
Preclinical PET Imaging of Granzyme B Shows Promotion of Immunological Response Following Combination Paclitaxel and Immune Checkpoint Inhibition in Triple Negative Breast Cancer

 , ,
, ,
Abstract
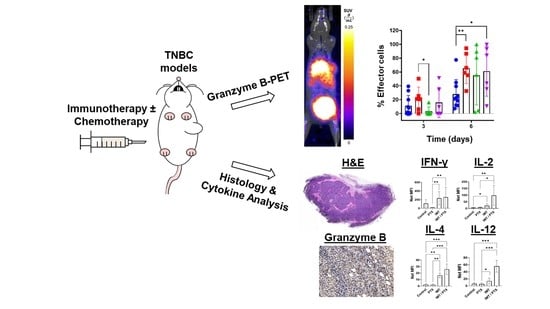
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Syngeneic Mouse Tumor Models
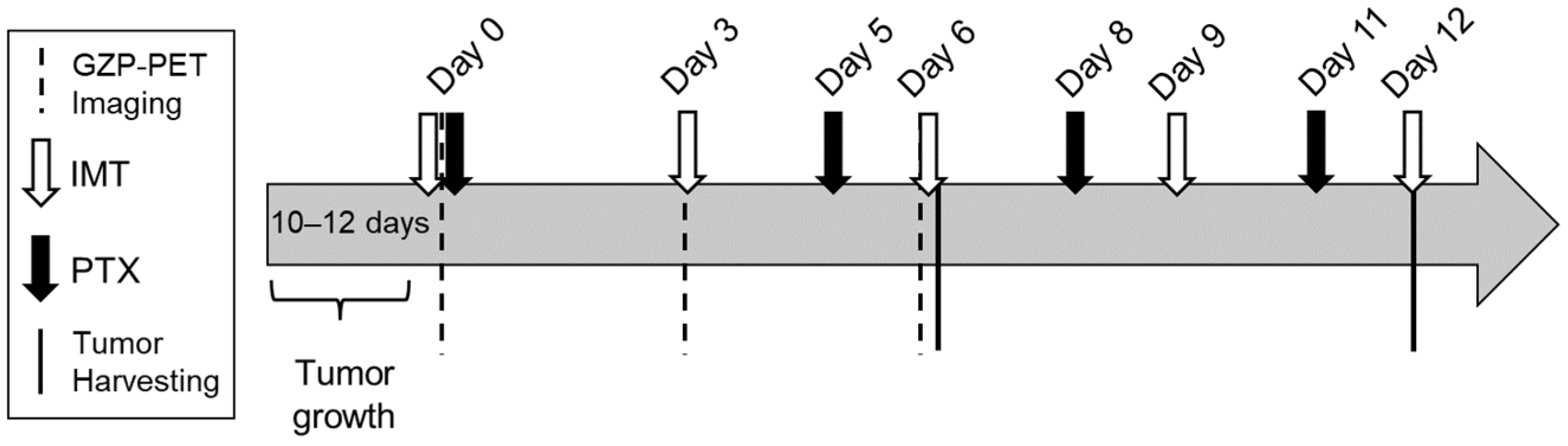
2.3. Treatments
2.4. NOTA-GZP Synthesis and 68Ga Radiolabeling
2.5. GZP-PET/CT Imaging and Analysis
2.6. Determination of Th1 and Th2 Expressed Cytokine Levels in Serum
2.7. Histological Analysis
2.8. Statistics
3. Results
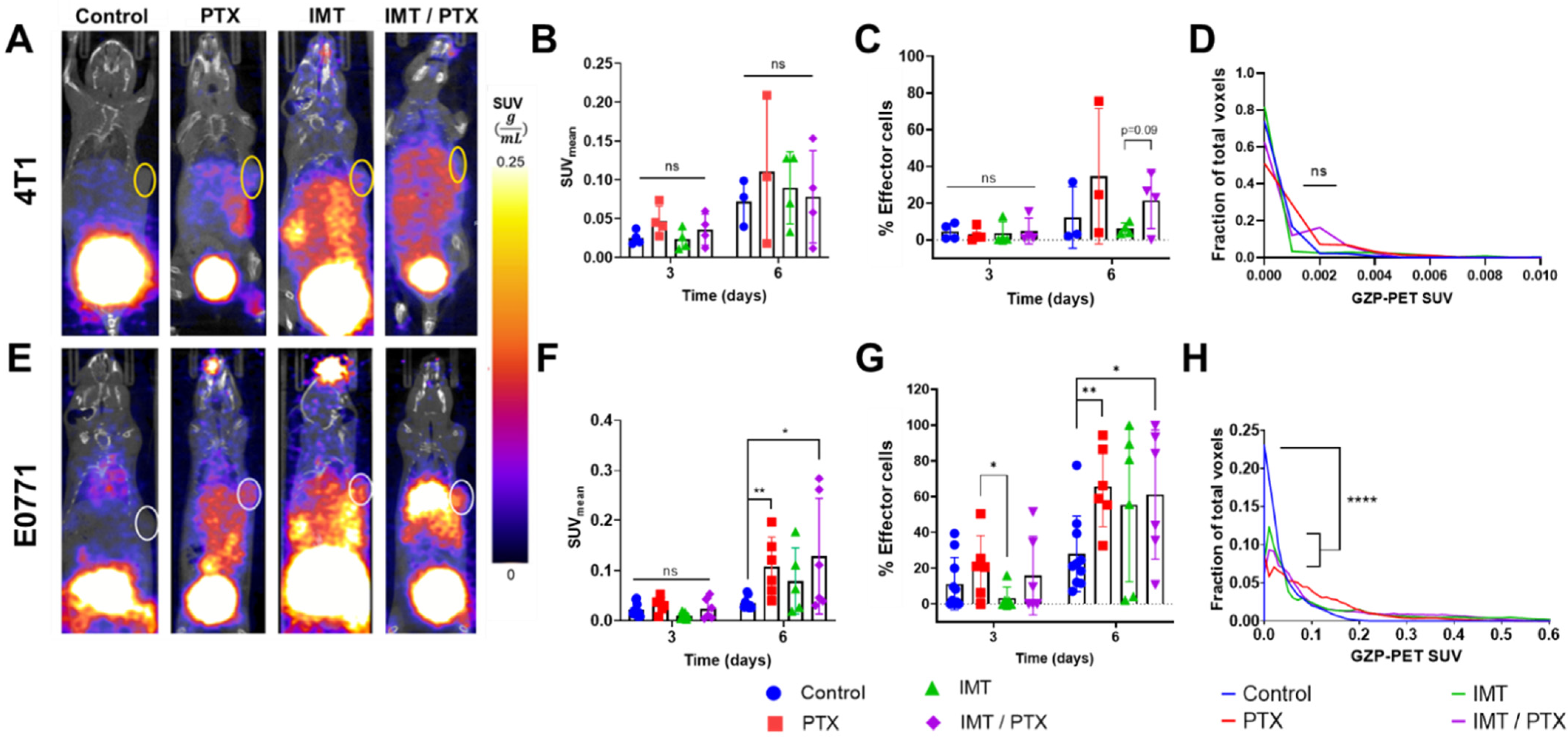
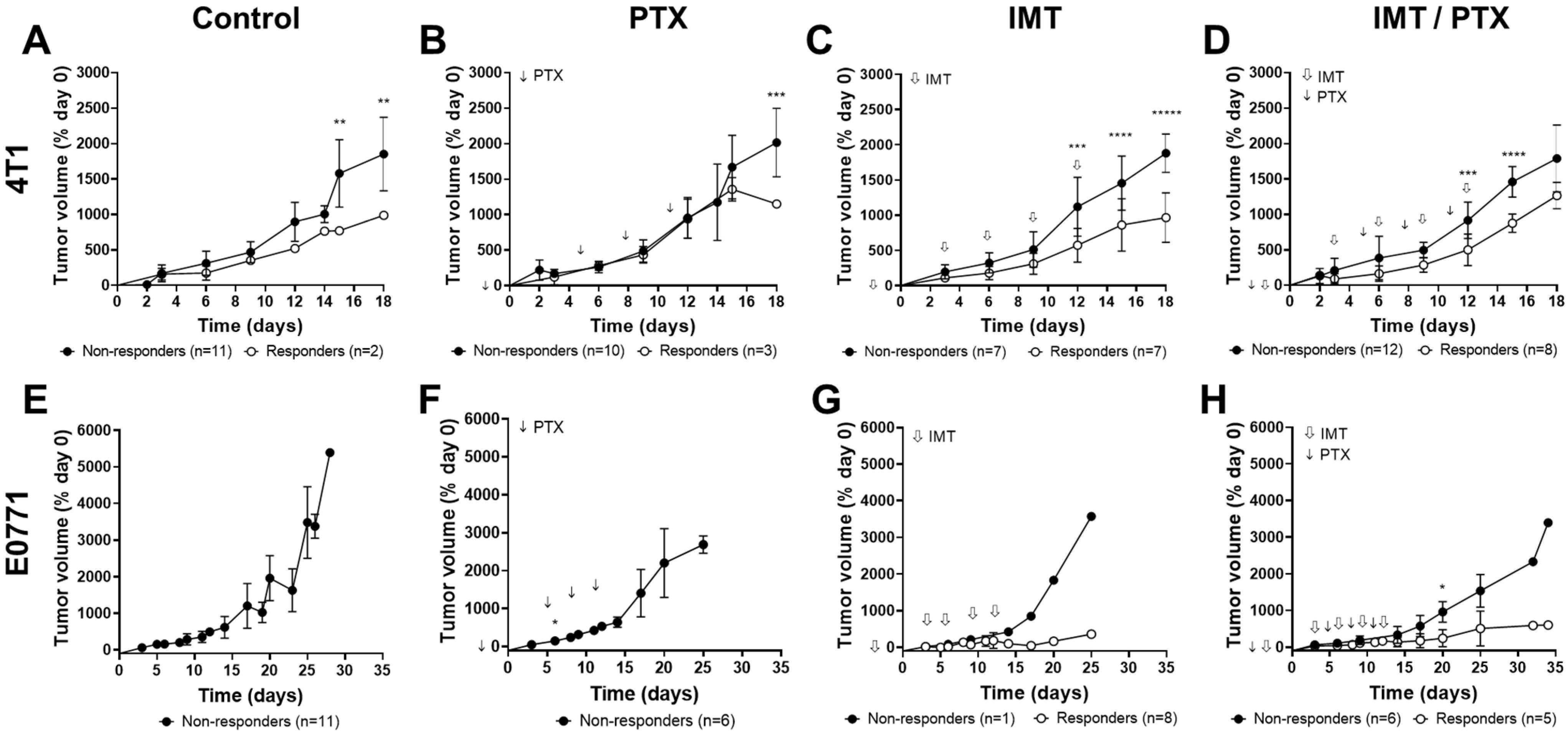
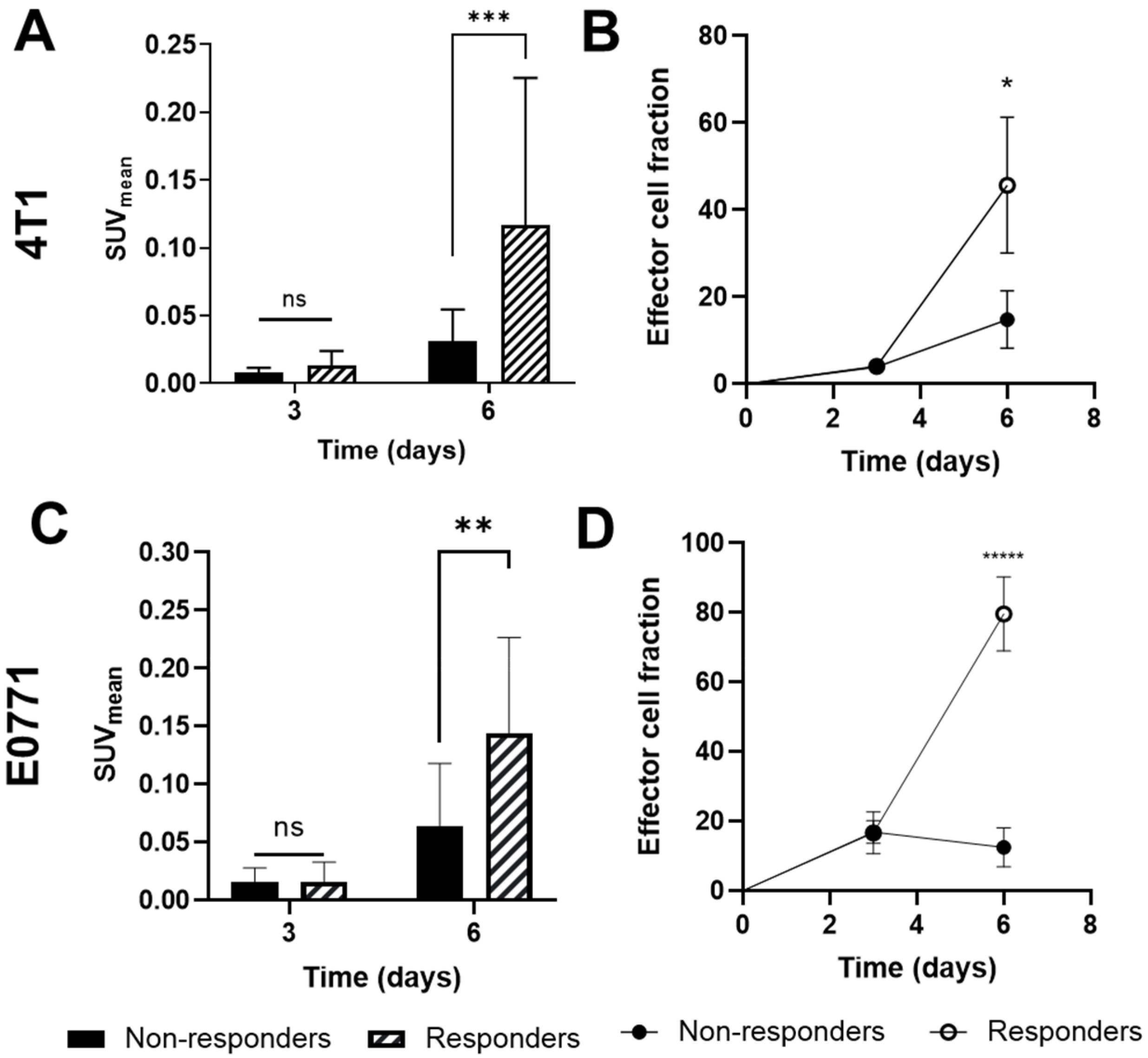
3.1. PTX Alone and in Combination with IMT Increases Effector Cell Activation and Intratumoral Heterogeneity
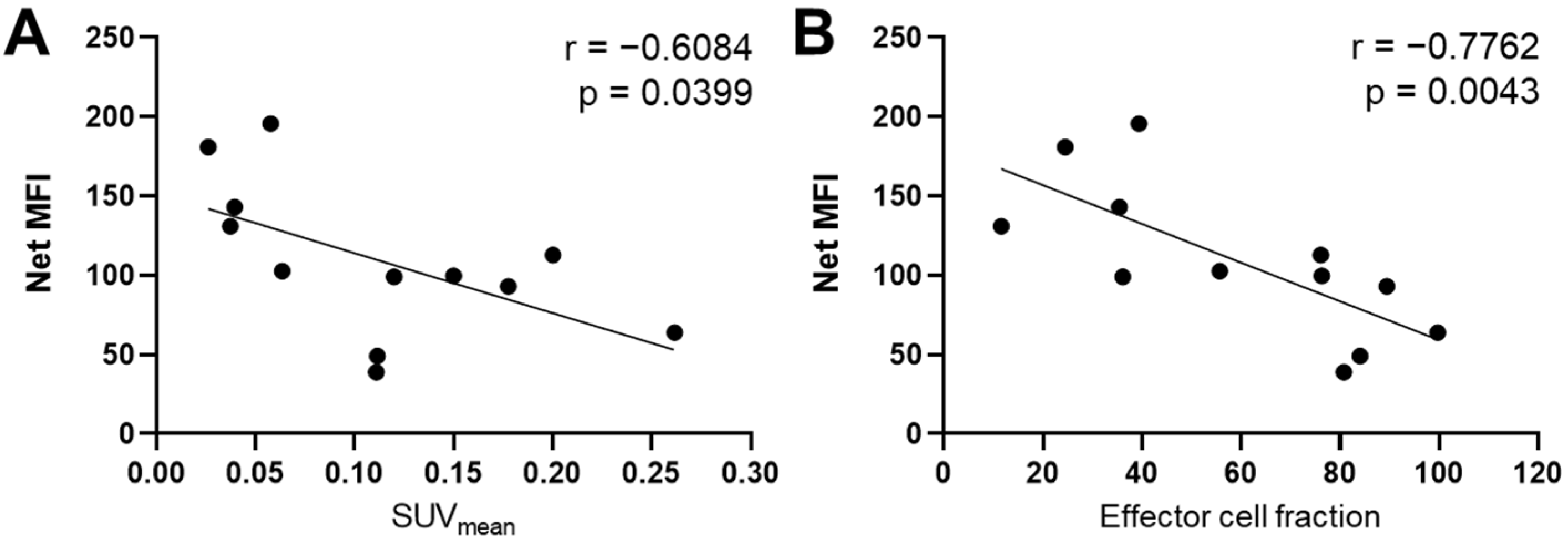
3.2. GZP-PET Has Predictive Value for Response to IMT plus PTX
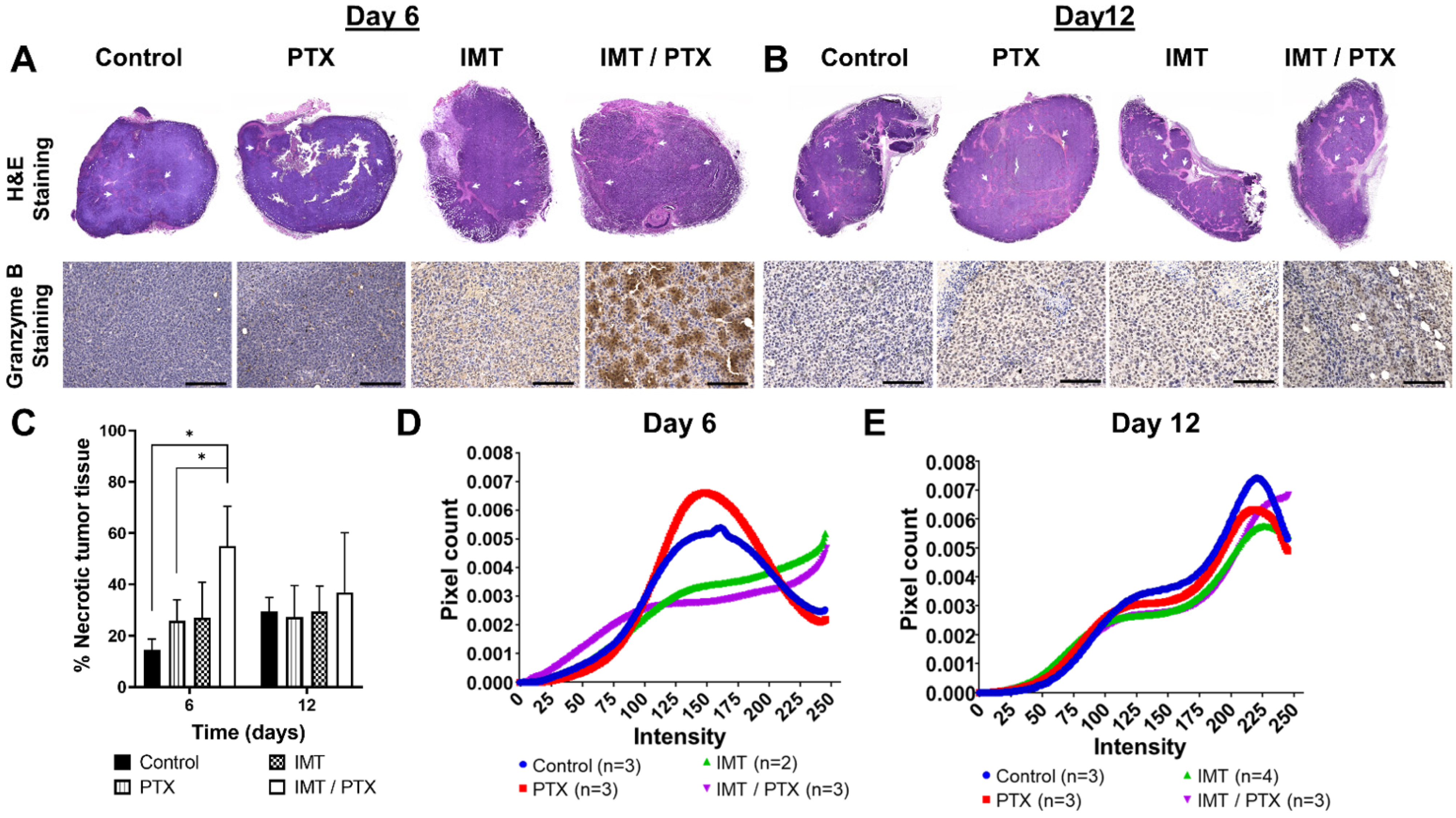
3.3. IMT Increases Necrosis and Granzyme B Expression in TNBC Tumors
3.4. IMT plus PTX Increases IFN-γ, IL-2, and IL-12 Expression, and Th2 Response in E0771 Tumors
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, K.-L.; Kuo, Y.-C.; Ho, Y.-S.; Huang, Y.-H. Triple-Negative Breast Cancer: Current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness. Cancers (Basel) 2019, 11, 1334. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Aggarwal, R. An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Plasilova, M.L.; Hayse, B.; Killelea, B.K.; Horowitz, N.R.; Chagpar, A.B.; Lannin, D.R. Features of triple-negative breast cancer: Analysis of 38,813 cases from the national cancer database. Med. (Baltim. ) 2016, 95, e4614. [Google Scholar] [CrossRef]
- Chaudhary, L.N.; Wilkinson, K.H.; Kong, A. Triple-Negative Breast Cancer: Who Should Receive Neoadjuvant Chemotherapy? Surg. Oncol. Clin. N. Am. 2018, 27, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagni, F.; Guerini-Rocco, E.; Schultheis, A.M.; Grazia, G.; Rijavec, E.; Ghidini, M.; Lopez, G.; Venetis, K.; Croci, G.A.; Malapelle, U.; et al. Targeting Immune-Related Biological Processes in Solid Tumors: We do Need Biomarkers. Int. J. Mol. Sci. 2019, 20, 5452. [Google Scholar] [CrossRef] [Green Version]
