
Antifungal and Antibiofilm Activity of Cyclic Temporin L Peptide Analogues against Albicans and Non-Albicans Candida Species

 , , ,
, , ,  ,
,  ,
,  ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Peptide Synthesis
2.3. Fungal Culture Conditions
2.4. Determination of Minimal Inhibitory Concentration (MIC) and Minimal Fungicidal Concentration (MFC)
2.5. Fungal Biofilm Formation
2.6. Antibiofilm Activity
2.7. Bright-Field Microscopy
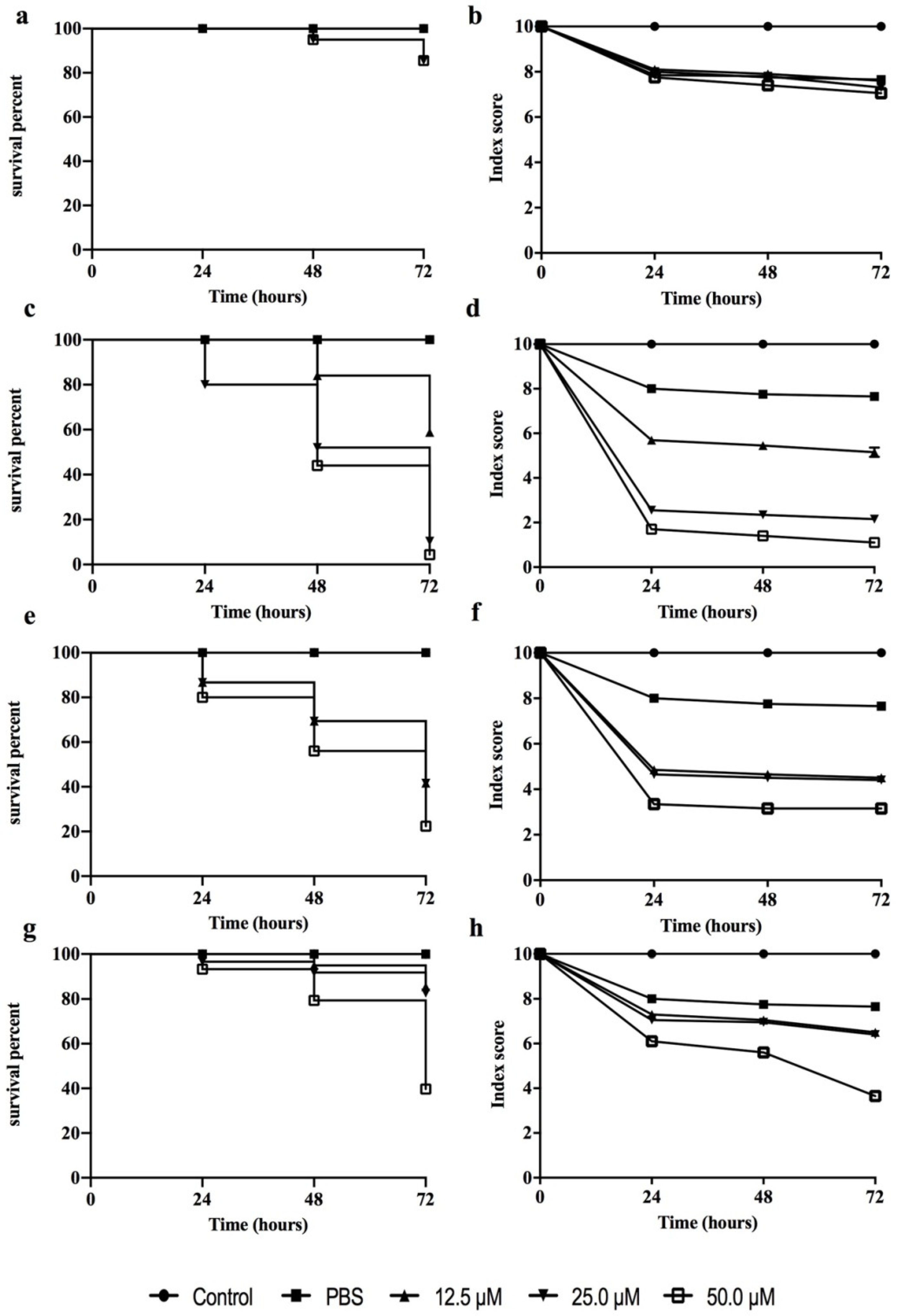
2.8. Effect of Peptide Toxicity on G. mellonella
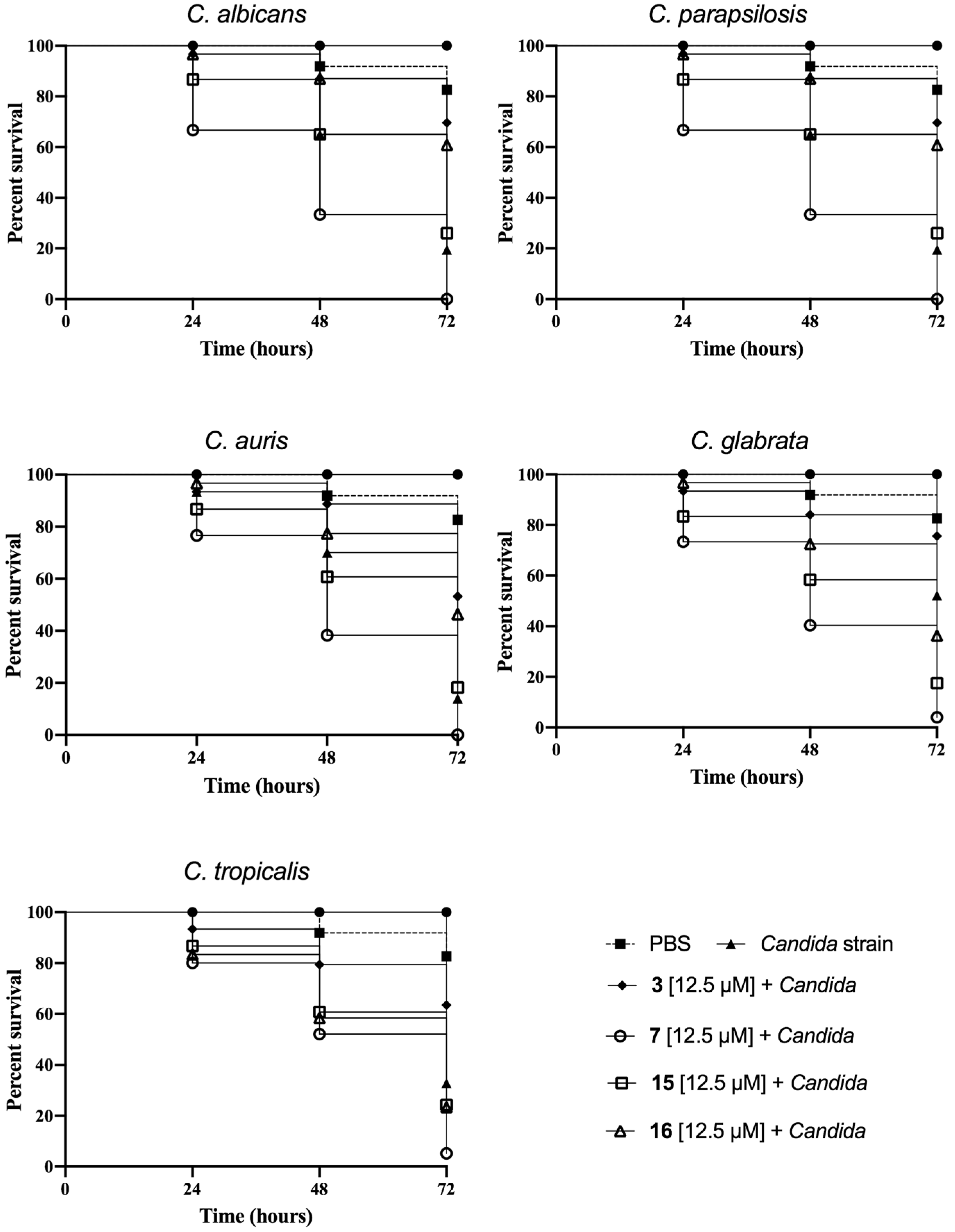
2.9. Kaplan–Meier Survival Analysis
2.10. Mutagenicity with S. typhimurium (Fluctuation Ames Test)
2.11. ANTS/DPX Leakage
2.12. Zeta Potential Measured by Dynamic Light Scattering
2.13. Critical Aggregation Concentration Calculated Using Nile Red as a Probe
2.14. Peptide Aggregation Monitored by a Thioflavin T (ThT) Fluorescence Assay
2.15. Secondary Structural Analysis
2.16. Statistical Analysis
2.17. Atomic Force Microscopy Imaging
3. Results
3.1. Peptide Synthesis
3.2. Peptide Screening by MIC for Candida Species
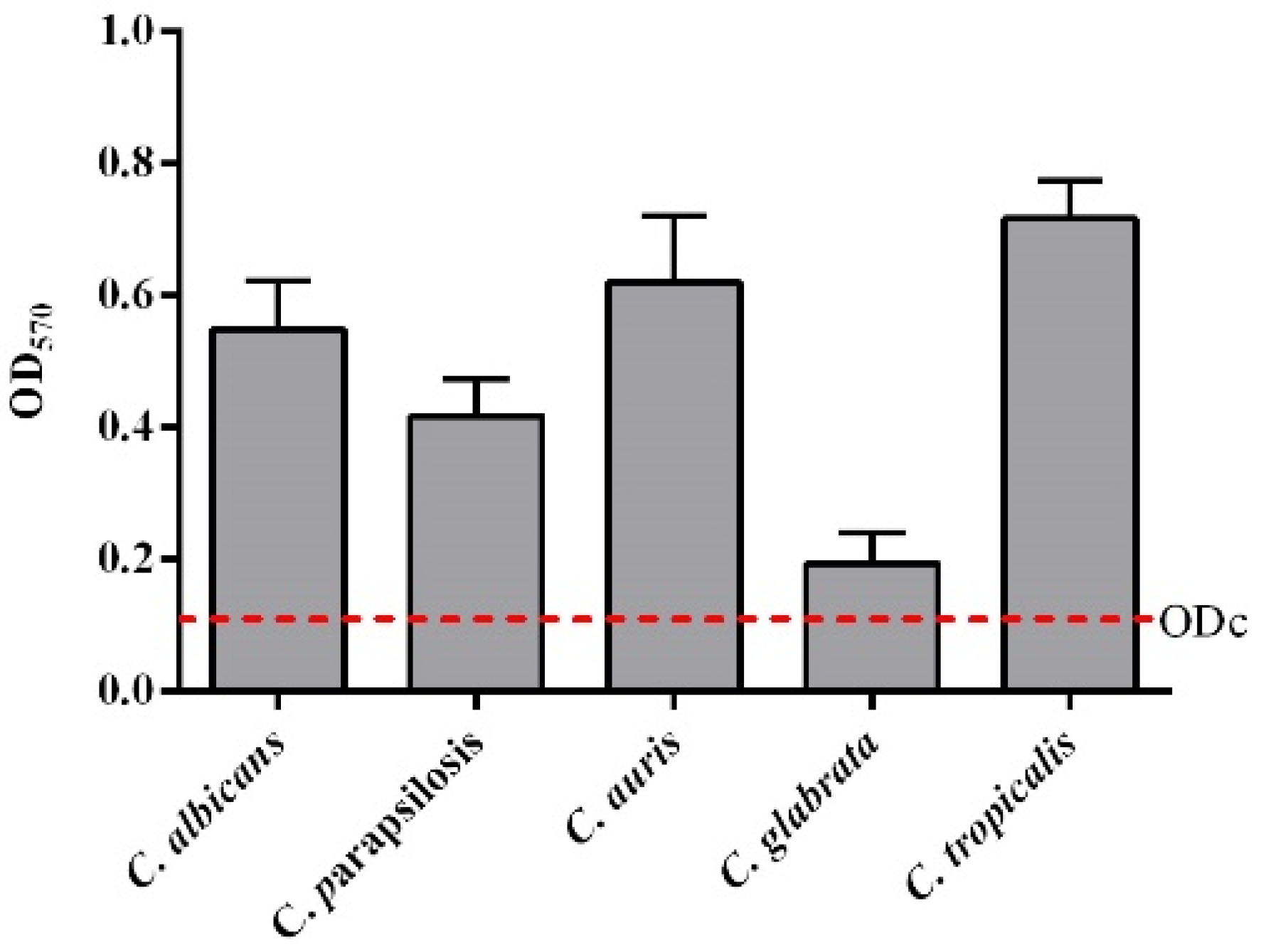
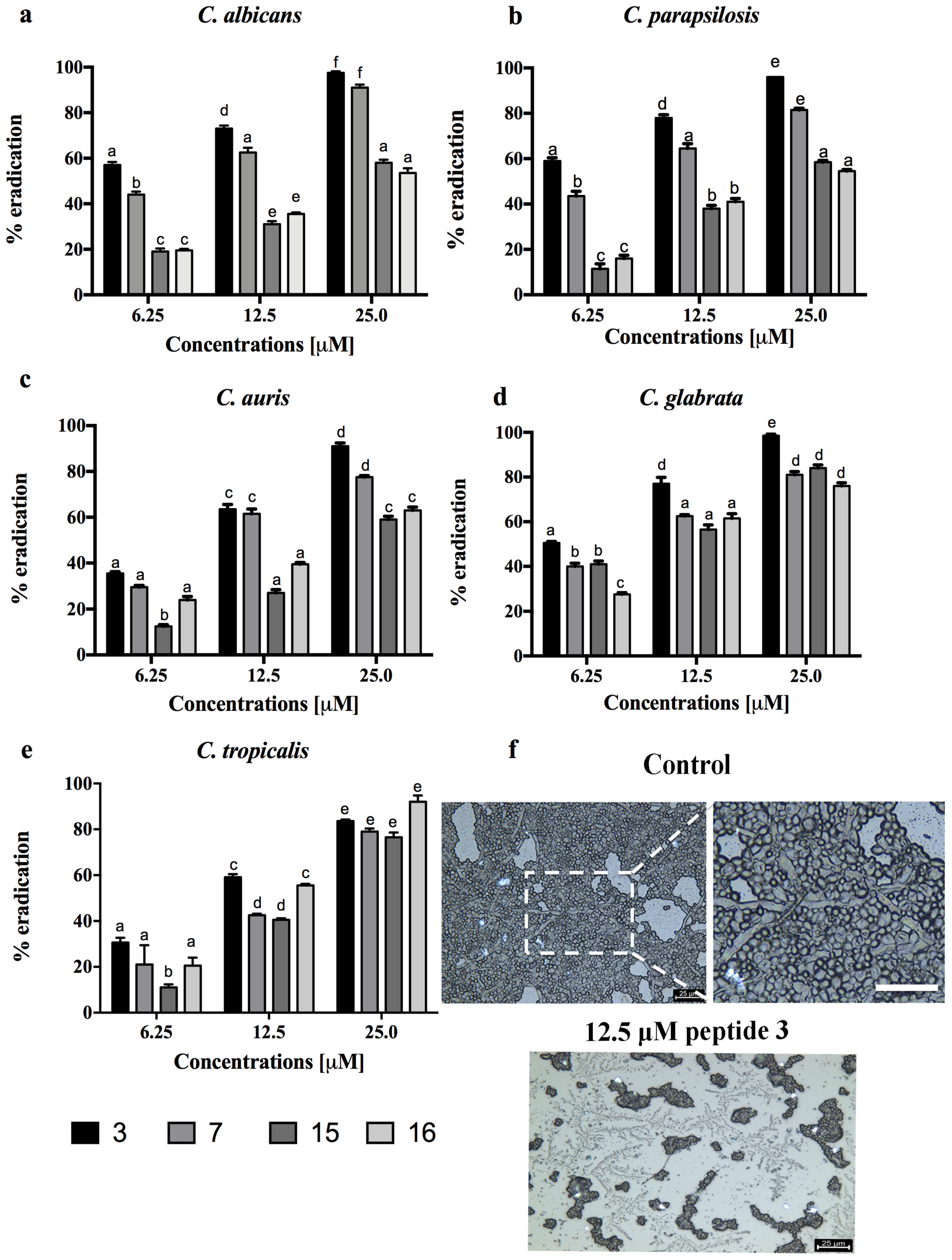
3.3. Activity against Candida Species Biofilm for a Selection of Peptides 3, 7, 15 and 16
3.4. Peptide Toxicity against G. mellonella Larvae
3.5. Peptide Effect on Infected G. mellonella Larvae
3.6. Peptides Did Not Induce Mutagenesis in Ames Test
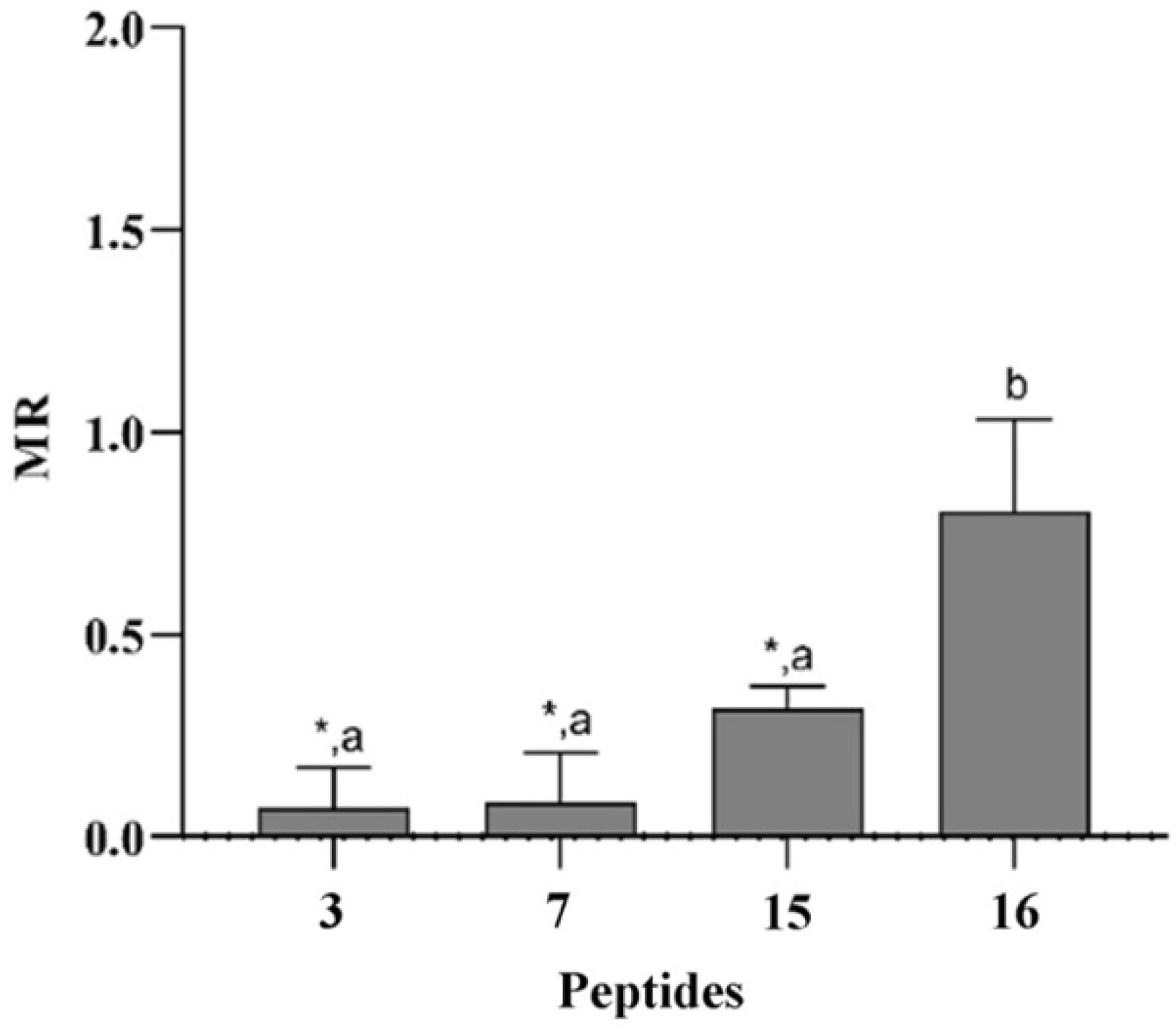
3.7. Correlation between Structural Features and Eukaryotic Toxicity
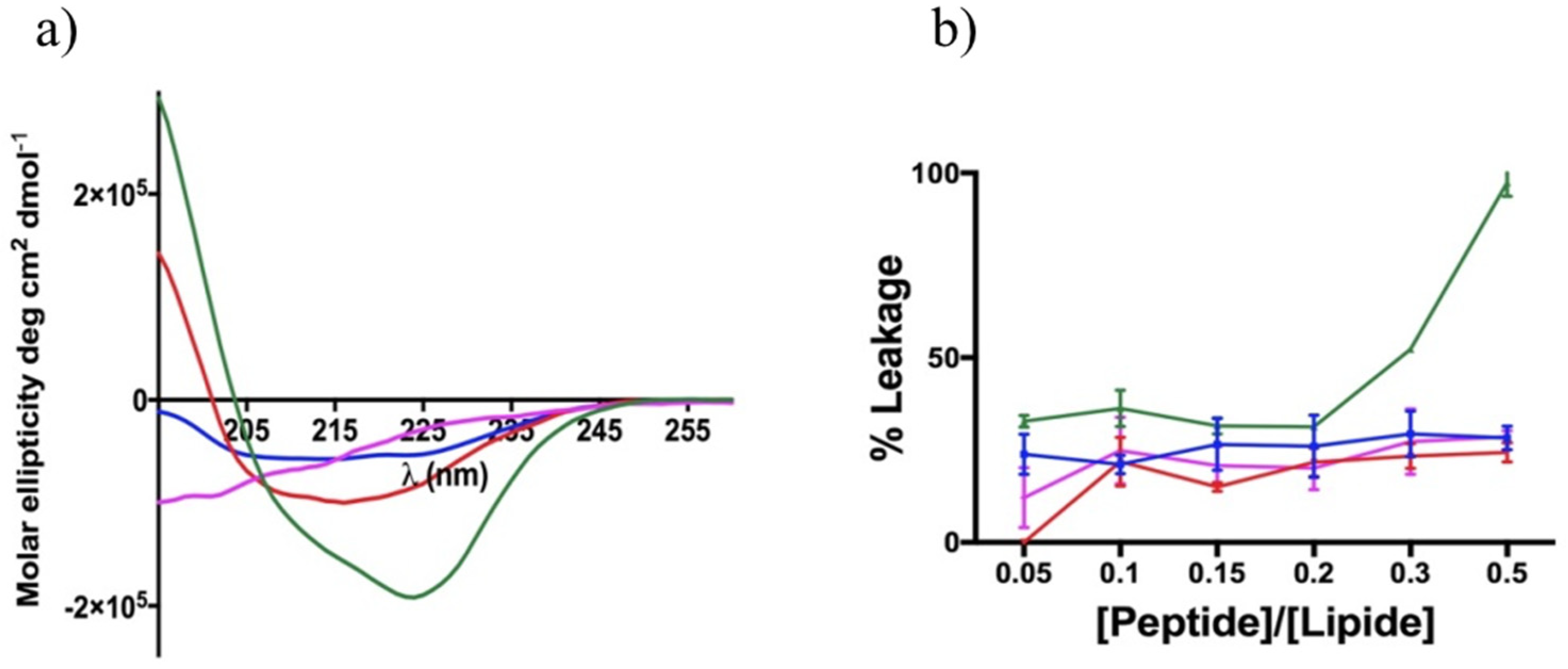
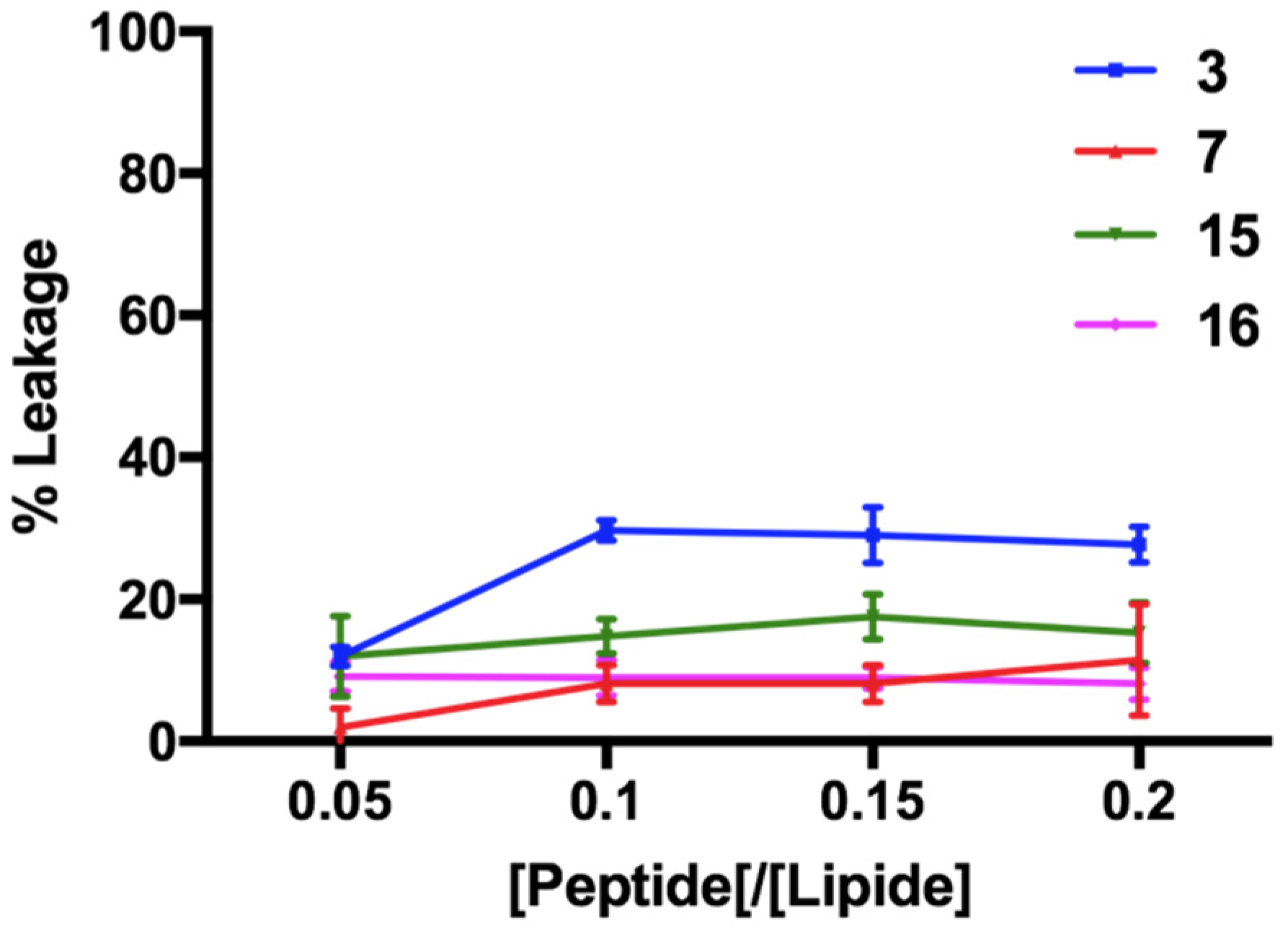
3.8. Non-Permeabilizing Action of Stapled Peptides in Fungal Membranes
3.9. Peptide Interaction with Lipid Fungal Membranes
3.10. Peptide Aggregation in Different Environments
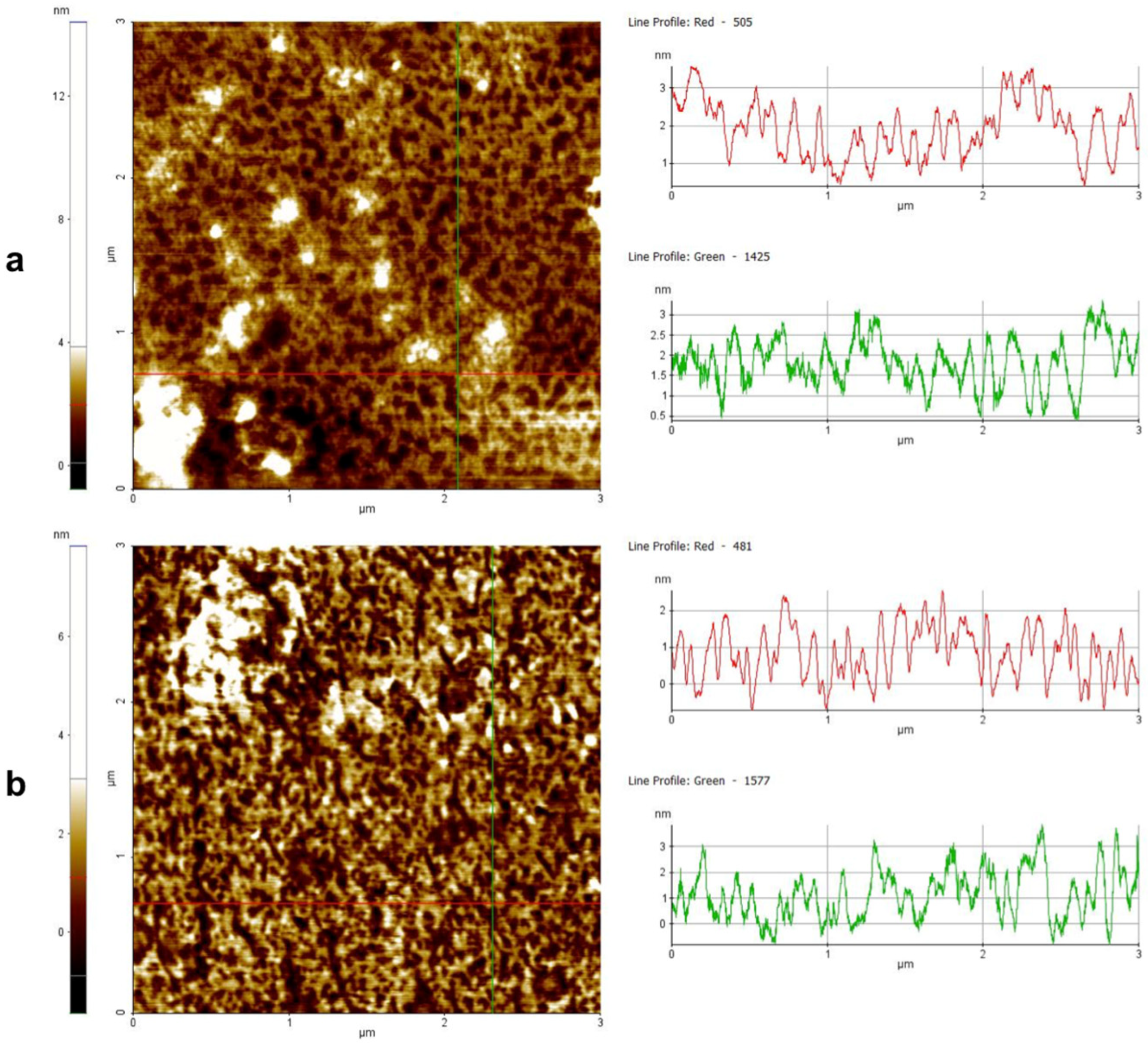
3.11. Peptide Aggregation Imaged by AFM
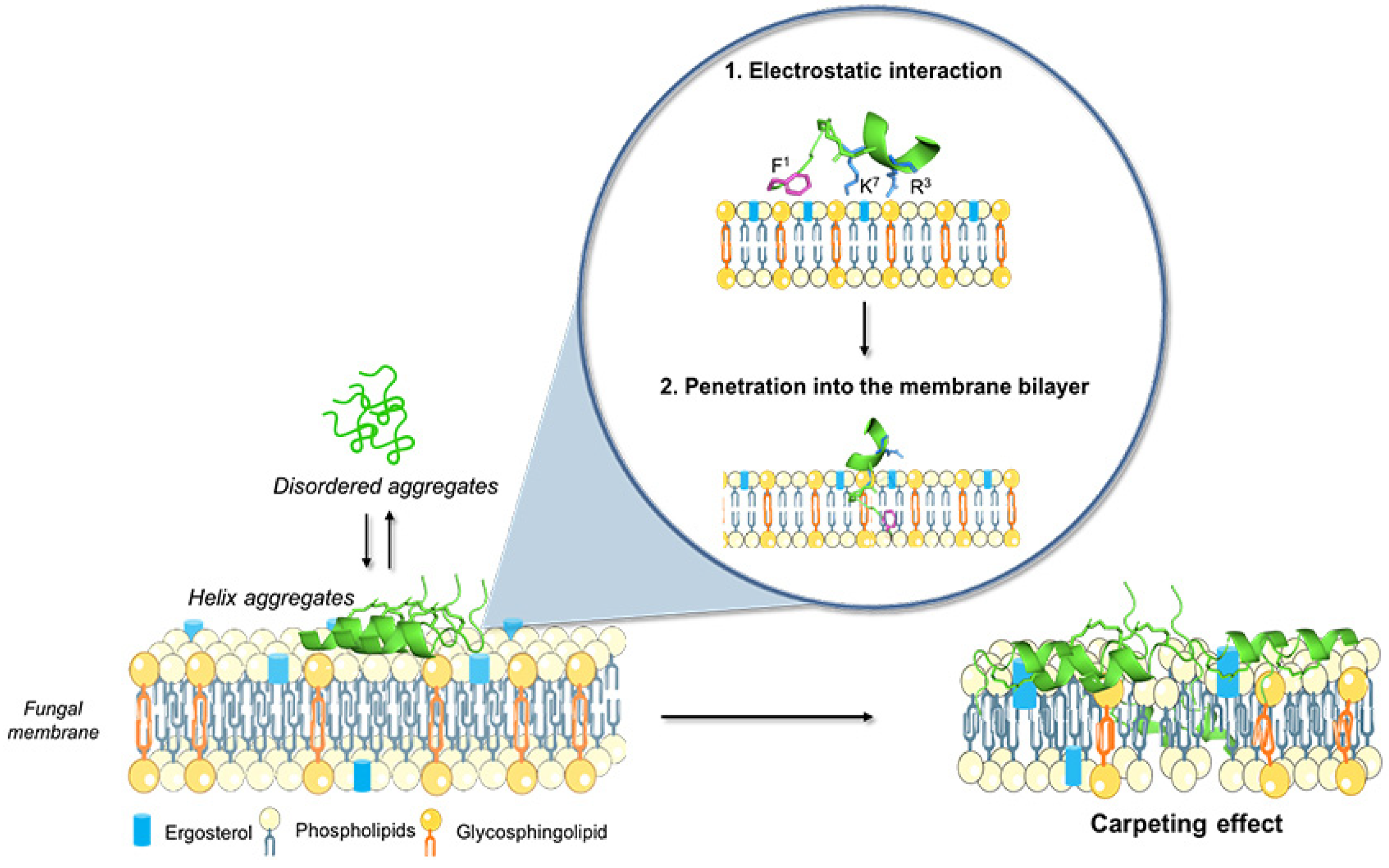
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kojic, E.M.; Darouiche, R.O. Candida infections of medical devices. Clin. Microbiol. Rev. 2004, 17, 255–267. [Google Scholar] [CrossRef] [Green Version]
