
Effects of Ultramicronized N-Palmitoylethanolamine Supplementation on Tramadol and Oxycodone Analgesia and Tolerance Prevention

 , , , , , and
, , , , , and 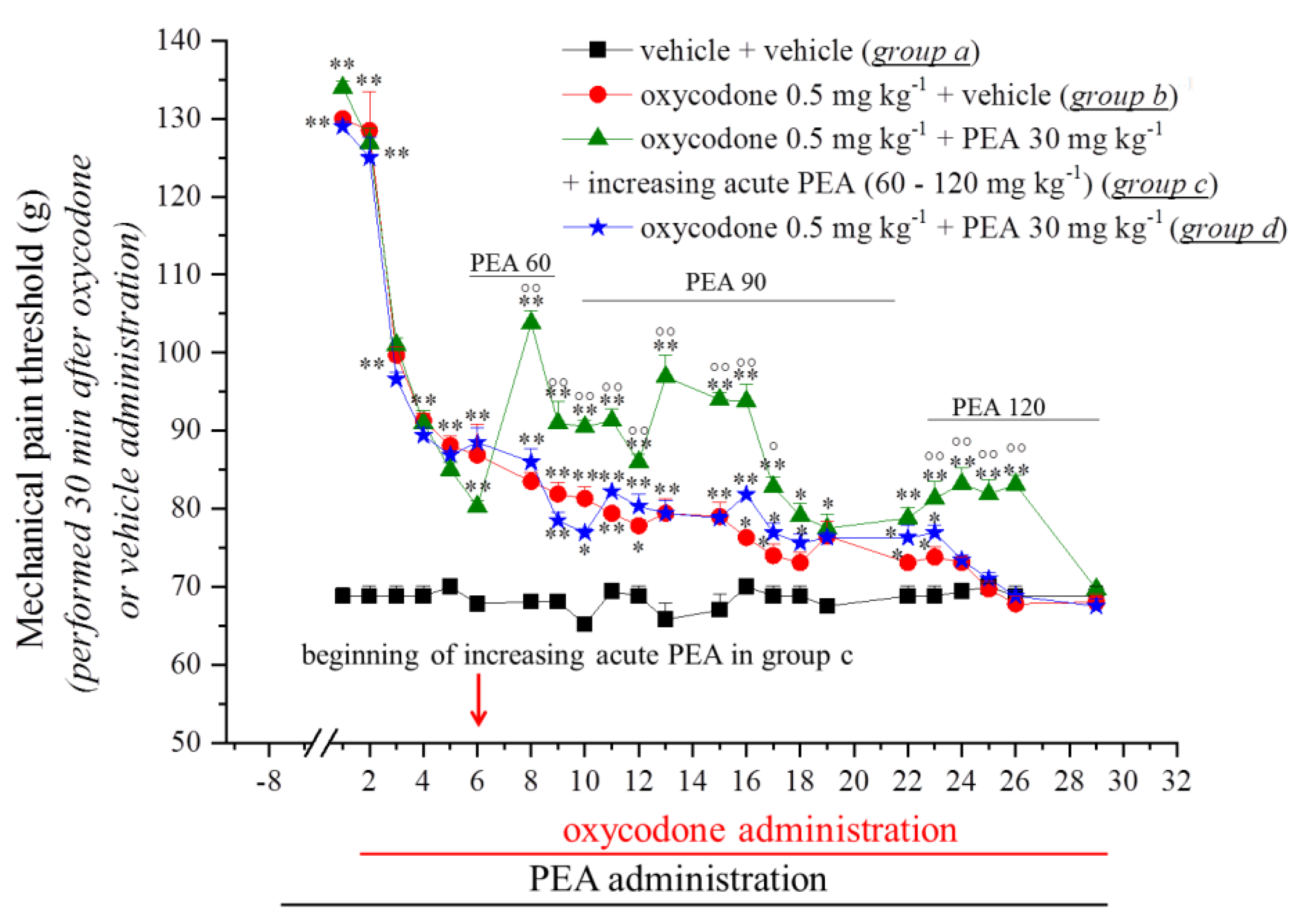{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
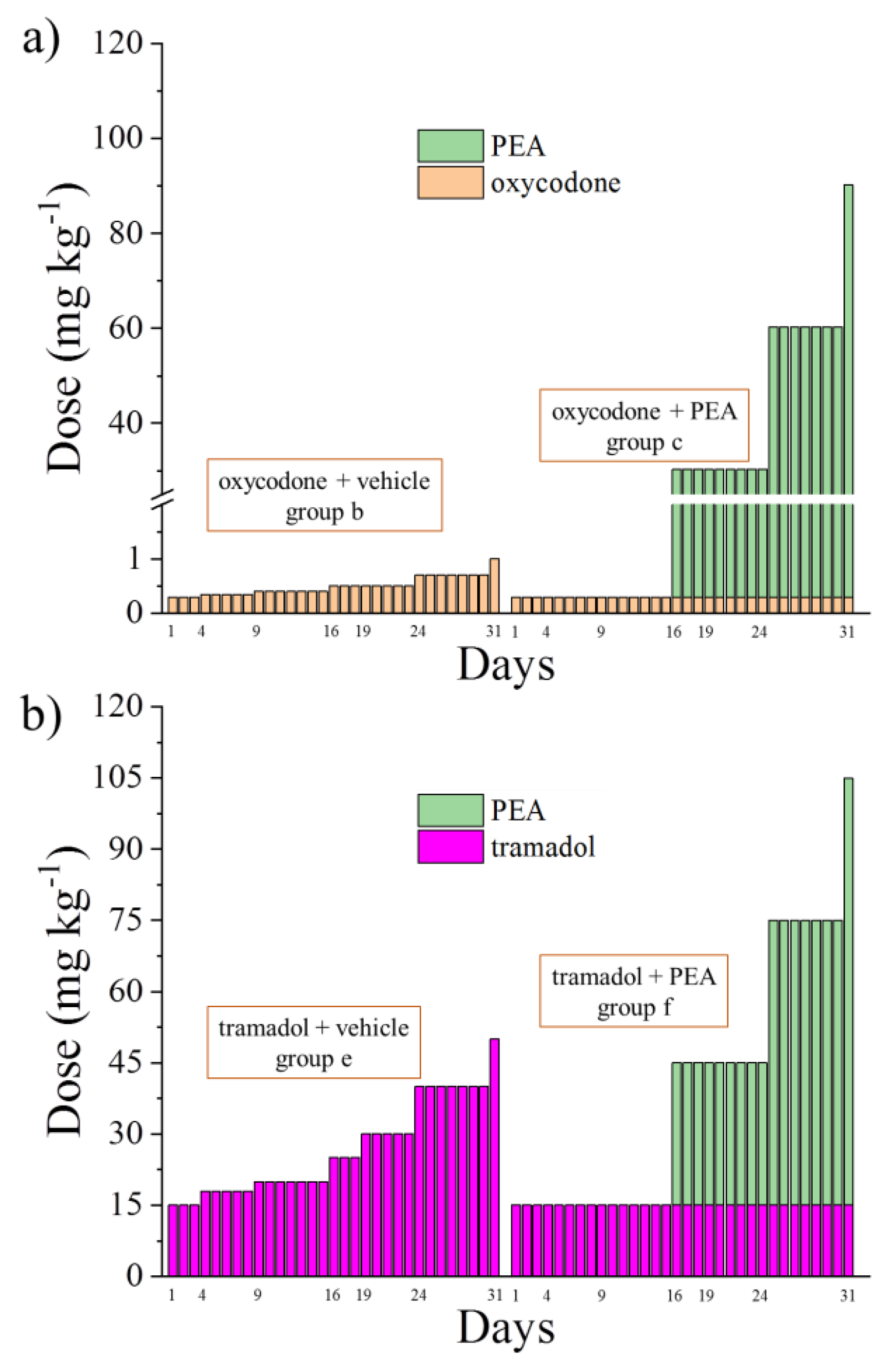
2.2. Study Design
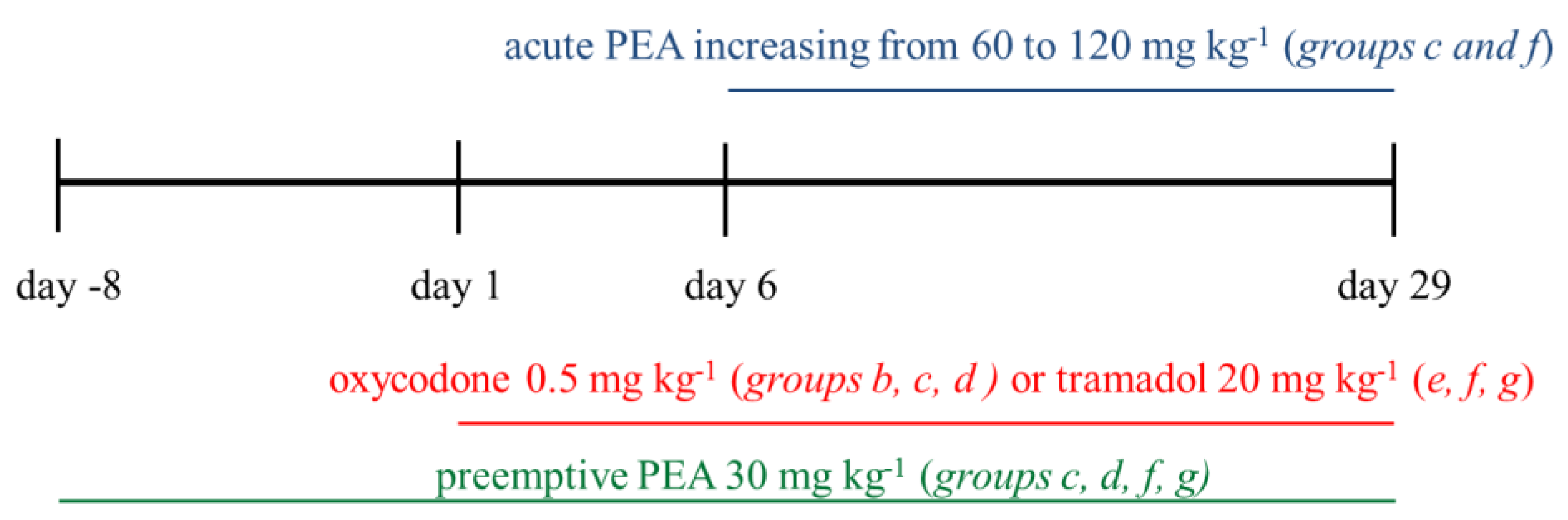
- Group a: vehicle + vehicle (from Day −8 to Day 29);
- Group b: 0.5 mg kg−1 oxycodone (from Day 1 to Day 29) + vehicle (from Day −8 to Day 29);
