA Encoding Survivin-T34A by Lipoplex for Efficient Colon Cancer Gene Therapy. Int. J. Nanomed. 2019, 14, 2733–2751. [Google Scholar] [CrossRef] [Green Version]
- Blakney, A.K.; Deletic, P.; McKay, P.F.; Bouton, C.R.; Ashford, M.; Shattock, R.J.; Sabirsh, A. Effect of Complexing Lipids on Cellular Uptake and Expression of Messenger RNA in Human Skin Explants. J. Control. Release Off. J. Control. Release Soc. 2021, 330, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Ziller, A.; Nogueira, S.S.; Hühn, E.; Funari, S.S.; Brezesinski, G.; Hartmann, H.; Sahin, U.; Haas, H.; Langguth, P. Incorporation of MRNA in Lamellar Lipid Matrices for Parenteral Administration. Mol. Pharm. 2018, 15, 642–651. [Google Scholar] [CrossRef] [Green Version]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA Delivery to Dendritic Cells Exploits Antiviral Defence for Cancer Immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Michel, T.; Luft, D.; Abraham, M.-K.; Reinhardt, S.; Medina, M.L.S.; Kurz, J.; Schaller, M.; Avci-Adali, M.; Schlensak, C.; Peter, K.; et al. Cationic Nanoliposomes Meet MRNA: Efficient Delivery of Modified MRNA Using Hemocompatible and Stable Vectors for Therapeutic Applications. Mol. Ther. Nucleic Acids 2017, 8, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Hoecke, L.; Roose, K.; Ballegeer, M.; Zhong, Z.; Sanders, N.N.; de Koker, S.; Saelens, X.; van Lint, S. The Opposing Effect of Type I IFN on the T Cell Response by Non-Modified MRNA-Lipoplex Vaccines Is Determined by the Route of Administration. Mol. Ther. Nucleic Acids 2020, 22, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Salomon, N.; Vascotto, F.; Selmi, A.; Vormehr, M.; Quinkhardt, J.; Bukur, T.; Schrörs, B.; Löewer, M.; Diken, M.; Türeci, Ö.; et al. A Liposomal RNA Vaccine Inducing Neoantigen-Specific CD4+ T Cells Augments the Antitumor Activity of Local Radiotherapy in Mice. OncoImmunology 2020, 9, 1771925. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, C.; Yang, S.; Xiao, W.; Zheng, Q.; Song, X. The Investigation of MRNA Vaccines Formulated in Liposomes Administrated in Multiple Routes against SARS-CoV-2. J. Control. Release 2021, 335, 449–456. [Google Scholar] [CrossRef]
- Dhaliwal, H.K.; Fan, Y.; Kim, J.; Amiji, M.M. Intranasal Delivery and Transfection of MRNA Therapeutics in the Brain Using Cationic Liposomes. Mol. Pharm. 2020, 17, 1996–2005. [Google Scholar] [CrossRef]
- Zhang, R.; Tang, L.; Tian, Y.; Ji, X.; Hu, Q.; Zhou, B.; Ding, Z.; Xu, H.; Yang, L. DP7-C-Modified Liposomes Enhance Immune Responses and the Antitumor Effect of a Neoantigen-Based MRNA Vaccine. J. Control. Release 2020, 328, 210–221. [Google Scholar] [CrossRef]
- Blakney, A.K.; McKay, P.F.; Hu, K.; Samnuan, K.; Jain, N.; Brown, A.; Thomas, A.; Rogers, P.; Polra, K.; Sallah, H.; et al. Polymeric and Lipid Nanoparticles for Delivery of Self-Amplifying RNA Vaccines. J. Control. Release 2021, 338, 201–210. [Google Scholar] [CrossRef]
- le Moignic, A.; Malard, V.; Benvegnu, T.; Lemiègre, L.; Berchel, M.; Jaffrès, P.-A.; Baillou, C.; Delost, M.; Macedo, R.; Rochefort, J.; et al. Preclinical Evaluation of MRNA Trimannosylated Lipopolyplexes as Therapeutic Cancer Vaccines Targeting Dendritic Cells. J. Control. Release 2018, 278, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xiao, W.; Elbahnasawy, M.A.; Bao, X.; Zheng, Q.; Gong, L.; Zhou, Y.; Yang, S.; Fang, A.; Farag, M.M.S.; et al. Optimization of the Linker Length of Mannose-Cholesterol Conjugates for Enhanced MRNA Delivery to Dendritic Cells by Liposomes. Front. Pharmacol. 2018, 9, 980. [Google Scholar] [CrossRef]
- Ganta, S.; Talekar, M.; Singh, A.; Coleman, T.P.; Amiji, M.M. Nanoemulsions in Translational Research—Opportunities and Challenges in Targeted Cancer Therapy. AAPS PharmSciTech 2014, 15, 694–708. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-López, E.; Guerra, M.; Dias-Ferreira, J.; Lopez-Machado, A.; Ettcheto, M.; Cano, A.; Espina, M.; Camins, A.; Garcia, M.L.; Souto, E.B. Current Applications of Nanoemulsions in Cancer Therapeutics. Nanomaterials 2019, 9, 821. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, H.F.; Bruxel, F.; Fraga, M.; Schuh, R.S.; Zorzi, G.K.; Matte, U.; Fattal, E. Cationic Nanoemulsions as Nucleic Acids Delivery Systems. Int. J. Pharm. 2017, 534, 356–367. [Google Scholar] [CrossRef]