- Kwapisz, D. Pembrolizumab and atezolizumab in triple-negative breast cancer. Cancer Immunol. Immunother. 2021, 70, 607–617. [Google Scholar] [CrossRef]
- Edechi, C.A.; Ikeogu, N.; Uzonna, J.E.; Myal, Y. Regulation of Immunity in Breast Cancer. Cancers (Basel) 2019, 11, 1080. [Google Scholar] [CrossRef] [Green Version]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [Green Version]
- Santoni, M.; Romagnoli, E.; Saladino, T.; Foghini, L.; Guarino, S.; Capponi, M.; Giannini, M.; Cognigni, P.D.; Ferrara, G.; Battelli, N. Triple negative breast cancer: Key role of Tumor-Associated Macrophages in regulating the activity of anti-PD-1/PD-L1 agents. Biochim. Biophys. Acta (BBA) – Rev. Cancer 2018, 1869, 78–84. [Google Scholar] [CrossRef]
- Marra, A.; Viale, G.; Curigliano, G. Recent advances in triple negative breast cancer: The immunotherapy era. BMC Med. 2019, 17, 90. [Google Scholar] [CrossRef] [Green Version]
- Soularue, E.; Lepage, P.; Colombel, J.F.; Coutzac, C.; Faleck, D.; Marthey, L.; Collins, M.; Chaput, N.; Robert, C.; Carbonnel, F. Enterocolitis due to immune checkpoint inhibitors: A systematic review. Gut 2018, 67, 2056. [Google Scholar] [CrossRef]
- Force, J.; Leal, J.H.S.; McArthur, H.L. Checkpoint Blockade Strategies in the Treatment of Breast Cancer: Where We Are and Where We Are Heading. Curr. Treat. Opt. Oncol. 2019, 20, 35. [Google Scholar] [CrossRef]
- Cyprian, F.S.; Akhtar, S.; Gatalica, Z.; Vranic, S. Targeted immunotherapy with a checkpoint inhibitor in combination with chemotherapy: A new clinical paradigm in the treatment of triple-negative breast cancer. Bosn. J. Basic Med. Sci. 2019, 19, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Nolan, E.; Savas, P.; Policheni, A.N.; Darcy, P.K.; Vaillant, F.; Mintoff, C.P.; Dushyanthen, S.; Mansour, M.; Pang, J.-M.B.; Fox, S.B.; et al. Combined immune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer. Sci. Transl. Med. 2017, 9, eaal4922. [Google Scholar] [CrossRef]
- Zheng, S.; Zou, Y.; Liang, J.Y.; Xiao, W.; Yang, A.; Meng, T.; Lu, S.; Luo, Z.; Xie, X. Identification and validation of a combined hypoxia and immune index for triple-negative breast cancer. Mol. Oncol. 2020, 14, 2814–2833. [Google Scholar] [CrossRef]