- Cavalheiro, M.; Teixeira, M.C. Candida biofilms: Threats, challenges, and promising strategies. Front. Med. (Lausanne) 2018, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Douglas, L.J. Medical importance of biofilms in candida infections. Rev. Iberoam. Micol. 2002, 19, 139–143. [Google Scholar]
- Silva, S.; Negri, M.; Henriques, M.; Oliveira, R.; Williams, D.W.; Azeredo, J. Candida glabrata, candida parapsilosis and candida tropicalis: Biology, epidemiology, pathogenicity and antifungal resistance. FEMS Microbiol. Rev. 2012, 36, 288–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, S.; Henriques, M.; Martins, A.; Oliveira, R.; Williams, D.; Azeredo, J. Biofilms of non-candida albicans candida species: Quantification, structure and matrix composition. Med. Mycol. 2009, 47, 681–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardi, J.C.O.; Scorzoni, L.; Bernardi, T.; Fusco-Almeida, A.M.; Mendes Giannini, M.J.S. Candida species: Current epidemiology, pathogenicity, biofilm formation, natural antifungal products and new therapeutic options. J. Med. Microbiol. 2013, 62, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Nobile, C.J.; Johnson, A.D. Candida albicans biofilms and human disease. Annu. Rev. Microbiol. 2015, 69, 71–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, S.; Henriques, M.; Oliveira, R.; Williams, D.; Azeredo, J. In vitro biofilm activity of non-candida albicans candida species. Curr. Microbiol. 2010, 61, 534–540. [Google Scholar] [CrossRef] [Green Version]
- Bidaud, A.L.; Chowdhary, A.; Dannaoui, E. Candida auris: An emerging drug resistant yeast-a mini-review. J. Mycol. Med. 2018, 28, 568–573. [Google Scholar] [CrossRef]
- Tóth, R.; Nosek, J.; Mora-Montes, H.M.; Gabaldon, T.; Bliss, J.M.; Nosanchuk, J.D.; Turner, S.A.; Butler, G.; Vágvölgyi, C.; Gácser, A. Candida parapsilosis: From genes to the bedside. Clin. Microbiol. Rev. 2019, 32, e00111-18. [Google Scholar] [CrossRef] [Green Version]
- Sasso, M.; Roger, C.; Lachaud, L. Rapid emergence of fks mutations in candida glabrata isolates in a peritoneal candidiasis. Med. Mycol. Case Rep. 2017, 16, 28–30. [Google Scholar] [CrossRef]