- McClements, D.J. Nanoemulsions versus Microemulsions: Terminology, Differences, and Similarities. Soft Matter 2012, 8, 1719–1729. [Google Scholar] [CrossRef]
- Mazza, M.; Alonso-Sande, M.; Jones, M.-C.; de la Fuente, M. The Potential of Nanoemulsions in Biomedicine. In Fundamentals of Pharmaceutical Nanoscience; Uchegbu, I.F., Schätzlein, A.G., Cheng, W.P., Lalatsa, A., Eds.; Springer: New York, NY, USA, 2013; pp. 117–158. ISBN 978-1-4614-9164-4. [Google Scholar]
- Bouzo, B.L.; Lores, S.; Jatal, R.; Alijas, S.; Alonso, M.J.; Conejos-Sánchez, I.; de la Fuente, M. Sphingomyelin Nanosystems Loaded with Uroguanylin and Etoposide for Treating Metastatic Colorectal Cancer. Sci. Rep. 2021, 11, 17213. [Google Scholar] [CrossRef]
- Pirhadi, S.; Amani, A. Molecular Dynamics Simulation of SiRNA Loading into a Nanoemulsion as a Potential Carrier. J. Mol. Modeling 2020, 26, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Azambuja, J.H.; Schuh, R.S.; Michels, L.R.; Gelsleichter, N.E.; Beckenkamp, L.R.; Iser, I.C.; Lenz, G.S.; de Oliveira, F.H.; Venturin, G.; Greggio, S.; et al. Nasal Administration of Cationic Nanoemulsions as CD73-SiRNA Delivery System for Glioblastoma Treatment: A New Therapeutical Approach. Mol. Neurobiol. 2020, 57, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.; Laux, M.; Zandoná, B.; Santos, G.R.; dos Santos Giuberti, C.; de Oliveira, M.C.; Matte, U.; Teixeira, H.F. Optimization of Stearylamine-Based Nanoemulsions Obtained by Spontaneous Emulsification Process as Nucleic Acids Delivery Systems. J. Drug Deliv. Sci. Technol. 2008, 18, 398–403. [Google Scholar] [CrossRef]
- Fraga, M.; de Carvalho, T.G.; Bidone, J.; Schuh, R.S.; Matte, U.; Teixeira, H.F. Factors Influencing Transfection Efficiency of PIDUA/Nanoemulsion Complexes in a Mucopolysaccharidosis Type I Murine Model. Int. J. Nanomed. 2017, 12, 2061–2067. [Google Scholar] [CrossRef] [Green Version]
- Fraga, M.; Bruxel, F.; Diel, D.; de Carvalho, T.G.; Perez, C.A.; Magalhães-Paniago, R.; Malachias, Â.; Oliveira, M.C.; Matte, U.; Teixeira, H.F. PEGylated Cationic Nanoemulsions Can Efficiently Bind and Transfect PIDUA in a Mucopolysaccharidosis Type I Murine Model. J. Control. Release Off. J. Control. Release Soc. 2015, 209, 37–46. [Google Scholar] [CrossRef]
- Nagachinta, S.; Bouzo, B.L.; Vazquez-Rios, A.J.; Lopez, R.; Fuente, M. de la Sphingomyelin-Based Nanosystems (SNs) for the Development of Anticancer MiRNA Therapeutics. Pharmaceutics 2020, 12, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A Cationic Nanoemulsion for the Delivery of Next-Generation RNA Vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito, L.A.; Kommareddy, S.; Maione, D.; Uematsu, Y.; Giovani, C.; Scorza, F.B.; Otten, G.R.; Yu, D.; Mandl, C.W.; Mason, P.W.; et al. Chapter Seven—Self-Amplifying MRNA Vaccines. In Advances in Genetics; Elsevier BV: Amsterdam, The Netherlands, 2015; Volume 89, pp. 179–233. [Google Scholar] [CrossRef]
- Bogers, W.M.; Oostermeijer, H.; Mooij, P.; Koopman, G.; Verschoor, E.J.; Davis, D.; Ulmer, J.B.; Brito, L.A.; Cu, Y.; Banerjee, K.; et al. Potent Immune Responses in Rhesus Macaques Induced by Nonviral Delivery of a Self-Amplifying RNA Vaccine Expressing HIV Type 1 Envelope with a Cationic Nanoemulsion. J. Infect. Dis. 2015, 211, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Brazzoli, M.; Magini, D.; Bonci, A.; Buccato, S.; Giovani, C.; Kratzer, R.; Zurli, V.; Mangiavacchi, S.; Casini, D.; Brito, L.M.; et al. Induction of Broad-Based Immunity and Protective Efficacy by Self-Amplifying MRNA Vaccines Encoding Influenza Virus Hemagglutinin. J. Virol. 2015, 90, 332–344. [Google Scholar] [CrossRef] [Green Version]