- Larimer, B.M.; Wehrenberg-Klee, E.; Dubois, F.; Mehta, A.; Kalomeris, T.; Flaherty, K.; Boland, G.; Mahmood, U. Granzyme B PET Imaging as a Predictive Biomarker of Immunotherapy Response. Cancer Res. 2017, 77, 2318–2327. [Google Scholar] [CrossRef] [Green Version]
- Larimer, B.M.; Bloch, E.; Nesti, S.; Austin, E.E.; Wehrenberg-Klee, E.; Boland, G.; Mahmood, U. The Effectiveness of Checkpoint Inhibitor Combinations and Administration Timing Can Be Measured by Granzyme B PET Imaging. Clin. Cancer Res. 2019, 25, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Bardine, C.; Lourenço, A.L.; Wang, Y.-h.; Huang, Y.; Cleary, S.J.; Wilson, D.M.; Oh, D.Y.; Fong, L.; Looney, M.R.; et al. In Vivo Measurement of Granzyme Proteolysis from Activated Immune Cells with PET. ACS Cent. Sci. 2021, 7, 1638–1649. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, D.; Zhao, S.; Suo, C.; Shi, J.; Xue, M.Z.; Ruan, M.; Wang, H.; Zhao, J.; Li, Q.; et al. Multi-Omics Profiling Reveals Distinct Microenvironment Characterization and Suggests Immune Escape Mechanisms of Triple-Negative Breast Cancer. Clin. Cancer Res. 2019, 25, 5002–5014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudis, C.A.; Gianni, L. Triple-Negative Breast Cancer: An Unmet Medical Need. Oncologist 2011, 16, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Ali, A.M.; Ansari, J.A.K.; El-Aziz, N.M.A.; Abozeed, W.N.; Warith, A.M.A.; Alsaleh, K.; Nabholtz, J.M. Triple Negative Breast Cancer: A Tale of Two Decades. Anticancer Agents Med. Chem. 2017, 17, 491–499. [Google Scholar] [CrossRef]
- Sorace, A.G.; Elkassem, A.A.; Galgano, S.J.; Lapi, S.E.; Larimer, B.M.; Partridge, S.C.; Quarles, C.C.; Reeves, K.; Napier, T.S.; Song, P.N.; et al. Imaging for Response Assessment in Cancer Clinical Trials. Semin. Nucl. Med. 2020, 50, 488–504. [Google Scholar] [CrossRef]
- Vaidyanathan, S.; Patel, C.N.; Scarsbrook, A.F.; Chowdhury, F.U. FDG PET/CT in infection and inflammation—Current and emerging clinical applications. Clin. Radiol. 2015, 70, 787–800. [Google Scholar] [CrossRef]