- Santos, F.; Leite-Andrade, M.C.; Brandão, I.S.; Alves, A.; Buonafina, M.D.S.; Nunes, M.; Araújo-Neto, L.N.; Freitas, M.A.; Brayner, F.A.; Alves, L.C.; et al. Anti-biofilm effect by the combined action of fluconazole and acetylsalicylic acid against species of candida parapsilosis complex. Infect. Genet. Evol. 2020, 84, 104378. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, A.; Sharma, C.; Meis, J.F. Azole-resistant aspergillosis: Epidemiology, molecular mechanisms, and treatment. J. Infect. Dis. 2017, 216, S436–S444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramage, G.; Rajendran, R.; Sherry, L.; Williams, C. Fungal biofilm resistance. Int. J. Microbiol. 2012, 2012, 528521. [Google Scholar] [CrossRef] [PubMed]
- Pierce, C.G.; Srinivasan, A.; Uppuluri, P.; Ramasubramanian, A.K.; López-Ribot, J.L. Antifungal therapy with an emphasis on biofilms. Curr. Opin. Pharmacol. 2013, 13, 726–730. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, T.; Mullick, J.B.; Bir, R.; Roy, j.; Sil, S.K. Determination of virulence factors and biofilm formation among isolates of vulvovaginal candidiasis. J. Med. Sci. 2016, 36, 53–58. [Google Scholar] [CrossRef]
- Buda De Cesare, G.; Cristy, S.A.; Garsin, D.A.; Lorenz, M.C. Antimicrobial peptides: A new frontier in antifungal therapy. mBio 2020, 11, e02123-20. [Google Scholar] [CrossRef] [PubMed]
- Struyfs, C.; Cammue, B.P.A.; Thevissen, K. Membrane-interacting antifungal peptides. Front. Cell Dev. Biol. 2021, 9, 649875. [Google Scholar] [CrossRef]
- Galdiero, S.; Falanga, A.; Berisio, R.; Grieco, P.; Morelli, G.; Galdiero, M. Antimicrobial peptides as an opportunity against bacterial diseases. Curr. Med. Chem. 2015, 22, 1665–1677. [Google Scholar] [CrossRef]
- Wang, K.; Dang, W.; Xie, J.; Zhu, R.; Sun, M.; Jia, F.; Zhao, Y.; An, X.; Qiu, S.; Li, X.; et al. Antimicrobial peptide protonectin disturbs the membrane integrity and induces ros production in yeast cells. Biochim. Biophys. Acta 2015, 1848, 2365–2373. [Google Scholar] [CrossRef] [Green Version]