- Samsa, M.M.; Dupuy, L.C.; Beard, C.W.; Six, C.M.; Schmaljohn, C.S.; Mason, P.W.; Geall, A.J.; Ulmer, J.B.; Yu, D. Self-Amplifying RNA Vaccines for Venezuelan Equine Encephalitis Virus Induce Robust Protective Immunogenicity in Mice. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 850–865. [Google Scholar] [CrossRef] [Green Version]
- Stokes, A.; Pion, J.; Binazon, O.; Laffont, B.; Bigras, M.; Dubois, G.; Blouin, K.; Young, J.K.; Ringenberg, M.A.; Abdeljelil, N.B.; et al. Nonclinical Safety Assessment of Repeated Administration and Biodistribution of a Novel Rabies Self-Amplifying MRNA Vaccine in Rats. Regul. Toxicol. Pharmacol. 2020, 113, 104648. [Google Scholar] [CrossRef]
- Luisi, K.; Morabito, K.M.; Burgomaster, K.E.; Sharma, M.; Kong, W.-P.; Foreman, B.M.; Patel, S.; Fisher, B.; Aleshnick, M.A.; Laliberte, J.; et al. Development of a Potent Zika Virus Vaccine Using Self-Amplifying Messenger RNA. Sci. Adv. 2020, 6, eaba5068. [Google Scholar] [CrossRef]
- Blakney, A.K.; Zhu, Y.; McKay, P.F.; Bouton, C.R.; Yeow, J.; Tang, J.; Hu, K.; Samnuan, K.; Grigsby, C.L.; Shattock, R.J.; et al. Big Is Beautiful: Enhanced SaRNA Delivery and Immunogenicity by a Higher Molecular Weight, Bioreducible, Cationic Polymer. ACS Nano 2020, 14, 5711–5727. [Google Scholar] [CrossRef] [Green Version]
- Alameh, M.-G.; Tombácz, I.; Bettini, E.; Lederer, K.; Sittplangkoon, C.; Wilmore, J.R.; Gaudette, B.T.; Soliman, O.Y.; Pine, M.; Hicks, P.; et al. Lipid Nanoparticles Enhance the Efficacy of MRNA and Protein Subunit Vaccines by Inducing Robust T Follicular Helper Cell and Humoral Responses. Immunity 2021, 54, 2877–2892.e7. [Google Scholar] [CrossRef] [PubMed]
- Kremsner, P.G.; Guerrero, R.A.A.; Arana, E.; Aroca Martinez, G.J.; Bonten, M.J.M.; Chandler, R.; Corral, G.; de Block, E.J.L.; Ecker, L.; Gabor, J.J.; et al. Efficacy and Safety of the CVnCoV SARS-CoV-2 MRNA Vaccine Candidate: Results from Herald, a Phase 2b/3, Randomised, Observer-Blinded, Placebo-Controlled Clinical Trial in Ten Countries in Europe and Latin America. SSRN Electron. J. 2021. [Google Scholar] [CrossRef]
- Kremsner, P.; Mann, P.; Bosch, J.; Fendel, R.; Gabor, J.J.; Kreidenweiss, A.; Kroidl, A.; Leroux-Roels, I.; Leroux-Roels, G.; Schindler, C.; et al. Phase 1 Assessment of the Safety and Immunogenicity of an MRNA- Lipid Nanoparticle Vaccine Candidate Against SARS-CoV-2 in Human Volunteers. medRxiv 2022. [Google Scholar] [CrossRef]
- Hoffmann, D.; Corleis, B.; Rauch, S.; Roth, N.; Mühe, J.; Halwe, N.J.; Ulrich, L.; Fricke, C.; Schön, J.; Kraft, A.; et al. CVnCoV and CV2CoV Protect Human ACE2 Transgenic Mice from Ancestral B BavPat1 and Emerging B.1.351 SARS-CoV-2. Nat. Commun. 2021, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kalnin, K.V.; Plitnik, T.; Kishko, M.; Zhang, J.; Zhang, D.; Beauvais, A.; Anosova, N.G.; Tibbitts, T.; DiNapoli, J.; Ulinski, G.; et al. Immunogenicity and Efficacy of MRNA COVID-19 Vaccine MRT5500 in Preclinical Animal Models. NPJ Vaccines 2021, 6, 61. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Khandhar, A.P.; O’Connor, M.A.; Walls, A.C.; Hemann, E.A.; Murapa, P.; Archer, J.; Leventhal, S.; Fuller, J.T.; Lewis, T.B.; et al. An Alphavirus -Derived Replicon RNA Vaccine Induces SARS-CoV-2 Neutralizing Antibody and T Cell Responses in Mice and Nonhuman Primates. Sci. Transl. Med. 2020, 12, eabc9396. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magné, R.; Gomard, E.; Guillet, J.-G.; Lévy, J.-P.; Meulien, P. Induction of Virus-Specific Cytotoxic T Lymphocytesin Vivo by Liposome-Entrapped MRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. MRNA Vaccines for Infectious Diseases: Principles, Delivery and Clinical Translation. Nat. Rev. Drug Discov. 2021, 20, 817–838. [Google Scholar] [CrossRef]
- Dybul, M.; Attoye, T.; Baptiste, S.; Cherutich, P.; Dabis, F.; Deeks, S.G.; Dieffenbach, C.; Doehle, B.; Goodenow, M.M.; Jiang, A.; et al. The Case for an HIV Cure and How to Get There. Lancet HIV 2021, 8, e51–e58. [Google Scholar] [CrossRef]
