
Increased Targeting Area in Tumors by Dual-Ligand Modification of Liposomes with RGD and TAT Peptides

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
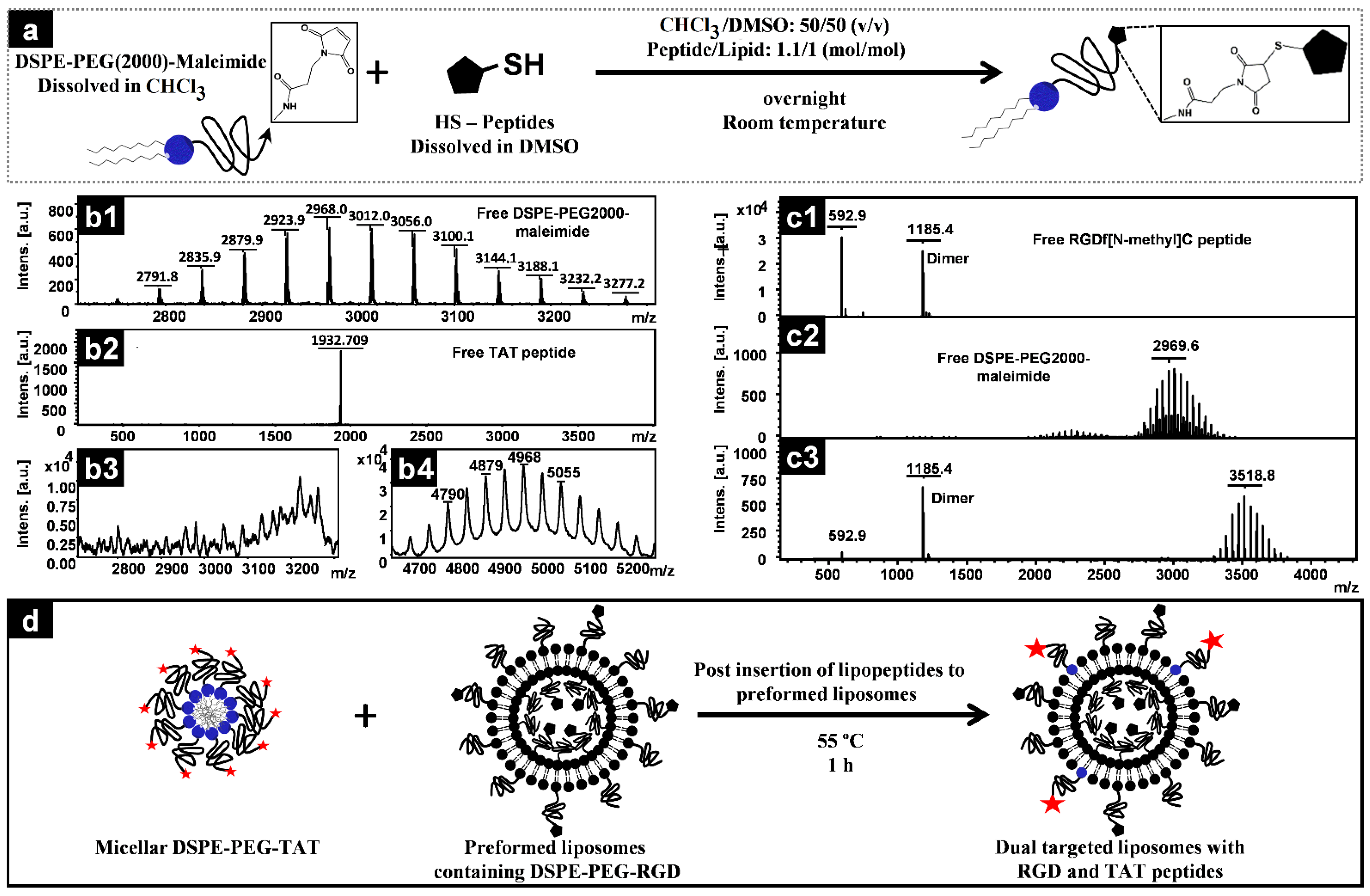
2.2. Conjugation of Peptides to DSPE–PEG–Maleimide
2.3. MALDI-TOF Measurements
2.4. Preparation and Characterization of Liposomes
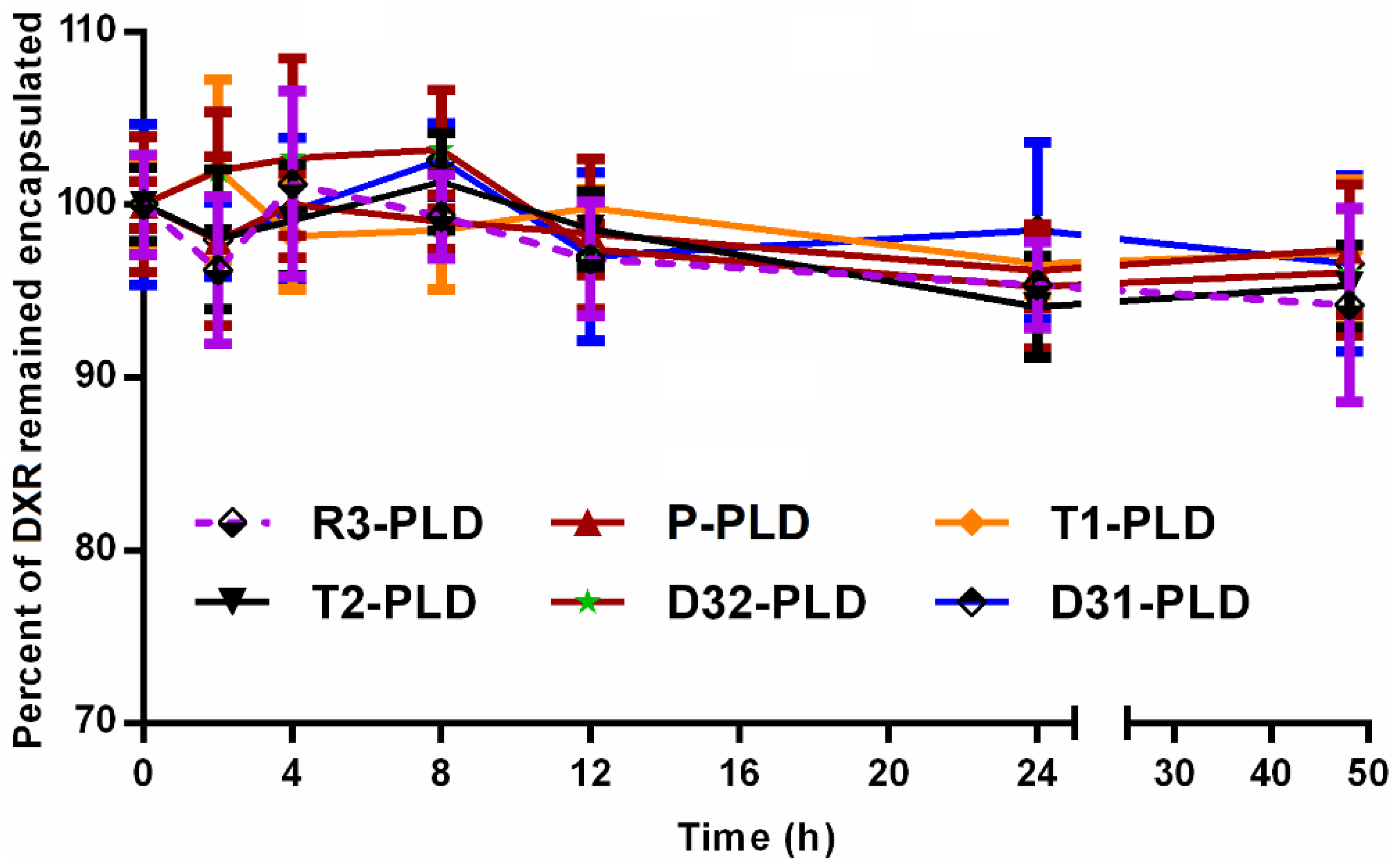
2.5. Leakage Stability Assessments
2.6. Cell Culturing
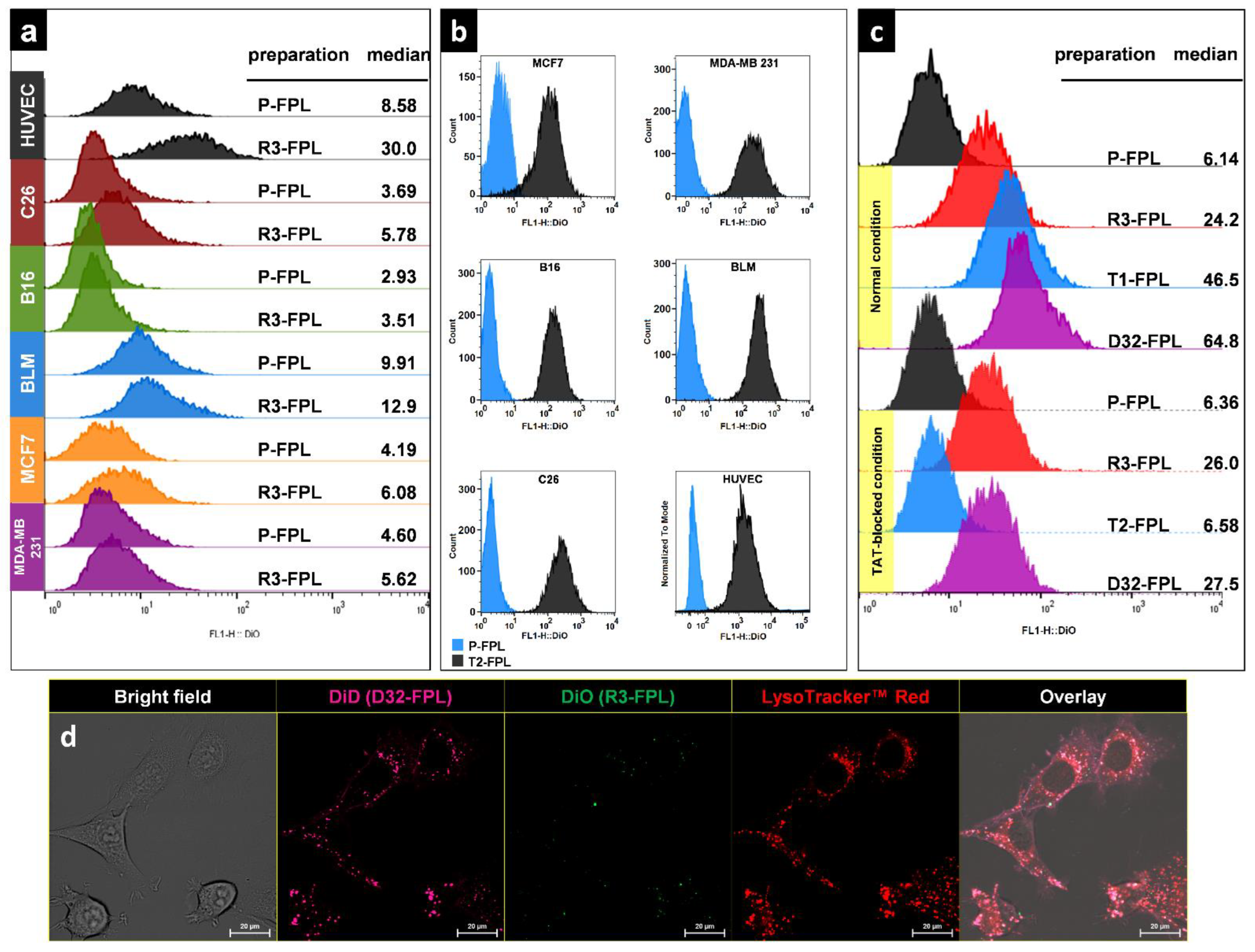
2.7. In Vitro Cellular Association of Peptide-Modified Liposomes
2.8. Intravital Imaging
2.8.1. Tumor Model
2.8.2. In Vivo Imaging
2.9. In Vivo Biodistribution
2.10. Therapeutic Efficacy
2.11. Statistical Analysis
3. Results
3.1. Conjugation of Peptides to PEG-Lipids
3.2. Liposome Characterization and Stability Assessments
3.3. In Vitro Cellular Association of Peptide-Modified Liposomes
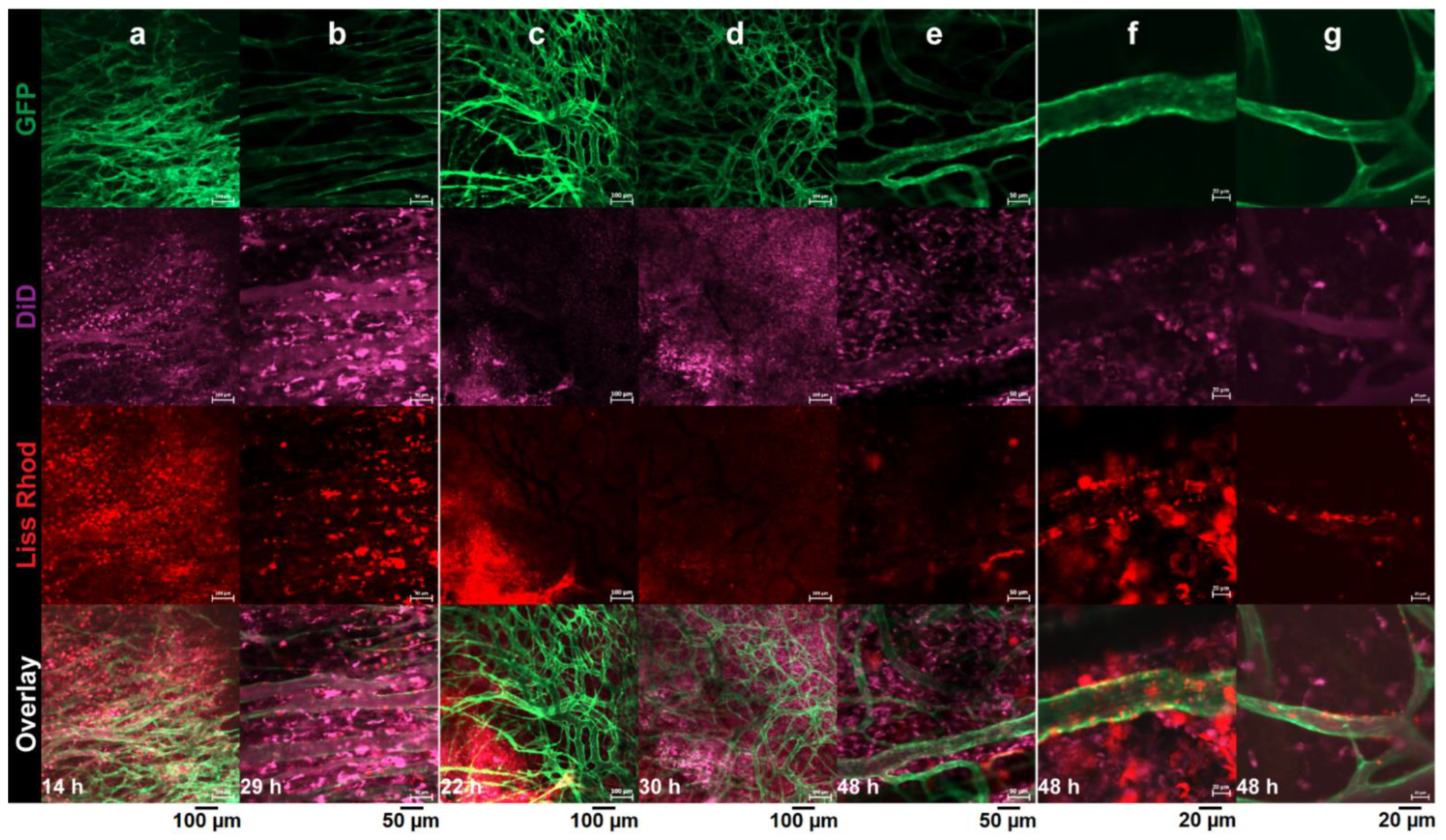
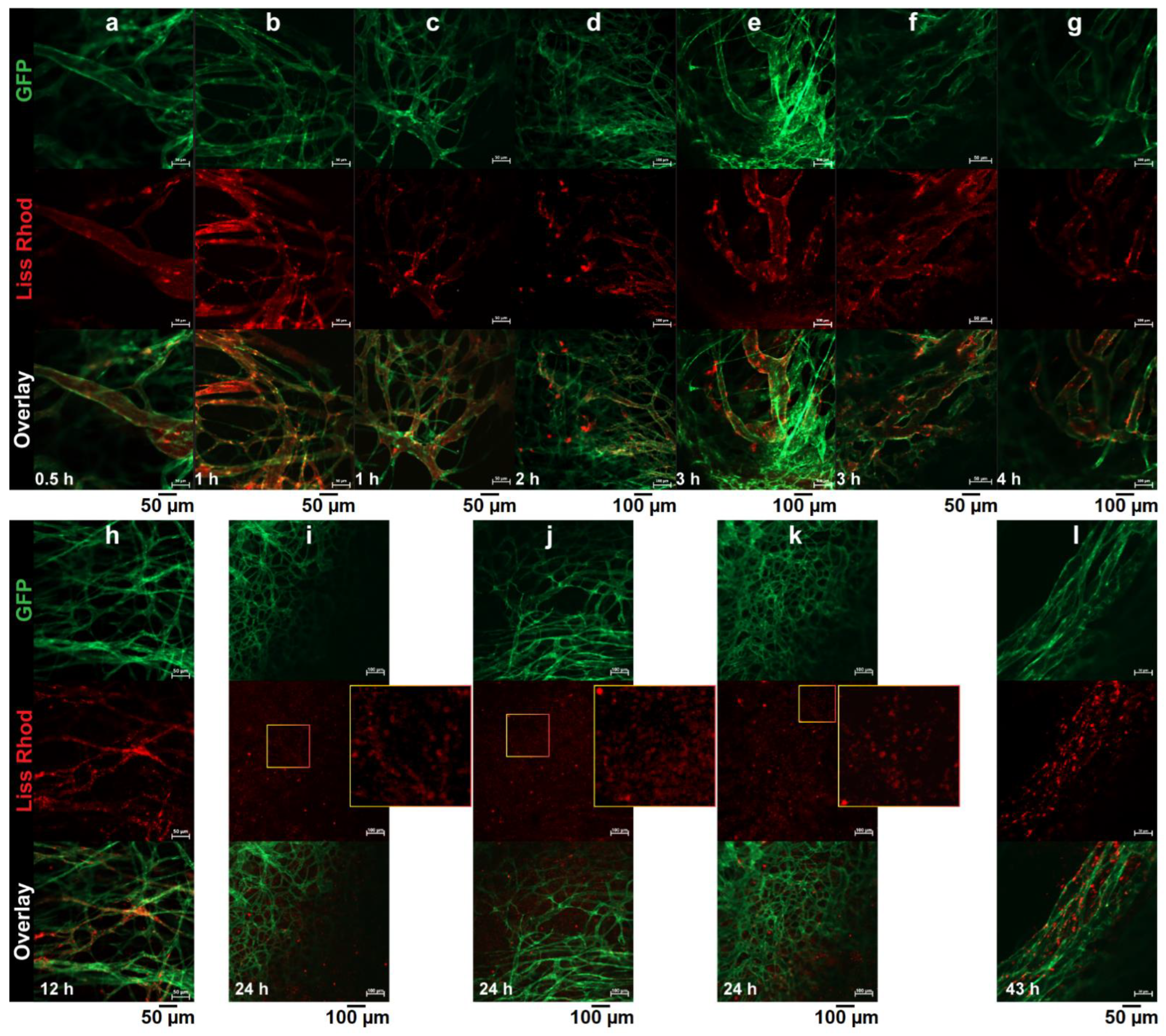
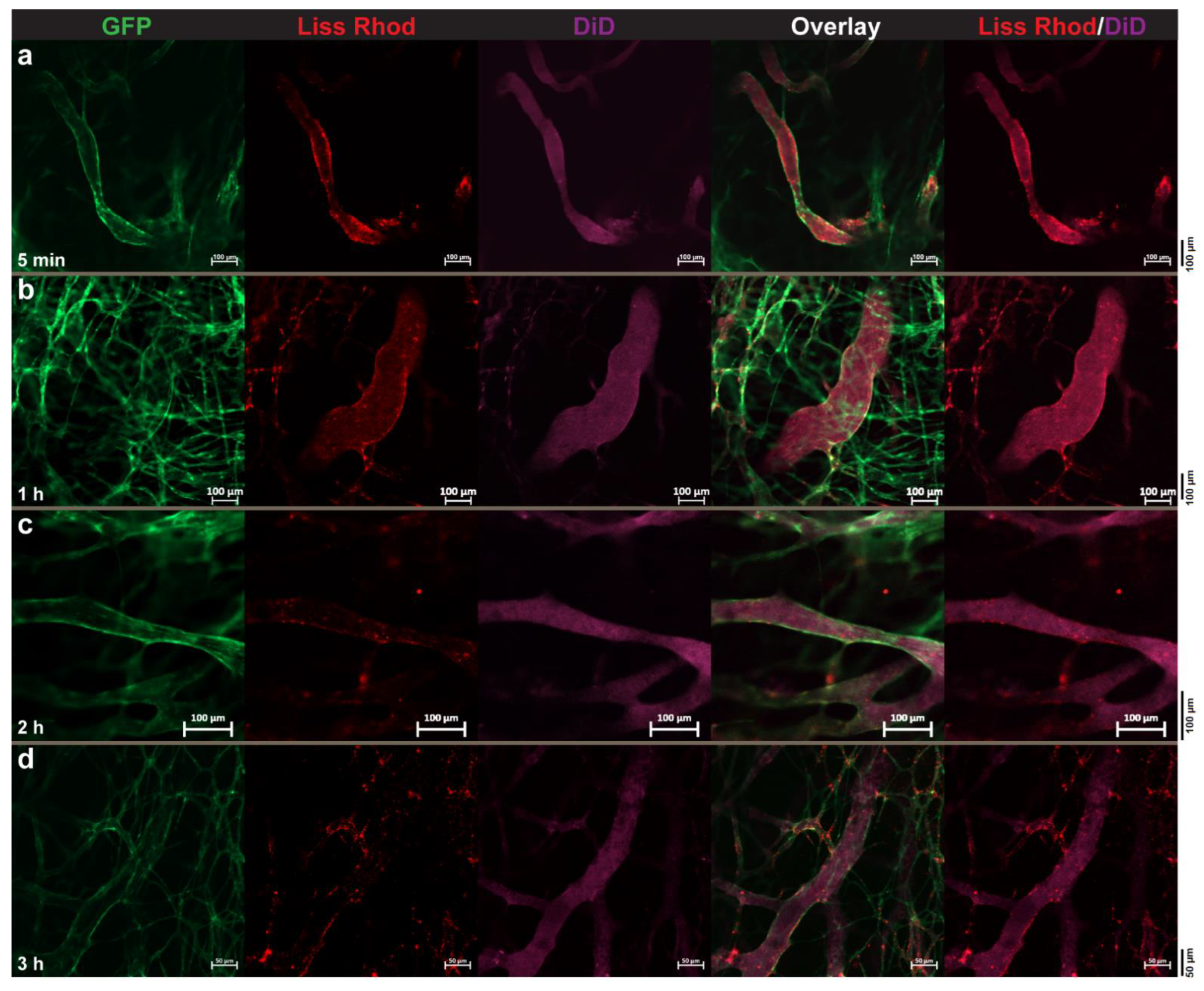
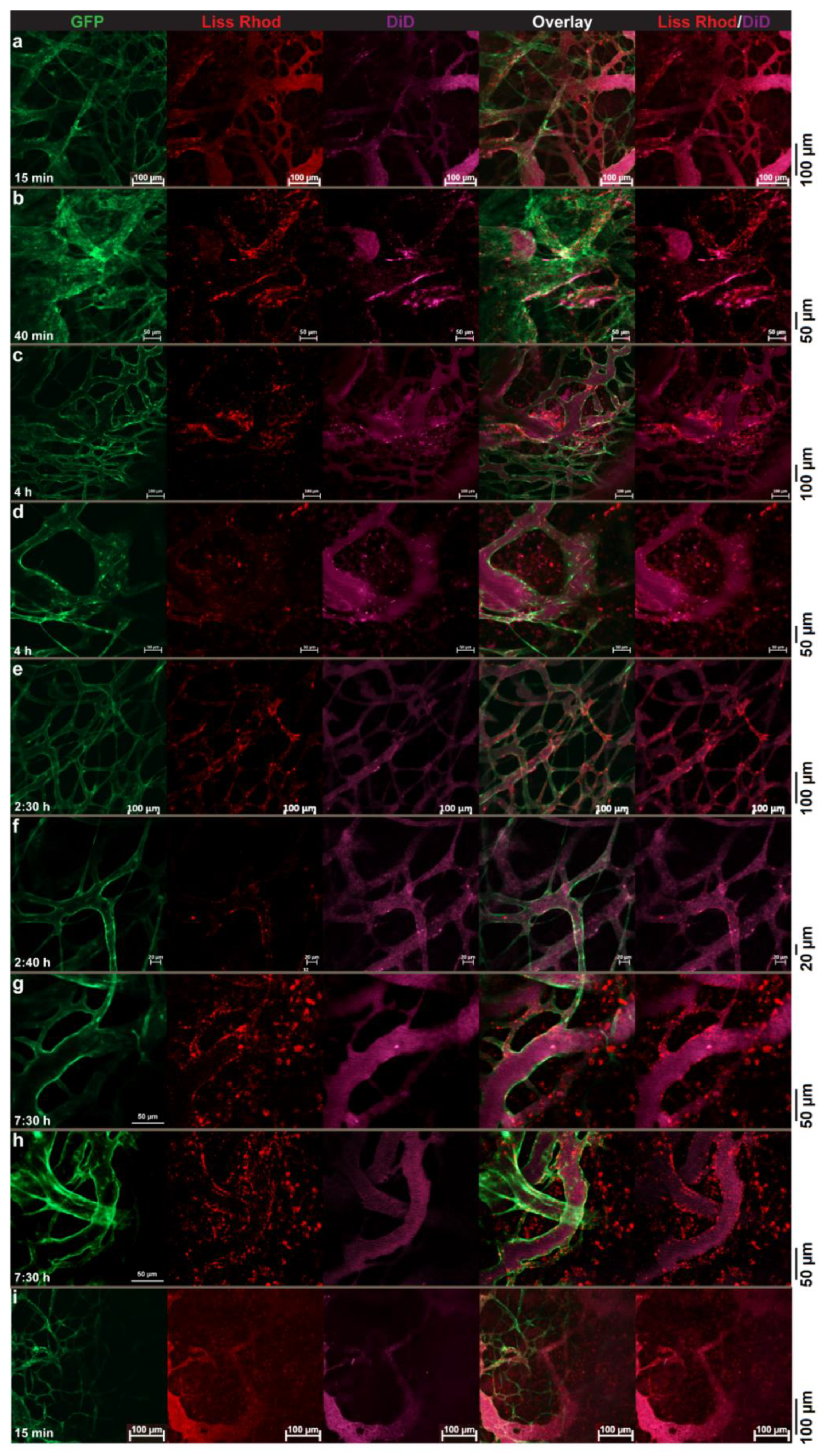
3.4. Intravital Microscopy
3.4.1. RGD-Liposomes vs. TAT-Liposomes
3.4.2. Dual-Targeted Liposomes
3.4.3. Dual-Targeted Liposomes vs. Single-Targeted Liposomes
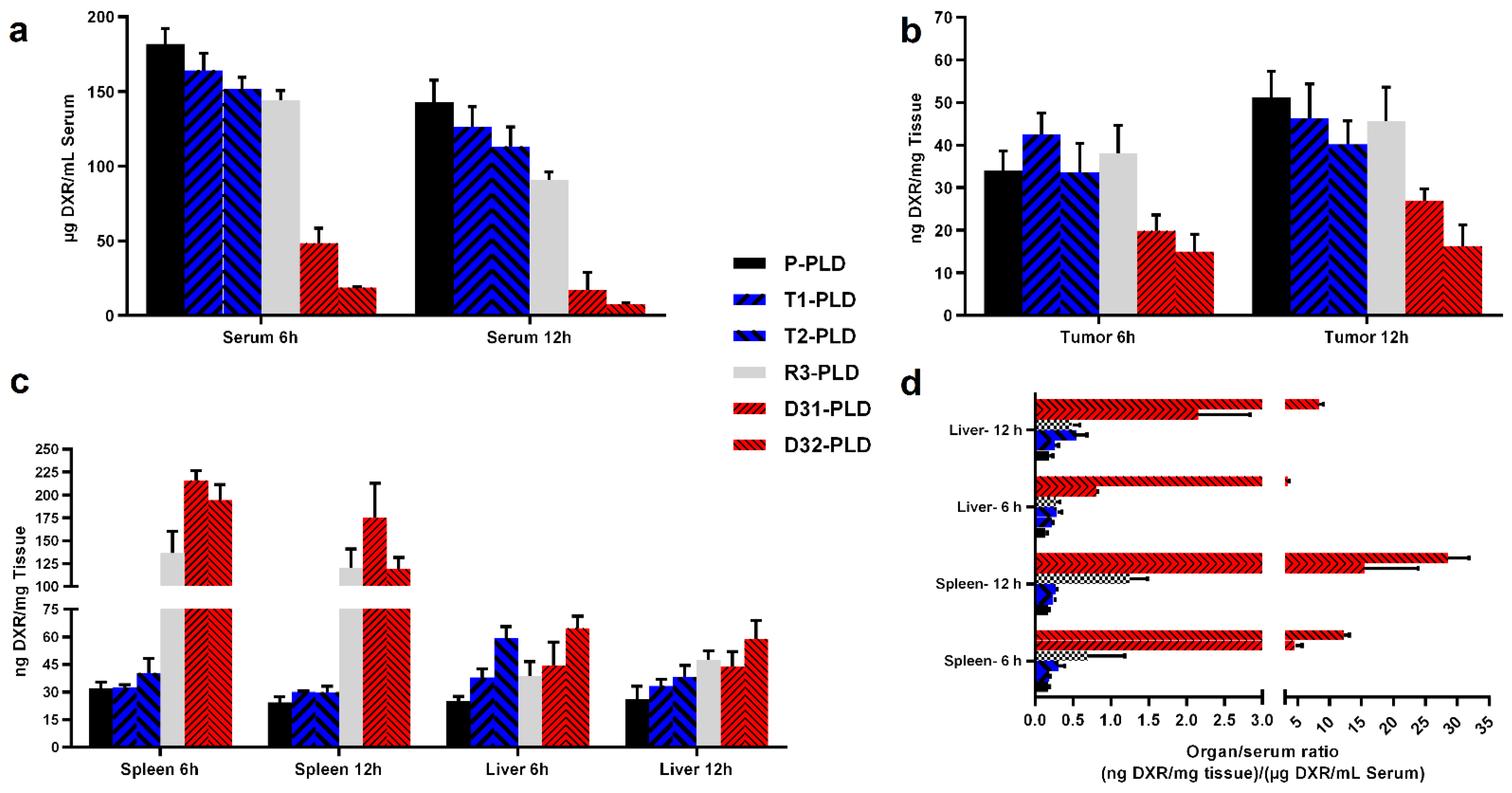
3.5. Biodistribution of PLDs in Tumor-Bearing Mice
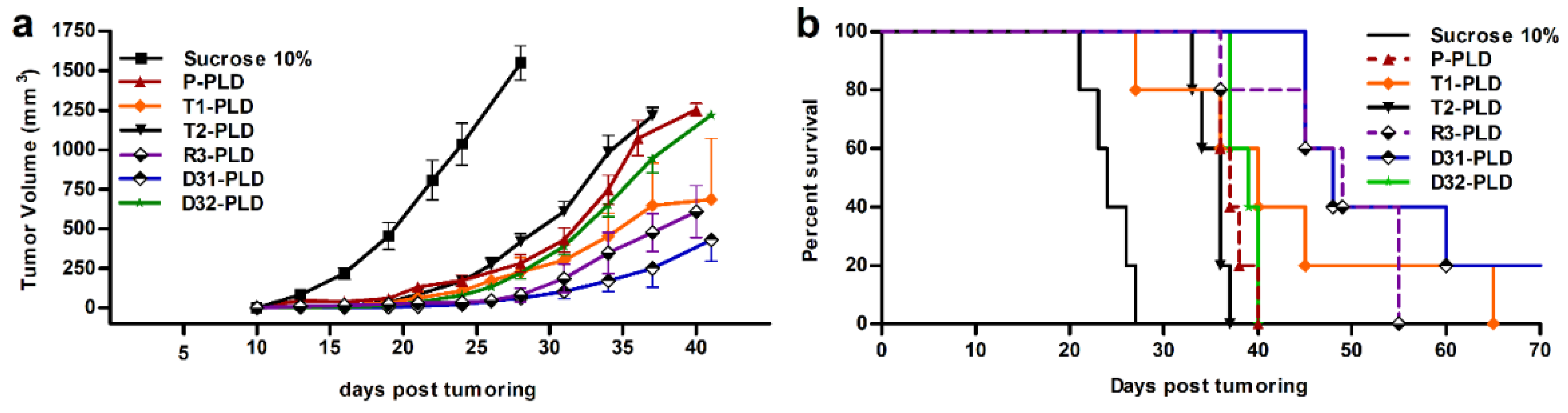
3.6. Therapeutic Efficacy
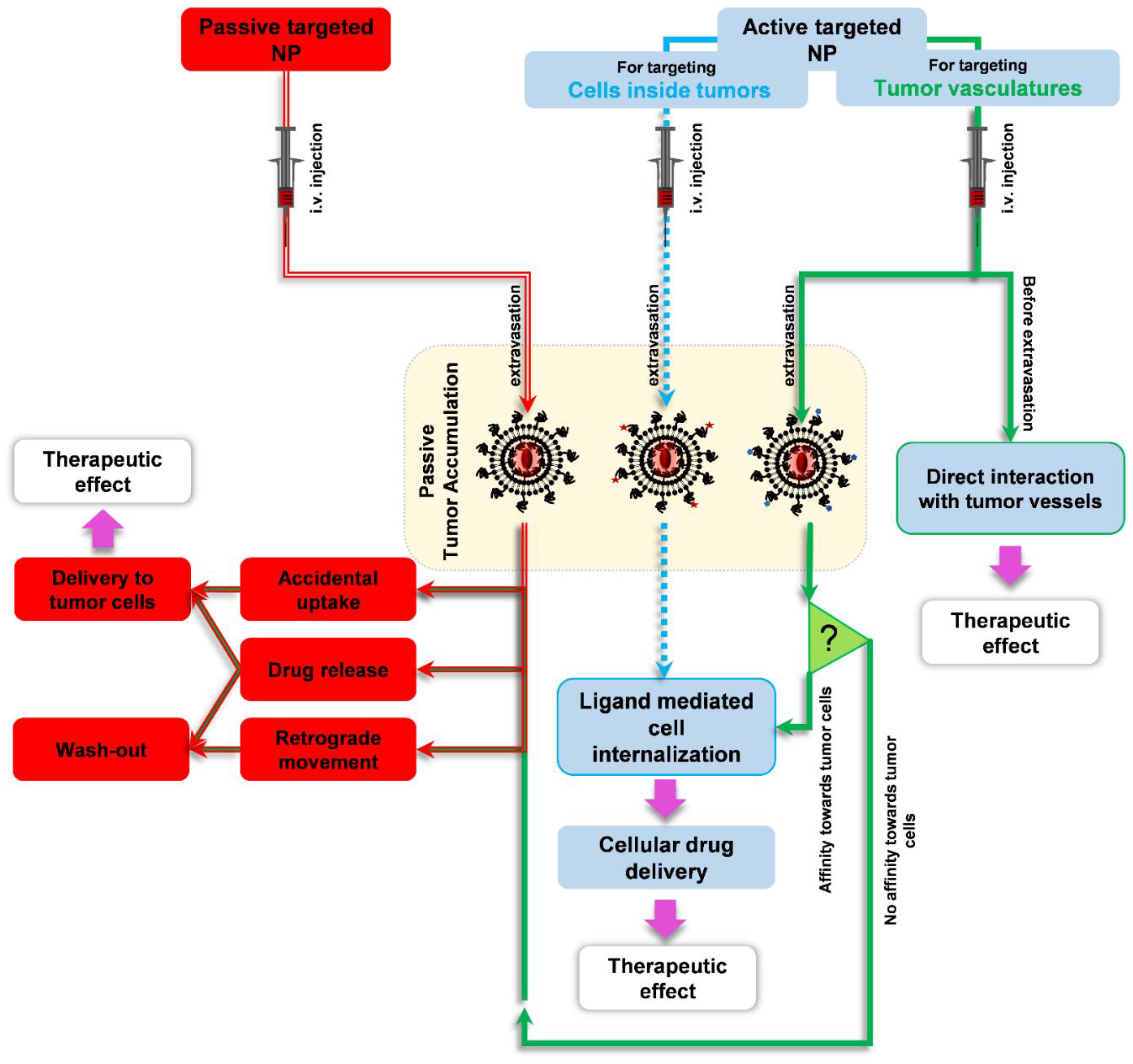
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Gregoriadis, G. Liposomology: Delivering the message. J. Liposome Res. 2018, 28, 1–4. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil(r)--the first fda-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Gabizon, A.; Shmeeda, H.; Barenholz, Y. Pharmacokinetics of pegylated liposomal doxorubicin: Review of animal and human studies. Clin. Pharm. 2003, 42, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Meshorer, A.; Barenholz, Y. Comparative long-term study of the toxicities of free and liposome-associated doxorubicin in mice after intravenous administration. J. Natl. Cancer Inst. 1986, 77, 459–469. [Google Scholar]
- Gabizon, A.; Shiota, R.; Papahadjopoulos, D. Pharmacokinetics and tissue distribution of doxorubicin encapsulated in stable liposomes with long circulation times. J. Natl. Cancer Inst. 1989, 81, 1484–1488. [Google Scholar] [CrossRef]