- Scarsini, M.; Tomasinsig, L.; Arzese, A.; D’Este, F.; Oro, D.; Skerlavaj, B. Antifungal activity of cathelicidin peptides against planktonic and biofilm cultures of candida species isolated from vaginal infections. Peptides 2015, 71, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Romero, S.M.; Cardillo, A.B.; Martínez Ceron, M.C.; Camperi, S.A.; Giudicessi, S.L. Temporins: An approach of potential pharmaceutic candidates. Surg. Infect. (Larchmt) 2020, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Simmaco, M.; Mignogna, G.; Canofeni, S.; Miele, R.; Mangoni, M.L.; Barra, D. Temporins, antimicrobial peptides from the european red frog rana temporaria. Eur. J. Biochem. 1996, 242, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L.; Papo, N.; Barra, D.; Simmaco, M.; Bozzi, A.; Di Giulio, A.; Rinaldi, A.C. Effects of the antimicrobial peptide temporin L on cell morphology, membrane permeability and viability of escherichia coli. Biochem. J. 2004, 380, 859–865. [Google Scholar] [CrossRef] [Green Version]
- Carotenuto, A.; Malfi, S.; Saviello, M.R.; Campiglia, P.; Gomez-Monterrey, I.; Mangoni, M.L.; Gaddi, L.M.; Novellino, E.; Grieco, P. A different molecular mechanism underlying antimicrobial and hemolytic actions of temporins A and L. J. Med. Chem. 2008, 51, 2354–2362. [Google Scholar] [CrossRef]
- Merlino, F.; Carotenuto, A.; Casciaro, B.; Martora, F.; Loffredo, M.R.; Di Grazia, A.; Yousif, A.M.; Brancaccio, D.; Palomba, L.; Novellino, E.; et al. Glycine-replaced derivatives of [Pro3,DLeu9]TL, a temporin l analogue: Evaluation of antimicrobial, cytotoxic and hemolytic activities. Eur. J. Med. Chem. 2017, 139, 750–761. [Google Scholar] [CrossRef]
- Buommino, E.; Carotenuto, A.; Antignano, I.; Bellavita, R.; Casciaro, B.; Loffredo, M.R.; Merlino, F.; Novellino, E.; Mangoni, M.L.; Nocera, F.P.; et al. The outcomes of decorated prolines in the discovery of antimicrobial peptides from temporin-L. ChemMedChem 2019, 14, 1283–1290. [Google Scholar] [CrossRef]