- Mascola, J.R. The Modern Era of HIV-1 Vaccine Development. Science 2015, 349, 139–140. [Google Scholar] [CrossRef]
- Pollard, C.; Rejman, J.; de Haes, W.; Verrier, B.; van Gulck, E.; Naessens, T.; de Smedt, S.; Bogaert, P.; Grooten, J.; Vanham, G.; et al. Type I IFN Counteracts the Induction of Antigen-Specific Immune Responses by Lipid-Based Delivery of MRNA Vaccines. Mol. Ther. 2013, 21, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Narayanan, E.; Liu, Q.; Tsybovsky, Y.; Boswell, K.; Ding, S.; Hu, Z.; Follmann, D.; Lin, Y.; Miao, H.; et al. A Multiclade Env–Gag VLP MRNA Vaccine Elicits Tier-2 HIV-1-Neutralizing Antibodies and Reduces the Risk of Heterologous SHIV Infection in Macaques. Nat. Med. 2021, 27, 2234–2245. [Google Scholar] [CrossRef]
- Pardi, N.; Secreto, A.J.; Shan, X.; Debonera, F.; Glover, J.; Yi, Y.; Muramatsu, H.; Ni, H.; Mui, B.L.; Tam, Y.K.; et al. Administration of Nucleoside-Modified MRNA Encoding Broadly Neutralizing Antibody Protects Humanized Mice from HIV-1 Challenge. Nat. Commun. 2017, 8, 14630. [Google Scholar] [CrossRef]
- Kaner, J.; Schaack, S. Understanding Ebola: The 2014 Epidemic. Glob. Health 2016, 12, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Huang, E.; Yuzhakov, O.; Ramanathan, P.; Ciaramella, G.; Bukreyev, A. Modified MRNA-Based Vaccines Elicit Robust Immune Responses and Protect Guinea Pigs From Ebola Virus Disease. J. Infect. Dis. 2018, 217, 451–455. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Recommends Groundbreaking Malaria Vaccine for Children at Risk; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Baeza Garcia, A.; Siu, E.; Sun, T.; Exler, V.; Brito, L.; Hekele, A.; Otten, G.; Augustijn, K.; Janse, C.J.; Ulmer, J.B.; et al. Neutralization of the Plasmodium-Encoded MIF Ortholog Confers Protective Immunity against Malaria Infection. Nat. Commun. 2018, 9, 2714. [Google Scholar] [CrossRef]
- Sun, T.; Holowka, T.; Song, Y.; Zierow, S.; Leng, L.; Chen, Y.; Xiong, H.; Griffith, J.; Nouraie, M.; Thuma, P.E.; et al. A Plasmodium-Encoded Cytokine Suppresses T-Cell Immunity during Malaria. Proc. Natl. Acad. Sci. USA 2012, 109, E2117–E2126. [Google Scholar] [CrossRef] [Green Version]
- Raj, D.K.; das Mohapatra, A.; Jnawali, A.; Zuromski, J.; Jha, A.; Cham-Kpu, G.; Sherman, B.; Rudlaff, R.M.; Nixon, C.E.; Hilton, N.; et al. Anti-PfGARP Activates Programmed Cell Death of Parasites and Reduces Severe Malaria. Nature 2020, 582, 104–108. [Google Scholar] [CrossRef]
- Goodall, G.J.; Wickramasinghe, V.O. RNA in Cancer. Nat. Rev. Cancer 2021, 21, 22–36. [Google Scholar] [CrossRef]
- Pan, C.; Liu, H.; Robins, E.; Song, W.; Liu, D.; Li, Z.; Zheng, L. Next-Generation Immuno-Oncology Agents: Current Momentum Shifts in Cancer Immunotherapy. J. Hematol. Oncol. 2020, 13, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhaya, S.; Hubbard-Lucey, V.M.; Yu, J.X. Immuno-Oncology Drug Development Forges on despite COVID-19. Nat. Rev. Drug Discov. 2020, 19, 751–752. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, S.; Renmans, D.; Broos, K.; Goethals, L.; Maenhout, S.; Benteyn, D.; Goyvaerts, C.; Four, S.; van der Jeught, K.; Bialkowski, L.; et al. Intratumoral Delivery of TriMix MRNA Results in T-Cell Activation by Cross-Presenting Dendritic Cells. Cancer Immunol. Res. 2016, 4, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Irvine, D.J.; Dane, E.L. Enhancing Cancer Immunotherapy with Nanomedicine. Nat. Rev. Immunol. 2020, 20, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F.; Berraondo, P.; Etxeberria, I.; Frederick, J.; Sahin, U.; Gilboa, E.; Melero, I. An RNA Toolbox for Cancer Immunotherapy. Nat. Rev. Drug Discov. 2018, 17, 751–767. [Google Scholar] [CrossRef]
- Blass, E.; Ott, P.A. Advances in the Development of Personalized Neoantigen-Based Therapeutic Cancer Vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Y.; Huang, L. MRNA Vaccine for Cancer Immunotherapy. Mol. Cancer 2021, 20, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and Challenges towards Targeted Delivery of Cancer Therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [Green Version]