- Gabizon, A.; Catane, R.; Uziely, B.; Kaufman, B.; Safra, T.; Cohen, R.; Martin, F.; Huang, A.; Barenholz, Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar]
- Harrington, K.J.; Mohammadtaghi, S.; Uster, P.S.; Glass, D.; Peters, A.M.; Vile, R.G.; Stewart, J.S. Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylated liposomes. Clin. Cancer Res. 2001, 7, 243–254. [Google Scholar]
- Uziely, B.; Jeffers, S.; Isacson, R.; Kutsch, K.; Wei-Tsao, D.; Yehoshua, Z.; Libson, E.; Muggia, F.M.; Gabizon, A. Liposomal doxorubicin: Antitumor activity and unique toxicities during two complementary phase i studies. J. Clin. Oncol. 1995, 13, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Hong, R.L.; Huang, C.J.; Tseng, Y.L.; Pang, V.F.; Chen, S.T.; Liu, J.J.; Chang, F.H. Direct comparison of liposomal doxorubicin with or without polyethylene glycol coating in c-26 tumor-bearing mice: Is surface coating with polyethylene glycol beneficial? Clin. Cancer Res. 1999, 5, 3645–3652. [Google Scholar] [PubMed]
- Parr, M.J.; Masin, D.; Cullis, P.R.; Bally, M.B. Accumulation of liposomal lipid and encapsulated doxorubicin in murine lewis lung carcinoma: The lack of beneficial effects by coating liposomes with poly(ethylene glycol). J. Pharmacol. Exp. Ther. 1997, 280, 1319–1327. [Google Scholar] [PubMed]
- Eberhard, A.; Kahlert, S.; Goede, V.; Hemmerlein, B.; Plate, K.H.; Augustin, H.G. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: Implications for antiangiogenic tumor therapies. Cancer Res. 2000, 60, 1388–1393. [Google Scholar] [PubMed]
- El Emir, E.; Qureshi, U.; Dearling, J.L.; Boxer, G.M.; Clatworthy, I.; Folarin, A.A.; Robson, M.P.; Nagl, S.; Konerding, M.A.; Pedley, R.B. Predicting response to radioimmunotherapy from the tumor microenvironment of colorectal carcinomas. Cancer Res. 2007, 67, 11896–11905. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Shields, A.F.; Siegel, B.A.; Miller, K.D.; Krop, I.; Ma, C.X.; LoRusso, P.M.; Munster, P.N.; Campbell, K.; Gaddy, D.F.; et al. (64)cu-mm-302 positron emission tomography quantifies variability of enhanced permeability and retention of nanoparticles in relation to treatment response in patients with metastatic breast cancer. Clin. Cancer Res. 2017, 23, 4190–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hare, J.I.; Lammers, T.; Ashford, M.B.; Puri, S.; Storm, G.; Barry, S.T. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Adv. Drug Deliv. Rev. 2017, 108, 25–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammers, T.; Kiessling, F.; Ashford, M.; Hennink, W.; Crommelin, D.; Storm, G. Cancer nanomedicine: Is targeting our target? Nat. Rev. Mater. 2016, 1, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Islam, W.; Maeda, H. Exploiting the dynamics of the epr effect and strategies to improve the therapeutic effects of nanomedicines by using epr effect enhancers. Adv. Drug Deliv. Rev. 2020, 157, 142–160. [Google Scholar] [CrossRef]
- Golombek, S.K.; May, J.N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via epr: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef]
- Wu, J. The enhanced permeability and retention (epr) effect: The significance of the concept and methods to enhance its application. J. Pers. Med. 2021, 11, 771. [Google Scholar] [CrossRef]
- Shi, Y.; van der Meel, R.; Chen, X.; Lammers, T. The epr effect and beyond: Strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics 2020, 10, 7921–7924. [Google Scholar] [CrossRef]
- Kobayashi, H.; Watanabe, R.; Choyke, P.L. Improving conventional enhanced permeability and retention (epr) effects; what is the appropriate target? Theranostics 2013, 4, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denekamp, J. Vascular endothelium as the vulnerable element in tumours. Acta Radiol. Oncol. 1984, 23, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Takeuchi, Y.; Takada, M.; Sadzuka, Y.; Namba, Y.; Oku, N. Anti-neovascular therapy by use of tumor neovasculature-targeted long-circulating liposome. J. Control Release 2004, 100, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Oku, N.; Asai, T.; Watanabe, K.; Kuromi, K.; Nagatsuka, M.; Kurohane, K.; Kikkawa, H.; Ogino, K.; Tanaka, M.; Ishikawa, D.; et al. Anti-neovascular therapy using novel peptides homing to angiogenic vessels. Oncogene 2002, 21, 2662–2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corti, A.; Pastorino, F.; Curnis, F.; Arap, W.; Ponzoni, M.; Pasqualini, R. Targeted drug delivery and penetration into solid tumors. Med. Res. Rev. 2012, 32, 1078–1091. [Google Scholar] [CrossRef]