- Bellavita, R.; Falanga, A.; Buommino, E.; Merlino, F.; Casciaro, B.; Cappiello, F.; Mangoni, M.L.; Novellino, E.; Catania, M.R.; Paolillo, R.; et al. Novel temporin l antimicrobial peptides: Promoting self-assembling by lipidic tags to tackle superbugs. J. Enzym. Inhib. Med. Chem. 2020, 35, 1751–1764. [Google Scholar] [CrossRef]
- Roscetto, E.; Bellavita, R.; Paolillo, R.; Merlino, F.; Molfetta, N.; Grieco, P.; Buommino, E.; Catania, M.R. Antimicrobial activity of a lipidated temporin L analogue against carbapenemase-producing klebsiella pneumoniae clinical isolates. Antibiotics 2021, 10, 1312. [Google Scholar] [CrossRef]
- Grieco, P.; Carotenuto, A.; Auriemma, L.; Saviello, M.R.; Campiglia, P.; Gomez-Monterrey, I.M.; Marcellini, L.; Luca, V.; Barra, D.; Novellino, E.; et al. The effect of d-amino acid substitution on the selectivity of temporin L towards target cells: Identification of a potent anti-candida peptide. Biochim. Biophys. Acta 2013, 1828, 652–660. [Google Scholar] [CrossRef]
- Bellavita, R.; Casciaro, B.; Di Maro, S.; Brancaccio, D.; Carotenuto, A.; Falanga, A.; Cappiello, F.; Buommino, E.; Galdiero, S.; Novellino, E.; et al. First-in-class cyclic temporin L analogue: Design, synthesis, and antimicrobial assessment. J. Med. Chem. 2021, 64, 11675–11694. [Google Scholar] [CrossRef] [PubMed]
- Merlino, F.; Tomassi, S.; Yousif, A.M.; Messere, A.; Marinelli, L.; Grieco, P.; Novellino, E.; Cosconati, S.; Di Maro, S. Boosting fmoc solid-phase peptide synthesis by ultrasonication. Org. Lett. 2019, 21, 6378–6382. [Google Scholar] [CrossRef] [PubMed]
- CLSI. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, 3th ed.; CLSI standard M27; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2008. [Google Scholar]
- de Alteriis, E.; Maselli, V.; Falanga, A.; Galdiero, S.; Di Lella, F.M.; Gesuele, R.; Guida, M.; Galdiero, E. Efficiency of gold nanoparticles coated with the antimicrobial peptide indolicidin against biofilm formation and development of candida spp. clinical isolates. Infect. Drug Resist. 2018, 11, 915–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanović, S.; Vuković, D.; Hola, V.; Di Bonaventura, G.; Djukić, S.; Cirković, I.; Ruzicka, F. Quantification of biofilm in microtiter plates: Overview of testing conditions and practical recommendations for assessment of biofilm production by staphylococci. Apmis 2007, 115, 891–899. [Google Scholar] [CrossRef]