- Aide, N.; Hicks, R.J.; Le Tourneau, C.; Lheureux, S.; Fanti, S.; Lopci, E. FDG PET/CT for assessing tumour response to immunotherapy. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Jagoda, E.M.; Vasalatiy, O.; Basuli, F.; Opina, A.C.L.; Williams, M.R.; Wong, K.; Lane, K.C.; Adler, S.; Ton, A.T.; Szajek, L.P.; et al. Immuno-PET Imaging of the Programmed Cell Death-1 Ligand (PD-L1) Using a Zirconium-89 Labeled Therapeutic Antibody, Avelumab. Mol. Imaging 2019, 18, 1536012119829986. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Ehlerding, E.B.; Jiang, D.; Barnhart, T.E.; Chen, W.; Cao, T.; Engle, J.W.; Cai, W. In vivo characterization of PD-L1 expression in breast cancer by immuno-PET with (89)Zr-labeled avelumab. Am. J. Transl. Res. 2020, 12, 1862–1872. [Google Scholar]
- Tavaré, R.; Escuin-Ordinas, H.; Mok, S.; McCracken, M.N.; Zettlitz, K.A.; Salazar, F.B.; Witte, O.N.; Ribas, A.; Wu, A.M. An Effective Immuno-PET Imaging Method to Monitor CD8-Dependent Responses to Immunotherapy. Cancer Res. 2016, 76, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Foubert, F.; Gouard, S.; Saï-Maurel, C.; Chérel, M.; Faivre-Chauvet, A.; Goldenberg, D.M.; Barbet, J.; Bailly, C.; Bodet-Milin, C.; Carlier, T.; et al. Sensitivity of pretargeted immunoPET using (68)Ga-peptide to detect colonic carcinoma liver metastases in a murine xenograft model: Comparison with (18)FDG PET-CT. Oncotarget 2018, 9, 27502–27513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailly, C.; Gouard, S.; Lacombe, M.; Remaud-Le Saëc, P.; Chalopin, B.; Bourgeois, M.; Chouin, N.; Tripier, R.; Halime, Z.; Haddad, F.; et al. Comparison of Immuno-PET of CD138 and PET imaging with (64)CuCl(2) and (18)F-FDG in a preclinical syngeneic model of multiple myeloma. Oncotarget 2018, 9, 9061–9072. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, C.; Goldenberg, D.M.; Colombié, M.; Sébille, J.-C.; Meingan, P.; Ferrer, L.; Baumgartner, P.; Cerato, E.; Masson, D.; Campone, M.; et al. Initial Clinical Results of a Novel Immuno-PET Theranostic Probe in Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2020, 61, 1205–1211. [Google Scholar] [CrossRef]
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152. [Google Scholar] [CrossRef]