- Wu, P.H.; Opadele, A.E.; Onodera, Y.; Nam, J.M. Targeting integrins in cancer nanomedicine: Applications in cancer diagnosis and therapy. Cancers 2019, 11, 1783. [Google Scholar] [CrossRef] [Green Version]
- Dechantsreiter, M.A.; Planker, E.; Matha, B.; Lohof, E.; Holzemann, G.; Jonczyk, A.; Goodman, S.L.; Kessler, H. N-methylated cyclic rgd peptides as highly active and selective alpha(v)beta(3) integrin antagonists. J. Med. Chem. 1999, 42, 3033–3040. [Google Scholar] [CrossRef]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate. Design, synthesis and clinical evaluation. Anti Cancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [Green Version]
- Posey, J.A.; Khazaeli, M.B.; DelGrosso, A.; Saleh, M.N.; Lin, C.Y.; Huse, W.; LoBuglio, A.F. A pilot trial of vitaxin, a humanized anti-vitronectin receptor (anti alpha v beta 3) antibody in patients with metastatic cancer. Cancer Biother. Radiopharm. 2001, 16, 125–132. [Google Scholar] [CrossRef]
- Seguin, J.; Nicolazzi, C.; Mignet, N.; Scherman, D.; Chabot, G.G. Vascular density and endothelial cell expression of integrin alpha v beta 3 and e-selectin in murine tumours. Tumour Biol. 2012, 33, 1709–1717. [Google Scholar] [CrossRef] [Green Version]
- Amin, M.; Mansourian, M.; Koning, G.A.; Badiee, A.; Jaafari, M.R.; Ten Hagen, T.L. Development of a novel cyclic rgd peptide for multiple targeting approaches of liposomes to tumor region. J. Control Release 2015, 220, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Badiee, A.; Jaafari, M.R. Improvement of pharmacokinetic and antitumor activity of pegylated liposomal doxorubicin by targeting with n-methylated cyclic rgd peptide in mice bearing c-26 colon carcinomas. Int. J. Pharm. 2013, 458, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, K.E.; Kessler, H. The structures of integrins and integrin-ligand complexes: Implications for drug design and signal transduction. Angew. Chem. Int. Ed. Engl. 2002, 41, 3767–3774. [Google Scholar] [CrossRef]
- Wang, F.; Wang, Y.; Zhang, X.; Zhang, W.; Guo, S.; Jin, F. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J. Control Release 2014, 174, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Sarko, D.; Beijer, B.; Garcia Boy, R.; Nothelfer, E.M.; Leotta, K.; Eisenhut, M.; Altmann, A.; Haberkorn, U.; Mier, W. The pharmacokinetics of cell-penetrating peptides. Mol. Pharm. 2010, 7, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Bechara, C.; Sagan, S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef]