- Stepanovic, S.; Vukovic, D.; Dakic, I.; Savic, B.; Svabic-Vlahovic, M. A modified microtiter-plate test for quantification of staphylococcal biofilm formation. J. Microbiol. Methods 2000, 40, 175–179. [Google Scholar] [CrossRef]
- Maione, A.; de Alteriis, E.; Carraturo, F.; Galdiero, S.; Falanga, A.; Guida, M.; Di Cosmo, A.; Maselli, V.; Galdiero, E. The membranotropic peptide gh625 to combat mixed candida albicans/klebsiella pneumoniae biofilm: Correlation between in vitro anti-biofilm activity and in vivo antimicrobial protection. J. Fungi 2021, 7, 26. [Google Scholar] [CrossRef]
- Loh, J.M.; Adenwalla, N.; Wiles, S.; Proft, T. Galleria mellonella larvae as an infection model for group a streptococcus. Virulence 2013, 4, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Vertyporokh, L.; Wojda, I. Immune response of galleria mellonella after injection with non-lethal and lethal dosages of candida albicans. J. Invertebr. Pathol. 2020, 170, 107327. [Google Scholar] [CrossRef]
- Ladokhin, A.S.; Wimley, W.C.; White, S.H. Leakage of membrane vesicle contents: Determination of mechanism using fluorescence requenching. Biophys. J. 1995, 69, 1964–1971. [Google Scholar] [CrossRef] [Green Version]
- Bondar, O.V.; Saifullina, D.V.; Shakhmaeva, I.I.; Mavlyutova, I.I.; Abdullin, T.I. Monitoring of the zeta potential of human cells upon reduction in their viability and interaction with polymers. Acta Nat. 2012, 4, 78–81. [Google Scholar] [CrossRef]
- Lombardi, L.; Shi, Y.; Falanga, A.; Galdiero, E.; de Alteriis, E.; Franci, G.; Chourpa, I.; Azevedo, H.S.; Galdiero, S. Enhancing the potency of antimicrobial peptides through molecular engineering and self-assembly. Biomacromolecules 2019, 20, 1362–1374. [Google Scholar] [CrossRef]