- Group c: pre-emptive PEA (30 mg kg−1 from Day −8 to Day 29) + 0.5 mg kg−1 oxycodone (from Day 1 to Day 29) + increasing acute PEA (60–120 mg kg−1 from Day 6 to Day 29);
- Group d: pre-emptive PEA (30 mg kg−1 from Day −8 to Day 29) + 0.5 mg kg−1 oxycodone (from Day 1 to Day 29);
- Group e: 20 mg kg−1 tramadol (from Day 1 to Day 29) + vehicle (from Day −8 to Day 29);
- Group f: pre-emptive PEA (30 mg kg−1 from Day −8 to Day 29) + 20 mg kg−1 tramadol (from Day 1 to Day 29) + increasing acute PEA (60–120 mg kg−1 from Day 6 to Day 29);
- Group g: pre-emptive PEA (30 mg kg−1 from Day −8 to Day 29) + 20 mg kg−1 tramadol (from Day 1 to Day 29).
- Group a: vehicle + vehicle (from Day −8 to Day 31);
- Group b: increasing oxycodone (0.3–1 mg kg−1 from Day 1 to Day 31) + vehicle (from Day −8 to Day 31);
- Group c: pre-emptive PEA (30 mg kg−1 from Day −8 to Day 31) + 0.3 mg kg−1 oxycodone (from Day 1 to Day 31) + increasing acute PEA (30–90 mg kg−1 from Day 16 to Day 31);
- Group d: increasing tramadol (15–50 mg kg−1 from Day 1 to Day 31) + vehicle (from Day −8 to Day 31);
- Group e: pre-emptive PEA (30 mg kg−1 from Day −8 to Day 31) + 15 mg kg−1 tramadol (from Day 1 to Day 31) + increasing acute PEA (30–90 mg kg−1 from Day 16 to Day 31).