- Mittal, S.; Brown, N.J.; Holen, I. The breast tumor microenvironment: Role in cancer development, progression and response to therapy. Expert Rev. Mol. Diagn. 2018, 18, 227–243. [Google Scholar] [CrossRef]
- Gruosso, T.; Gigoux, M.; Manem, V.S.K.; Bertos, N.; Zuo, D.; Perlitch, I.; Saleh, S.M.I.; Zhao, H.; Souleimanova, M.; Johnson, R.M.; et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J. Clin. Investig. 2019, 129, 1785–1800. [Google Scholar] [CrossRef] [Green Version]
- Sorace, A.G.; Syed, A.K.; Barnes, S.L.; Quarles, C.C.; Sanchez, V.; Kang, H.; Yankeelov, T.E. Quantitative [18F]FMISO PET Imaging Shows Reduction of Hypoxia Following Trastuzumab in a Murine Model of HER2+ Breast Cancer. Mol. Imaging Biol. 2017, 19, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Qiao, F.; Su, X.; Zhang, D.; Wang, H.; Jiang, J.; Xu, H. 18F-HX4/18F-FMISO-based micro PET for imaging of tumor hypoxia and radiotherapy-associated changes in mice. Biomed. Pharmacother. 2019, 119, 109454. [Google Scholar] [CrossRef] [PubMed]
- Capaccione, K.M.; Doubrovin, M.; Bhatt, N.; Mintz, A.; Molotkov, A. Granzyme B PET Imaging of the Innate Immune Response. Molecules 2020, 25, 3102. [Google Scholar] [CrossRef]
- Goggi, J.L.; Hartimath, S.V.; Hwang, Y.; Tan, Y.X.; Khanapur, S.; Ramasamy, B.; Jiang, L.; Yong, F.F.; Cheng, P.; Tan, P.W.; et al. Examining Immunotherapy Response Using Multiple Radiotracers. Mol. Imaging Biol. 2020, 22, 993–1002. [Google Scholar] [CrossRef]
- Bloom, M.J.; Jarrett, A.M.; Triplett, T.A.; Syed, A.K.; Davis, T.; Yankeelov, T.E.; Sorace, A.G. Anti-HER2 induced myeloid cell alterations correspond with increasing vascular maturation in a murine model of HER2+ breast cancer. BMC Cancer 2020, 20, 359. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, M.; Massicano, A.V.F.; Song, P.N.; Mansur, A.; Heinzman, K.A.; Larimer, B.M.; Lapi, S.E.; Sorace, A.G. [(89)Zr]-Pertuzumab PET Imaging Reveals Paclitaxel Treatment Efficacy Is Positively Correlated with HER2 Expression in Human Breast Cancer Xenograft Mouse Models. Molecules 2021, 26, 1568. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.K.; Fröhlich, C.; Christensen, C.; Melander, M.C.; Poulsen, T.T.; Galler, G.R.; Lantto, J.; Horak, I.D.; Kragh, M.; Nielsen, C.H.; et al. CD4(+) and CD8a(+) PET imaging predicts response to novel PD-1 checkpoint inhibitor: Studies of Sym021 in syngeneic mouse cancer models. Theranostics 2019, 9, 8221–8238. [Google Scholar] [CrossRef] [PubMed]
- Kasten, B.B.; Houson, H.A.; Coleman, J.M.; Leavenworth, J.W.; Markert, J.M.; Wu, A.M.; Salazar, F.; Tavaré, R.; Massicano, A.V.F.; Gillespie, G.Y.; et al. Positron emission tomography imaging with 89Zr-labeled anti-CD8 cys-diabody reveals CD8+ cell infiltration during oncolytic virus therapy in a glioma murine model. Sci. Rep. 2021, 11, 15384. [Google Scholar] [CrossRef] [PubMed]
- Onyema, O.O.; Decoster, L.; Njemini, R.; Forti, L.N.; Bautmans, I.; De Waele, M.; Mets, T. Chemotherapy-induced Changes and Immunosenescence of CD8+ T-Cells in Patients with Breast Cancer. Anticancer Res. 2015, 35, 1481. [Google Scholar] [PubMed]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy induces enrichment of CD47+/CD73+/PDL1+ immune evasive triple-negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Garg, A.D.; Dudek-Peric, A.M.; Romano, E.; Agostinis, P. Immunogenic cell death. Int. J. Dev. Biol. 2015, 59, 131–140. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- LaSalle, T.; Austin, E.E.; Rigney, G.; Wehrenberg-Klee, E.; Nesti, S.; Larimer, B.; Mahmood, U. Granzyme B PET imaging of immune-mediated tumor killing as a tool for understanding immunotherapy response. J. Immunother. Cancer 2020, 8, e000291. [Google Scholar] [CrossRef]
- Vo, J.L.; Yang, L.; Kurtz, S.L.; Smith, S.G.; Koppolu, B.P.; Ravindranathan, S.; Zaharoff, D.A. Neoadjuvant immunotherapy with chitosan and interleukin-12 to control breast cancer metastasis. Oncoimmunology 2014, 3, e968001. [Google Scholar] [CrossRef] [Green Version]
- Akbaribazm, M.; Khazaei, M.R.; Khazaei, M. Trifolium pratense L. (red clover) extract and doxorubicin synergistically inhibits proliferation of 4T1 breast cancer in tumor-bearing BALB/c mice through modulation of apoptosis and increase antioxidant and anti-inflammatory related pathways. Food Sci. Nutr. 2020, 8, 4276–4290. [Google Scholar] [CrossRef] [PubMed]