- van Hoecke, L.; Roose, K. How MRNA Therapeutics Are Entering the Monoclonal Antibody Field. J. Transl. Med. 2019, 17, 54. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, S.L.; Bai, A.; Bailey, D.; Ichikawa, K.; Zielinski, J.; Karp, R.; Apte, A.; Arnold, K.; Zacharek, S.J.; Iliou, M.S.; et al. Durable Anticancer Immunity from Intratumoral Administration of IL-23, IL-36γ, and OX40L MRNAs. Sci. Transl. Med. 2019, 11, eaat9143. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Etxeberria, I.; Ponz-Sarvise, M.; Melero, I. Revisiting Interleukin-12 as a Cancer Immunotherapy Agent. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2716–2718. [Google Scholar] [CrossRef] [Green Version]
- Lai, I.; Swaminathan, S.; Baylot, V.; Mosley, A.; Dhanasekaran, R.; Gabay, M.; Felsher, D.W. Lipid Nanoparticles That Deliver IL-12 Messenger RNA Suppress Tumorigenesis in MYC Oncogene-Driven Hepatocellular Carcinoma. J. Immunother. Cancer 2018, 6, 125. [Google Scholar] [CrossRef]
- Li, Y.; Su, Z.; Zhao, W.; Zhang, X.; Momin, N.; Zhang, C.; Wittrup, K.D.; Dong, Y.; Irvine, D.J.; Weiss, R. Multifunctional Oncolytic Nanoparticles Deliver Self-Replicating IL-12 RNA to Eliminate Established Tumors and Prime Systemic Immunity. Nat. Cancer 2020, 1, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in Clinical Cancer Immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Rybakova, Y.; Kowalski, P.S.; Huang, Y.; Gonzalez, J.T.; Heartlein, M.W.; DeRosa, F.; Delcassian, D.; Anderson, D.G. MRNA Delivery for Therapeutic Anti-HER2 Antibody Expression In Vivo. Mol. Ther. 2019, 27, 1415–1423. [Google Scholar] [CrossRef]
- Stadler, C.R.; Bähr-Mahmud, H.; Celik, L.; Hebich, B.; Roth, A.S.; Roth, R.P.; Karikó, K.; Türeci, Ö.; Sahin, U. Elimination of Large Tumors in Mice by MRNA-Encoded Bispecific Antibodies. Nat. Med. 2017, 23, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, Y.; Han, W.-D. Chimeric Antigen Receptor Modified T-Cells for Cancer Treatment. Chronic Dis. Transl. Med. 2018, 4, 225–243. [Google Scholar] [CrossRef]
- Scarfò, I.; Maus, M.V. Current Approaches to Increase CAR T Cell Potency in Solid Tumors: Targeting the Tumor Microenvironment. J. ImmunoTherapy Cancer 2017, 5, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhard, K.; Rengstl, B.; Oehm, P.; Michel, K.; Billmeier, A.; Hayduk, N.; Klein, O.; Kuna, K.; Ouchan, Y.; Wöll, S.; et al. An RNA Vaccine Drives Expansion and Efficacy of Claudin-CAR-T Cells against Solid Tumors. Science 2020, 367, 446. [Google Scholar] [CrossRef]
- Berraondo, P.; Martini, P.G.; Avila, M.A.; Fontanellas, A. Messenger RNA Therapy for Rare Genetic Metabolic Diseases. Gut 2019, 68, 1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaques, R.; Shakeel, A.; Hoyle, C. Novel Therapeutic Approaches for the Management of Cystic Fibrosis. Multidiscip. Respir. Med. 2020, 15, 690. [Google Scholar] [CrossRef]
- Christopher Boyd, A.; Guo, S.; Huang, L.; Kerem, B.; Oren, Y.S.; Walker, A.J.; Hart, S.L. New Approaches to Genetic Therapies for Cystic Fibrosis. J. Cyst. Fibros. 2020, 19, S54–S59. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, A.D.S.; Paunovska, K.; Cristian, A.; Dahlman, J.E. Treating Cystic Fibrosis with MRNA and CRISPR. Hum. Gene Ther. 2020, 31, 940–955. [Google Scholar] [CrossRef]
- Bissell, D.M.; Anderson, K.E.; Bonkovsky, H.L. Porphyria. N. Engl. J. Med. 2017, 377, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.E.; Bloomer, J.R.; Bonkovsky, H.L.; Kushner, J.P.; Pierach, C.A.; Pimstone, N.R.; Desnick, R.J. Recommendations for the Diagnosis and Treatment of the Acute Porphyrias. Ann Intern Med. 2005, 142, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Berraondo, P.; Jericó, D.; Guey, L.T.; Sampedro, A.; Frassetto, A.; Benenato, K.E.; Burke, K.; Santamaría, E.; Alegre, M.; et al. Systemic Messenger RNA as an Etiological Treatment for Acute Intermittent Porphyria. Nat. Med. 2018, 24, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Park, J.-S.; Yin, L.; Laureano, R.; Jacquinet, E.; Yang, J.; Liang, S.; Frassetto, A.; Zhuo, J.; Yan, X.; et al. Dual MRNA Therapy Restores Metabolic Function in Long-Term Studies in Mice with Propionic Acidemia. Nat. Commun. 2020, 11, 5339. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.; Adam, D.N. A Review of Fabry Disease. Ski. Ther. Lett. 2018, 23, 4–6. [Google Scholar]