2.3. Paw Pressure Test
2.4. Tissue Collection
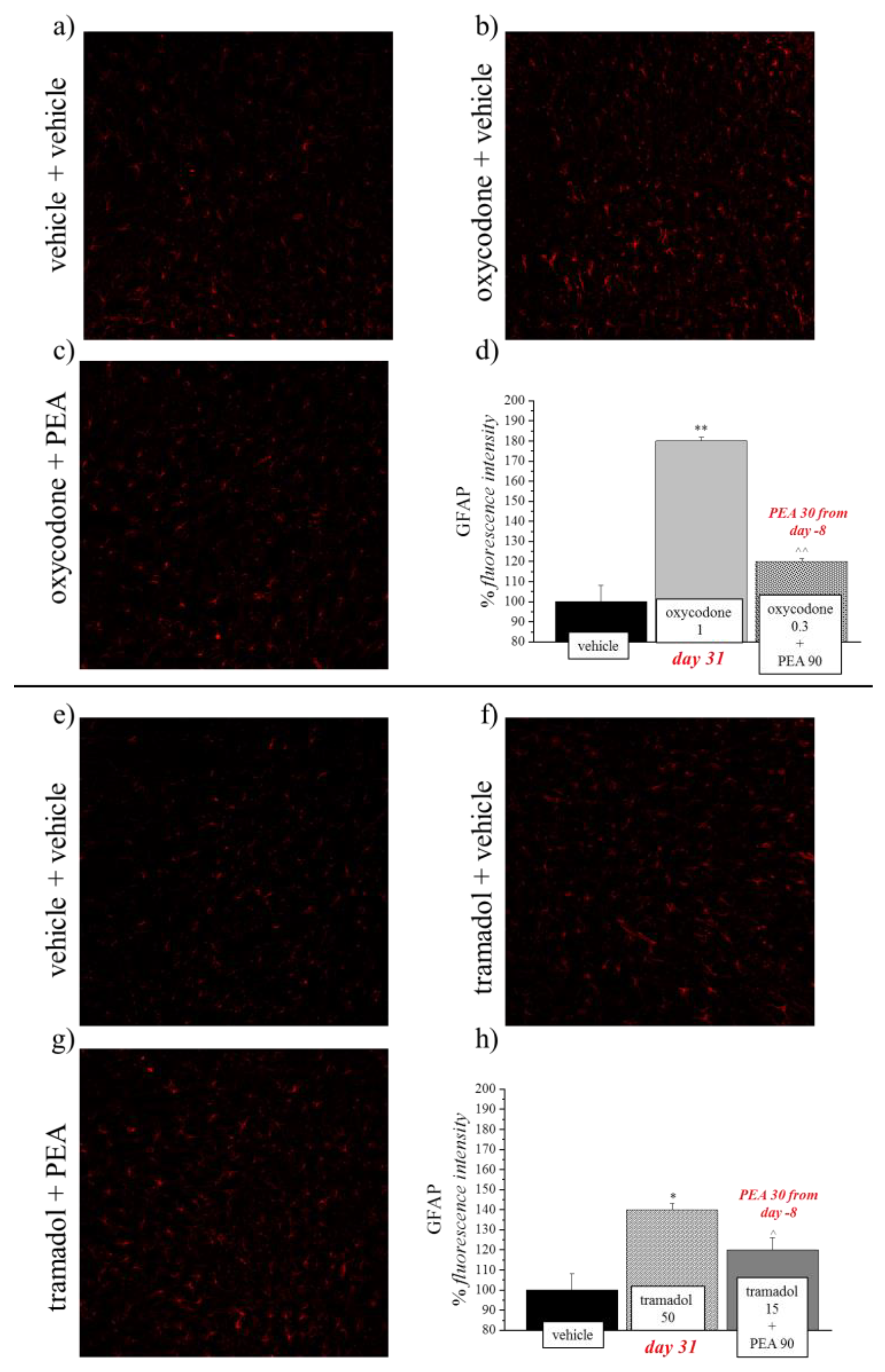
2.5. Immunohistochemistry of the Spinal Cord