- Ghouse, S.M.; Nguyen, H.-M.; Bommareddy, P.K.; Guz-Montgomery, K.; Saha, D. Oncolytic Herpes Simplex Virus Encoding IL12 Controls Triple-Negative Breast Cancer Growth and Metastasis. Front. Oncol. 2020, 10, 384. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yang, H.H.; Hu, Y.; Watson, P.H.; Liu, H.; Geiger, T.R.; Anver, M.R.; Haines, D.C.; Martin, P.; Green, J.E.; et al. Immunocompetent mouse allograft models for development of therapies to target breast cancer metastasis. Oncotarget 2017, 8, 30621–30643. [Google Scholar] [CrossRef] [Green Version]
- Parveen, S.; Siddharth, S.; Cheung, L.S.; Kumar, A.; Shen, J.; Murphy, J.R.; Sharma, D.; Bishai, W.R. Therapeutic targeting with DABIL-4 depletes myeloid suppressor cells in 4T1 triple-negative breast cancer model. Mol. Oncol. 2021, 15, 1330–1344. [Google Scholar] [CrossRef]
- Zhao, L.; Jin, Y.; Yang, C.; Li, C. HBV-specific CD8 T cells present higher TNF-α expression but lower cytotoxicity in hepatocellular carcinoma. Clin. Exp. Immunol. 2020, 201, 289–296. [Google Scholar] [CrossRef]
- Bertrand, F.; Rochotte, J.; Colacios, C.; Montfort, A.; Tilkin-Mariamé, A.-F.; Touriol, C.; Rochaix, P.; Lajoie-Mazenc, I.; Andrieu-Abadie, N.; Levade, T.; et al. Blocking Tumor Necrosis Factor α Enhances CD8 T-cell–Dependent Immunity in Experimental Melanoma. Cancer Res. 2015, 75, 2619–2628. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.Y.; Wolchok, J.D.; Bass, A.R. TNF in the era of immune checkpoint inhibitors: Friend or foe? Nat. Rev. Rheumatol. 2021, 17, 213–223. [Google Scholar] [CrossRef]
- Montfort, A.; Virazels, M.; Colacios, C.; Meyer, N.; Ségui, B. Combining TNF blockade with immune checkpoint inhibitors in patients with cancer. Nat. Rev. Rheumatol. 2021, 17, 577. [Google Scholar] [CrossRef]
- Weber, J.S.; Gibney, G.; Sullivan, R.J.; Sosman, J.A.; Slingluff, C.L., Jr.; Lawrence, D.P.; Logan, T.F.; Schuchter, L.M.; Nair, S.; Fecher, L.; et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): An open-label, randomised, phase 2 trial. Lancet Oncol. 2016, 17, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Messenheimer, D.J.; Jensen, S.M.; Afentoulis, M.E.; Wegmann, K.W.; Feng, Z.; Friedman, D.J.; Gough, M.J.; Urba, W.J.; Fox, B.A. Timing of PD-1 Blockade Is Critical to Effective Combination Immunotherapy with Anti-OX40. Clin. Cancer Res. 2017, 23, 6165. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef]