- Falanga, A.; Tarallo, R.; Vitiello, G.; Vitiello, M.; Perillo, E.; Cantisani, M.; D’Errico, G.; Galdiero, M.; Galdiero, S. Biophysical characterization and membrane interaction of the two fusion loops of glycoprotein b from herpes simplex type i virus. PLoS ONE 2012, 7, e32186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabartty, A.; Kortemme, T.; Baldwin, R.L. Helix propensities of the amino acids measured in alanine-based peptides without helix-stabilizing side-chain interactions. Protein Sci. 1994, 3, 843–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christenson, H.K.; Thomson, N.H. The nature of the air-cleaved mica surface. Surf. Sci. Rep. 2016, 71, 367–390. [Google Scholar] [CrossRef] [Green Version]
- Harding, C.R.; Schroeder, G.N.; Collins, J.W.; Frankel, G. Use of galleria mellonella as a model organism to study legionella pneumophila infection. J. Vis. Exp. 2013, 81, e50964. [Google Scholar]
- Mortelmans, K.; Zeiger, E. The ames salmonella/microsome mutagenicity assay. Mutat. Res. 2000, 455, 29–60. [Google Scholar] [CrossRef]
- Lau, S.Y.; Taneja, A.K.; Hodges, R.S. Synthesis of a model protein of defined secondary and quaternary structure. Effect of chain length on the stabilization and formation of two-stranded alpha-helical coiled-coils. J. Biol. Chem. 1984, 259, 13253–13261. [Google Scholar] [CrossRef]
- Oshiro, K.G.N.; Rodrigues, G.; Monges, B.E.D.; Cardoso, M.H.; Franco, O.L. Bioactive peptides against fungal biofilms. Front. Microbiol. 2019, 10, 2169. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Activity (MIC, µM) | |||
|---|---|---|---|
| Peptides | Sequence | C. albicans ATCC 90028 | C. parapsilosis DSM 5784 |
| 1 | F V P W F S K F [k G R I E] | >50 | >50 |
| 2 | F V P W F S K [K l G R E] L | >50 | >50 |
| 3 | F V P W F [K K F l E] R I L | 50 | 50 |
| 4 | F V P W [K S K F e] G R I L | >50 | >50 |
| 5 | F V P [K F S K E] l G R I L | >50 | >50 |
| 6 | F V P W F S K F [pra G R I Az] | >50 | >50 |
| 7 | F V P W F S K [Pra l G R Az] L | 50 | 50 |
| 8 | F V P W F [Pra K F l Az] R I L | >50 | >50 |
| 9 | F V P W [Pra S K F az] G R I L | >50 | >50 |
| 10 | F V P [Pra F S K Az] l G R I L | >50 | >50 |
| 11 | F V P W F [K F l G R I E] | >50 | >50 |