- Pastores, G.M. Agalsidase Alfa (Replagal) in the Treatment of Anderson-Fabry Disease. Biol. Targets Ther. 2007, 1, 291–300. [Google Scholar]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef]
- DeRosa, F.; Smith, L.; Shen, Y.; Huang, Y.; Pan, J.; Xie, H.; Yahalom, B.; Heartlein, M.W. Improved Efficacy in a Fabry Disease Model Using a Systemic MRNA Liver Depot System as Compared to Enzyme Replacement Therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Yin, L.; Theisen, M.; Zhuo, J.; Siddiqui, S.; Levy, B.; Presnyak, V.; Frassetto, A.; Milton, J.; Salerno, T.; et al. Systemic MRNA Therapy for the Treatment of Fabry Disease: Preclinical Studies in Wild-Type Mice, Fabry Mouse Model, and Wild-Type Non-Human Primates. Am. J. Hum. Genet. 2019, 104, 625–637. [Google Scholar] [CrossRef] [Green Version]
- Fraser, J.L.; Venditti, C.P. Methylmalonic and Propionic Acidemias: Clinical Management Update. Curr. Opin. Pediatrics 2016, 28, 682–693. [Google Scholar] [CrossRef]
- An, D.; Schneller, J.L.; Frassetto, A.; Liang, S.; Zhu, X.; Park, J.-S.; Theisen, M.; Hong, S.-J.; Zhou, J.; Rajendran, R.; et al. Systemic Messenger RNA Therapy as a Treatment for Methylmalonic Acidemia. Cell Rep. 2017, 21, 3548–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, M.-Z.; Li, X.-Z.; Mei, H.-F.; Sheng, H.-Y.; Yin, X.; Jiang, M.-Y.; Cai, Y.-N.; Su, L.; Lin, Y.-T.; Shao, Y.-X.; et al. Clinical and Biochemical Characteristics of Patients with Ornithine Transcarbamylase Deficiency. Clin. Biochem. 2020, 84, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Prieve, M.G.; Harvie, P.; Monahan, S.D.; Roy, D.; Li, A.G.; Blevins, T.L.; Paschal, A.E.; Waldheim, M.; Bell, E.C.; Galperin, A.; et al. Targeted MRNA Therapy for Ornithine Transcarbamylase Deficiency. J. Am. Soc. Gene Ther. 2018, 26, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome Editing. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Luther, D.C.; Lee, Y.W.; Nagaraj, H.; Scaletti, F.; Rotello, V.M. Delivery Approaches for CRISPR/Cas9 Therapeutics in Vivo: Advances and Challenges. null 2018, 15, 905–913. [Google Scholar] [CrossRef]
- Mali, P.; Esvelt, K.M.; Church, G.M. Cas9 as a Versatile Tool for Engineering Biology. Nat. Methods 2013, 10, 957–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sander, J.D.; Joung, J.K. CRISPR-Cas Systems for Editing, Regulating and Targeting Genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Song, C.-Q.; Dorkin, J.R.; Zhu, L.J.; Li, Y.; Wu, Q.; Park, A.; Yang, J.; Suresh, S.; Bizhanova, A.; et al. Therapeutic Genome Editing by Combined Viral and Non-Viral Delivery of CRISPR System Components in Vivo. Nat. Biotechnol. 2016, 34, 328–333. [Google Scholar] [CrossRef]
- Yin, H.; Song, C.-Q.; Suresh, S.; Wu, Q.; Walsh, S.; Rhym, L.H.; Mintzer, E.; Bolukbasi, M.F.; Zhu, L.J.; Kauffman, K.; et al. Structure-Guided Chemical Modification of Guide RNA Enables Potent Non-Viral in Vivo Genome Editing. Nat. Biotechnol. 2017, 35, 1179–1187. [Google Scholar] [CrossRef]
- Miller, J.B.; Zhang, S.; Kos, P.; Xiong, H.; Zhou, K.; Perelman, S.S.; Zhu, H.; Siegwart, D.J. Non-Viral CRISPR/Cas Gene Editing In Vitro and In Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 MRNA and SgRNA. Angew. Chem. 2017, 56, 1059–1063. [Google Scholar] [CrossRef] [Green Version]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef] [Green Version]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Intellia Therapeutics Intellia Therapeutics. Available online: https://ir.intelliatx.com/news-releases/news-release-details/intellia-therapeutics-receives-authorization-initiate-phase-12 (accessed on 16 October 2021).