- Vafaei, S.; Zekiy, A.O.; Khanamir, R.A.; Zaman, B.A.; Ghayourvahdat, A.; Azimizonuzi, H.; Zamani, M. Combination therapy with immune checkpoint inhibitors (ICIs); a new frontier. Cancer Cell Int. 2022, 22, 2. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DAY 6 | DAY 12 | |||||
|---|---|---|---|---|---|---|
| PTX | IMT | IMT/PTX | PTX | IMT | IMT/PTX | |
| GM-CSF | −2 | −41 | −12 | −57 | −78 | −50 |
| IFN-γ | −81 | 105 | 125 | −17 | 182 | 237 |
| IL-1β | −45 | 52 | −56 | 13 | 78 | 77 |
| IL-2 | 28 | 228 | 1366 | 54 | 69 | 172 |
| IL-4 | −13 | 588 | 983 | 400 | 418 | 3991 |
| IL-5 | 22 | 463 | 999 | 11 | 686 | 1892 |
| IL-6 | −50 | −81 | −50 | 23 | −56 | −17 |
| IL-12p70 | −17 | 108 | 732 | 20 | 14 | 394 |
| IL-13 | 61 | 524 | 1635 | 89 | 465 | 1770 |
| IL-18 | −32 | 25 | −24 | 47 | 36 | 67 |
| TNF-α | −24 | −42 | −13 | −37 | −49 | −45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Napier, T.S.; Hunter, C.L.; Song, P.N.; Larimer, B.M.; Sorace, A.G. Preclinical PET Imaging of Granzyme B Shows Promotion of Immunological Response Following Combination Paclitaxel and Immune Checkpoint Inhibition in Triple Negative Breast Cancer. Pharmaceutics 2022, 14, 440. https://doi.org/10.3390/pharmaceutics14020440
Napier TS, Hunter CL, Song PN, Larimer BM, Sorace AG. Preclinical PET Imaging of Granzyme B Shows Promotion of Immunological Response Following Combination Paclitaxel and Immune Checkpoint Inhibition in Triple Negative Breast Cancer. Pharmaceutics. 2022; 14(2):440. https://doi.org/10.3390/pharmaceutics14020440
Chicago/Turabian StyleNapier, Tiara S., Chanelle L. Hunter, Patrick N. Song, Benjamin M. Larimer, and Anna G. Sorace. 2022. "Preclinical PET Imaging of Granzyme B Shows Promotion of Immunological Response Following Combination Paclitaxel and Immune Checkpoint Inhibition in Triple Negative Breast Cancer" Pharmaceutics 14, no. 2: 440. https://doi.org/10.3390/pharmaceutics14020440