- Brooks, H.; Lebleu, B.; Vives, E. Tat peptide-mediated cellular delivery: Back to basics. Adv. Drug Deliv. Rev. 2005, 57, 559–577. [Google Scholar] [CrossRef]
- Amin, M.; Bagheri, M.; Mansourian, M.; Jaafari, M.R.; Ten Hagen, T.L. Regulation of in vivo behavior of tat-modified liposome by associated protein corona and avidity to tumor cells. Int. J. Nanomed. 2018, 13, 7441–7455. [Google Scholar] [CrossRef] [Green Version]
- Suckau, D.; Resemann, A.; Schuerenberg, M.; Hufnagel, P.; Franzen, J.; Holle, A. A novel maldi lift-tof/tof mass spectrometer for proteomics. Anal. Bioanal. Chem. 2003, 376, 952–965. [Google Scholar] [CrossRef]
- Bolotin, E.M.; Cohen, R.; Bar, L.K.; Emanuel, N.; Ninio, S.; Lasic, D.D.; Barenholz, Y. Ammonium sulfate gradients for efficient and stable remote loading of amphipathic weak bases into liposomes and ligandoliposomes. J. Liposome Res. 1993, 4, 455–479. [Google Scholar] [CrossRef]
- Jaffe, E.A.; Nachman, R.L.; Becker, C.G.; Minick, C.R. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J. Clin. Invest. 1973, 52, 2745–2756. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, A.; Nagel, J.H.A.; Smid, M.; Lam, S.; Elstrodt, F.; Wasielewski, M.; Ng, S.S.; French, P.J.; Peeters, J.K.; Rozendaal, M.J.; et al. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res. Tr. 2010, 121, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Van Muijen, G.N.; Cornelissen, L.M.; Jansen, C.F.; Figdor, C.G.; Johnson, J.P.; Brocker, E.B.; Ruiter, D.J. Antigen expression of metastasizing and non-metastasizing human melanoma cells xenografted into nude mice. Clin. Exp. Metastasis 1991, 9, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Seynhaeve, A.L.; Ten Hagen, T.L. High-resolution intravital microscopy of tumor angiogenesis. Methods Mol. Biol. 2016, 1464, 115–127. [Google Scholar] [PubMed]