- Rayner, F.; Isaacs, J.D. Therapeutic Tolerance in Autoimmune Disease. Semin. Arthritis Rheum. 2018, 48, 558–562. [Google Scholar] [CrossRef]
- Serra, P.; Santamaria, P. Antigen-Specific Therapeutic Approaches for Autoimmunity. Nat. Biotechnol. 2019, 37, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Krienke, C.; Kolb, L.; Diken, E.; Streuber, M.; Kirchhoff, S.; Bukur, T.; Akilli-Öztürk, Ö.; Kranz, L.M.; Berger, H.; Petschenka, J.; et al. A Noninflammatory MRNA Vaccine for Treatment of Experimental Autoimmune Encephalomyelitis. Science 2021, 371, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Magadum, A.; Kaur, K.; Zangi, L. MRNA-Based Protein Replacement Therapy for the Heart. Mol. Ther. 2019, 27, 785–793. [Google Scholar] [CrossRef] [Green Version]
- Kaur, K.; Zangi, L. Modified MRNA as a Therapeutic Tool for the Heart. Cardiovasc. Drugs Ther. 2020, 34, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Dilliard, S.A.; Cheng, Q.; Siegwart, D.J. On the Mechanism of Tissue-Specific MRNA Delivery by Selective Organ Targeting Nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2109256118. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Infectious Diseases Other than COVID-19 | |||||
|---|---|---|---|---|---|
| Therapeutic | Clinical Application | Formulation | Phase | NCT Number | Sponsor |
| mRNA-1653 | hMPV/PIV3 | Modified mRNA-LNPs | Phase I | NCT04144348 NCT03392389 | Moderna |
| mRNA-1345 | RSV | Modified mRNA-LNPs | Phase I | NCT04528719 | Moderna |
| mRNA-11777 | RSV | Modified mRNA-LNPs | Phase I | Unregistered | Moderna, Merck |
| mRNA-1172 | RSV | Modified mRNA-LNPs | Phase I | Unregistered | Moderna, Merck |
| mRNA-1893 | Zika | Modified mRNA-LNPs | Phase I | NCT04064905 | Moderna |
| mRNA-1325 | Zika | Modified mRNA-LNPs | Phase I | NCT03014089 | Moderna |
| mRNA-1647 | CMV | Modified mRNA-LNPs | Phase II | NCT04232280 | Moderna |
| mRNA-1647 | CMV | Modified mRNA-LNPs | Phase II | NCT03382405 | Moderna |
| mRNA-1443 | CMV | Modified mRNA-LNPs | Phase I | NCT03382405 | Moderna |
| MRT5400 | Influenza A (H3N2) | Not disclosed | Phase I | Unregistered | TranslateBio, Sanofi |
| MRT5401 | Influenza A (H3N2) | Not disclosed | Phase I | Unregistered | TranslateBio, Sanofi |
| mRNA-1010 | Influenza A, Influenza B | Not disclosed | Phase II/III | NCT04956575 | Moderna |
| mRNA-1851 | Influenza A (H7N9) | Modified mRNA-LNPs | Phase I | NCT03345043 | Moderna |
| mRNA-1440 | Influenza A (H7N8) | Modified mRNA-LNPs | Phase I | NCT03076385 | Moderna |
| mRNA-1388 | Chikungunya | Modified mRNA-LNPs | Phase I | NCT03829384 | Moderna |
| CV7201 | Rabies | Non-modified mRNA- in RNActive | Phase I | NCT02241135 | CureVac |
| CV7202 | Rabies | Unmodified mRNA-LNPs | Phase I | NCT03713086 | CureVac |
| GSK3903133A | Rabies | saRNA–cationic nanoemulsion | Phase I | NCT04062669 | GSK |
| Oncology | |||||
| Cancer Vaccines | |||||
| Shared Tumor Antigens | |||||
| BNT111 | Advanced Melanoma | Modified mRNA-LPX | Phase II | NCT04526899 | BioNTech |
| BNT112 | Prostate Cancer | Modified mRNA-LPX | Phase I/II | NCT04382898 | BioNTech |
| BNT113 | HPV16+ head and neck cancer | Modified mRNA-LPX | Phase II | NCT04534205 | BioNTech |
| BNT114 | Triple-Negative Breast Cancer | Modified mRNA-LPX | Phase I | NCT03815058 | BioNTech |