| 12 | F V P W [K S K F l G R E] L | >50 | >50 |
| 13 | F V P W F [Pra K F l G R I Az] | >50 | >50 |
| 14 | F V P W [Pra S K F l G R Az] L | >50 | >50 |
| 15 | F V P W F [E K F l K] R I L | >50 | 50 |
| 16 | F V P W F [C K F l C] R I L | 50 | 50 |
| 17 | F V P W F [S5 K F l S5] R I L | >50 | >50 |
| Peptides | ||||||||
|---|---|---|---|---|---|---|---|---|
| 3 | 7 | 15 | 16 | |||||
| MIC | MFC | MIC | MFC | MIC | MFC | MIC | MFC | |
| C. albicans ATCC 90028 | 50 | 50 | 50 | 50 | >50 | 50 | 50 | >50 |
| C. parapsilosis DSM 5784 | 50 | 50 | 50 | 50 | 50 | 50 | 50 | >50 |
| C. auris DSM 21092 | 50 | 50 | 50 | >50 | 50 | >50 | 50 | >50 |
| C. glabrata DSM 11226 | 50 | 50 | 50 | 50 | 50 | >50 | 25 | 50 |
| C. tropicalis DSM 11951 | 50 | 50 | 50 | 50 | 50 | >50 | 25 | 50 |
| Compound | Calculated Zeta Potential | Experimental Zeta Potential (ς, mV) |
|---|---|---|
| 3 | −3.2 ± 0.5 | 1.6 ± 0.3 |
| 7 | −3.2 ± 0.5 | −4.5 ± 1.6 |
| 15 | −3.2 ± 0.5 | 13.5 ± 3.4 |
| 16 | −3.2 ± 0.5 | −6.5 ± 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellavita, R.; Maione, A.; Merlino, F.; Siciliano, A.; Dardano, P.; De Stefano, L.; Galdiero, S.; Galdiero, E.; Grieco, P.; Falanga, A. Antifungal and Antibiofilm Activity of Cyclic Temporin L Peptide Analogues against Albicans and Non-Albicans Candida Species. Pharmaceutics 2022, 14, 454. https://doi.org/10.3390/pharmaceutics14020454
Bellavita R, Maione A, Merlino F, Siciliano A, Dardano P, De Stefano L, Galdiero S, Galdiero E, Grieco P, Falanga A. Antifungal and Antibiofilm Activity of Cyclic Temporin L Peptide Analogues against Albicans and Non-Albicans Candida Species. Pharmaceutics. 2022; 14(2):454. https://doi.org/10.3390/pharmaceutics14020454
Chicago/Turabian StyleBellavita, Rosa, Angela Maione, Francesco Merlino, Antonietta Siciliano, Principia Dardano, Luca De Stefano, Stefania Galdiero, Emilia Galdiero, Paolo Grieco, and Annarita Falanga. 2022. "Antifungal and Antibiofilm Activity of Cyclic Temporin L Peptide Analogues against Albicans and Non-Albicans Candida Species" Pharmaceutics 14, no. 2: 454. https://doi.org/10.3390/pharmaceutics14020454