- Seynhaeve, A.L.B.; Ten Hagen, T.L.M. An adapted dorsal skinfold model used for 4d intravital followed by whole-mount imaging to reveal endothelial cell-pericyte association. Sci. Rep. 2021, 11, 20389. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Szoka, F.C., Jr. Sterol-modified phospholipids: Cholesterol and phospholipid chimeras with improved biomembrane properties. J. Am. Chem. Soc. 2008, 130, 15702–15712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Jaafari, M.R.; Szoka, F.C., Jr. Disterolphospholipids: Nonexchangeable lipids and their application to liposomal drug delivery. Angew. Chem. Int. Ed. Engl. 2009, 48, 4146–4149. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, A.; Seelig, J. Interaction of the protein transduction domain of hiv-1 tat with heparan sulfate: Binding mechanism and thermodynamic parameters. Biophys. J. 2004, 86, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Marty, C.; Meylan, C.; Schott, H.; Ballmer-Hofer, K.; Schwendener, R.A. Enhanced heparan sulfate proteoglycan-mediated uptake of cell-penetrating peptide-modified liposomes. Cell. Mol. Life Sci. 2004, 61, 1785–1794. [Google Scholar] [CrossRef] [Green Version]
- Mei, L.; Fu, L.; Shi, K.; Zhang, Q.; Liu, Y.; Tang, J.; Gao, H.; Zhang, Z.; He, Q. Increased tumor targeted delivery using a multistage liposome system functionalized with rgd, tat and cleavable peg. Int. J. Pharm. 2014, 468, 26–38. [Google Scholar] [CrossRef]
- Saul, J.M.; Annapragada, A.V.; Bellamkonda, R.V. A dual-ligand approach for enhancing targeting selectivity of therapeutic nanocarriers. J. Control Release 2006, 114, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Kibria, G.; Hatakeyama, H.; Ohga, N.; Hida, K.; Harashima, H. Dual-ligand modification of pegylated liposomes shows better cell selectivity and efficient gene delivery. J. Control Release 2011, 153, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Zhang, L.; Liu, Y.; Zhang, Q.; Qin, Y.; Yin, Y.; Yuan, W.; Yang, Y.; Xie, Y.; Zhang, Z.; et al. Synergistic targeted delivery of payload into tumor cells by dual-ligand liposomes co-modified with cholesterol anchored transferrin and tat. Int. J. Pharm. 2013, 454, 31–40. [Google Scholar] [CrossRef]