| BNT115 | Ovarian Cancer | Modified mRNA-LPX | Phase I | NCT04163094 | BioNTech |
| mRNA-5671 | Carcinoma, NSCLC, Pancreatic and colorectal neoplasms | Non-disclosed | Phase I | NCT03948763 | Merck/Moderna |
| Personalized cancer vaccines | |||||
| mRNA-4157 | Melanoma | Modified mRNA-LNPs | Phase II | NCT03897881 | Moderna |
| BNT122 | Advanced Melanoma | Modified mRNA-LPX | Phase I | NCT03815058 | BioNTech |
| mRNA-4157 | Solid Tumors | Modified mRNA-LNPs | Phase I | NCT03313778 | Moderna |
| mRNA-2752 | Solid Tumors | Modified mRNA-LNPs | Phase I | NCT03739931 | Moderna |
| CV8102 | Solid Tumors | Not disclosed | Phase I | NCT03291002 | CureVac |
| V941 | Solid Tumors | Not disclosed | Phase I | NCT03948763 | Merck Sharp & Dohme Corp. |
| Medi1191 | Solid Tumors | Modified mRNA-LPNs | Phase I | NCT03946800 | MedImmuneLC |
| mRNA-2416 | Solid Tumors and Lymphoma | Modified mRNA-LNPs | Phase I/II | NCT03323398 | Moderna |
| mRNA-2752 | Solid Tumors and Lymphoma | Modified mRNA-LNPs | Phase I | NCT03739931 | Moderna |
| mRNA-2752 | Carcinoma | Modified mRNA-LNPs | Phase I | NCT02872025 | Moderna |
| BNT122 | Colorectal Cancer Stage II/III | Modified mRNA-LPX | Phase II | NCT04486378 | BioNTech |
| MVT-5873 | Pancreatic Cancer | Not disclosed | Phase I | NCT02672917 | BioNTech |
| BNT131 | Metastatic Neoplasm | Not disclosed | Phase I | NCT03871348 | BioNTech |
| The Liver as a Bioreactor | |||||
| Encoding Cytokines | |||||
| BNT152, BNT153 | Solid Tumor | Not disclosed | Phase I | NCT04710043 | BioNTech |
| BNT151 | Solid Tumor | Not disclosed | Phase I/II | NCT04455620 | BioNTech |
| mRNA-6231 | Autoimmune Disorders | Modified mRNA-LNPs | Phase I | NCT04916431 | Moderna |
| Encoding Antibodies | |||||
| BNT142 | Solid Tumor | Encoding Ab | Phase I/II | NCT04503278 | BioNTech |
| mRNA-1944 | Chikungunya | Modified mRNA-LNPs | Phase I | NCT03829384 | Moderna |
| mRNA Delivery for Protein Replacement Therapies | |||||
|---|---|---|---|---|---|
| mRNA-3927 | Propionic Acidemia | Modified mRNA-LNPs | Phase 1/2 | NCT04159103 | Moderna |
| MRT5005 | Cystic Fibrosis | Modified mRNA-LNPs | Phase 1/2 | NCT03375047 | Translate Bio |
| Gene Editing | |||||
|---|---|---|---|---|---|
| NTLA-2001 | Hereditary Transthyretin Amyloidosis | Cas9 mRNA and sg mRNA-LNPs | Phase 1 | NCT04601051 | Intellia Therapeutics |
| NTLA-2002 | Hereditary Angioedema | Cas9 mRNA and sg mRNA LNPs | Phase 1/2 | Not registered yet | Intellia Therapeutics |
| Cardiovascular Diseases | |||||
|---|---|---|---|---|---|
| AZD8601 | Heart Failure | Modified mRNA-LNPs | Phase II | NCT03370887 | AstraZeneca |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taina-González, L.; de la Fuente, M. The Potential of Nanomedicine to Unlock the Limitless Applications of mRNA. Pharmaceutics 2022, 14, 460. https://doi.org/10.3390/pharmaceutics14020460
Taina-González L, de la Fuente M. The Potential of Nanomedicine to Unlock the Limitless Applications of mRNA. Pharmaceutics. 2022; 14(2):460. https://doi.org/10.3390/pharmaceutics14020460
Chicago/Turabian StyleTaina-González, Laura, and María de la Fuente. 2022. "The Potential of Nanomedicine to Unlock the Limitless Applications of mRNA" Pharmaceutics 14, no. 2: 460. https://doi.org/10.3390/pharmaceutics14020460