- Li, X.C.; Diao, W.B.; Xue, H.T.; Wu, F.; Wang, W.Y.; Jiang, B.; Bai, J.K.; Lian, B.; Feng, W.G.; Sun, T.Y.; et al. Improved efficacy of doxorubicin delivery by a novel dual-ligand-modified liposome in hepatocellular carcinoma. Cancer Lett. 2020, 489, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Kluza, E.; Jacobs, I.; Hectors, S.J.; Mayo, K.H.; Griffioen, A.W.; Strijkers, G.J.; Nicolay, K. Dual-targeting of alphavbeta3 and galectin-1 improves the specificity of paramagnetic/fluorescent liposomes to tumor endothelium in vivo. J. Control Release 2012, 158, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Murase, Y.; Asai, T.; Katanasaka, Y.; Sugiyama, T.; Shimizu, K.; Maeda, N.; Oku, N. A novel dds strategy, “dual-targeting”, and its application for antineovascular therapy. Cancer Lett. 2010, 287, 165–171. [Google Scholar] [CrossRef]
- Sugiyama, T.; Asai, T.; Nedachi, Y.M.; Katanasaka, Y.; Shimizu, K.; Maeda, N.; Oku, N. Enhanced active targeting via cooperative binding of ligands on liposomes to target receptors. PLoS ONE 2013, 8, e67550. [Google Scholar] [CrossRef]
- Ying, X.; Wen, H.; Lu, W.L.; Du, J.; Guo, J.; Tian, W.; Men, Y.; Zhang, Y.; Li, R.J.; Yang, T.Y.; et al. Dual-targeting daunorubicin liposomes improve the therapeutic efficacy of brain glioma in animals. J. Control Release 2010, 141, 183–192. [Google Scholar] [CrossRef]
- Loureiro, J.A.; Gomes, B.; Fricker, G.; Cardoso, I.; Ribeiro, C.A.; Gaiteiro, C.; Coelho, M.A.; Pereira Mdo, C.; Rocha, S. Dual ligand immunoliposomes for drug delivery to the brain. Colloids Surf. B Biointerfaces 2015, 134, 213–219. [Google Scholar] [CrossRef]
- Rodrigues, B.D.; Kanekiyo, T.; Singh, J. In vitro and in vivo characterization of cpp and transferrin modified liposomes encapsulating pdna. Nanomed. Nanotechnol. Biol. Med. 2020, 28, 102225. [Google Scholar] [CrossRef]
- Zheng, C.Y.; Ma, C.Y.; Bai, E.Q.; Yang, K.; Xu, R.X. Transferrin and cell-penetrating peptide dual-functioned liposome for targeted drug delivery to glioma. Int. J. Clin. Exp. Med. 2015, 8, 1658–1668. [Google Scholar] [PubMed]
- Lakkadwala, S.; Rodrigues, B.D.; Sun, C.W.; Singh, J. Dual functionalized liposomes for efficient co-delivery of anti-cancer chemotherapeutics for the treatment of glioblastoma. J. Control. Release 2019, 307, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, P.; Jhaveri, A.; Pattni, B.; Biswas, S.; Torchilin, V. Transferrin and octaarginine modified dual-functional liposomes with improved cancer cell targeting and enhanced intracellular delivery for the treatment of ovarian cancer. Drug Deliv. 2018, 25, 517–532. [Google Scholar] [CrossRef]
- Zhu, Y.Q.; Feijen, J.; Zhong, Z.Y. Dual-targeted nanomedicines for enhanced tumor treatment. Nano Today 2018, 18, 65–85. [Google Scholar] [CrossRef]
- Belfiore, L.; Saunders, D.N.; Ranson, M.; Thurecht, K.J.; Storm, G.; Vine, K.L. Towards clinical translation of ligand-functionalized liposomes in targeted cancer therapy: Challenges and opportunities. J. Control Release 2018, 277, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boles, K.; Schmieder, A.; Caruthers, S.; Hu, G.; Scott, M.; Zhang, H.Y.; Reynolds, B.; Wickline, S.; Lanza, G. Mr angiogenesis imaging with robo4-versus alpha(v)beta(3)- targeted nanoparticles in the b16/f10 mouse melanoma model. FASEB J. 2010, 24. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preparation Name | Density of Surface-Inserted Peptides (Peptides/ Liposome) | ζ—Average (nm) | PDI 1 | ζ Potential (mv) | |
|---|---|---|---|---|---|
| RGD | TAT | ||||
| Plain-PLD | 0 | 0 | 105 ± 4.3 | 0.085 ± 0.01 | −27.1 ± 2.1 |
| T1-PLD | 0 | 100 | 102 ± 5.2 | 0.073 ± 0.01 | −25.3 ± 1.7 |
| T2-PLD | 0 | 200 | 106 ± 6.3 | 0.091 ± 0.01 | −24.2 ± 1.4 |
| R3-PLD | 300 | 0 | 101 ± 2.4 | 0.048 ± 0.01 | −26.3 ± 1.8 |
| D31-PLD | 300 | 100 | 100 ± 3.8 | 0.052 ± 0.01 | −24.9 ± 1.3 |
| D32-PLD | 300 | 200 | 104 ± 5.2 | 0.055 ± 0.02 | −22.8 ± 0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amin, M.; Mansourian, M.; Burgers, P.C.; Amin, B.; Jaafari, M.R.; ten Hagen, T.L.M. Increased Targeting Area in Tumors by Dual-Ligand Modification of Liposomes with RGD and TAT Peptides. Pharmaceutics 2022, 14, 458. https://doi.org/10.3390/pharmaceutics14020458
Amin M, Mansourian M, Burgers PC, Amin B, Jaafari MR, ten Hagen TLM. Increased Targeting Area in Tumors by Dual-Ligand Modification of Liposomes with RGD and TAT Peptides. Pharmaceutics. 2022; 14(2):458. https://doi.org/10.3390/pharmaceutics14020458
Chicago/Turabian StyleAmin, Mohamadreza, Mercedeh Mansourian, Peter C. Burgers, Bahareh Amin, Mahmoud Reza Jaafari, and Timo L. M. ten Hagen. 2022. "Increased Targeting Area in Tumors by Dual-Ligand Modification of Liposomes with RGD and TAT Peptides" Pharmaceutics 14, no. 2: 458. https://doi.org/10.3390/pharmaceutics14020458