2.6. Quantitative Analyses of GFAP Immunohistochemistry
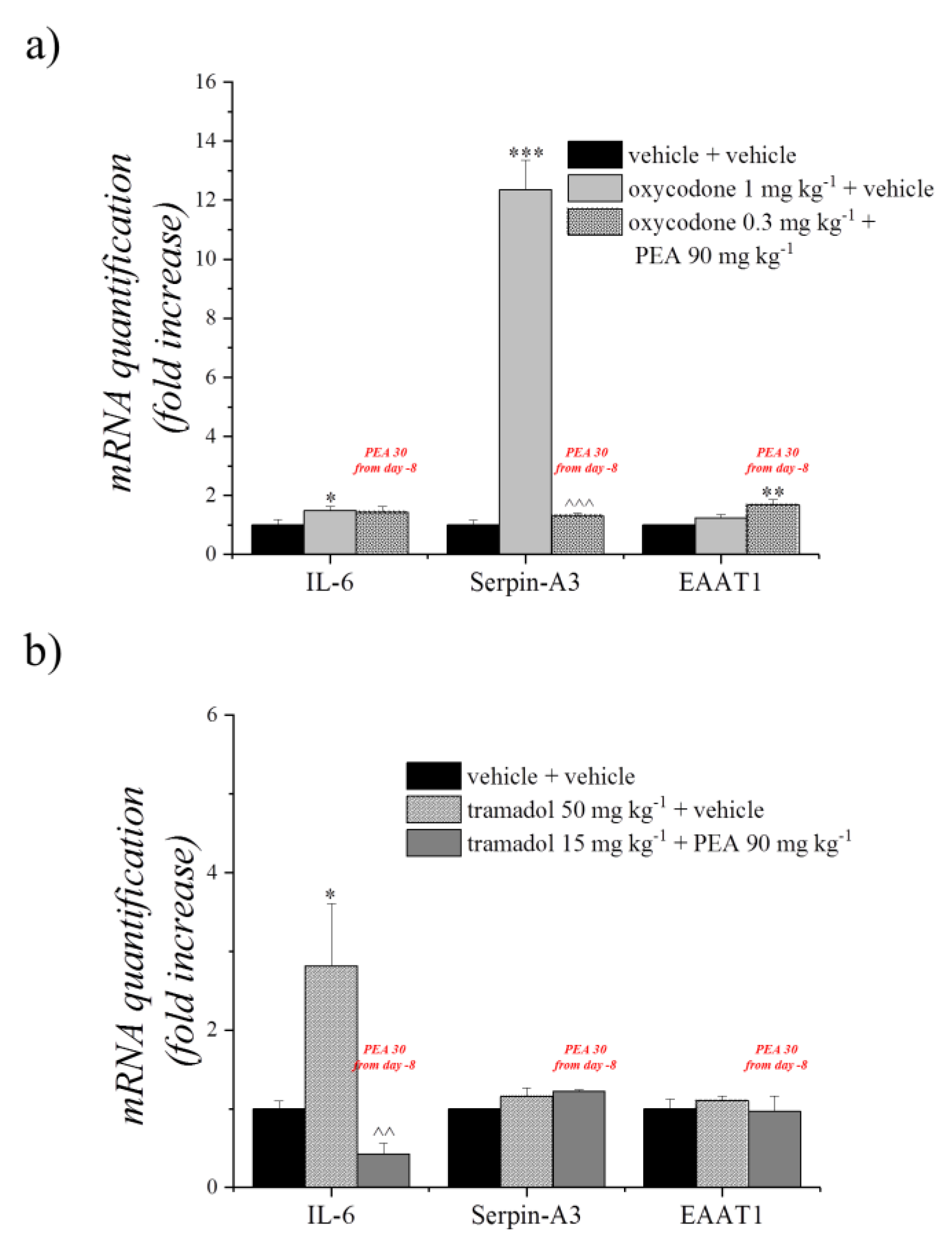
2.7. Real-Time Polymerase Chain Reaction (RT-PCR)
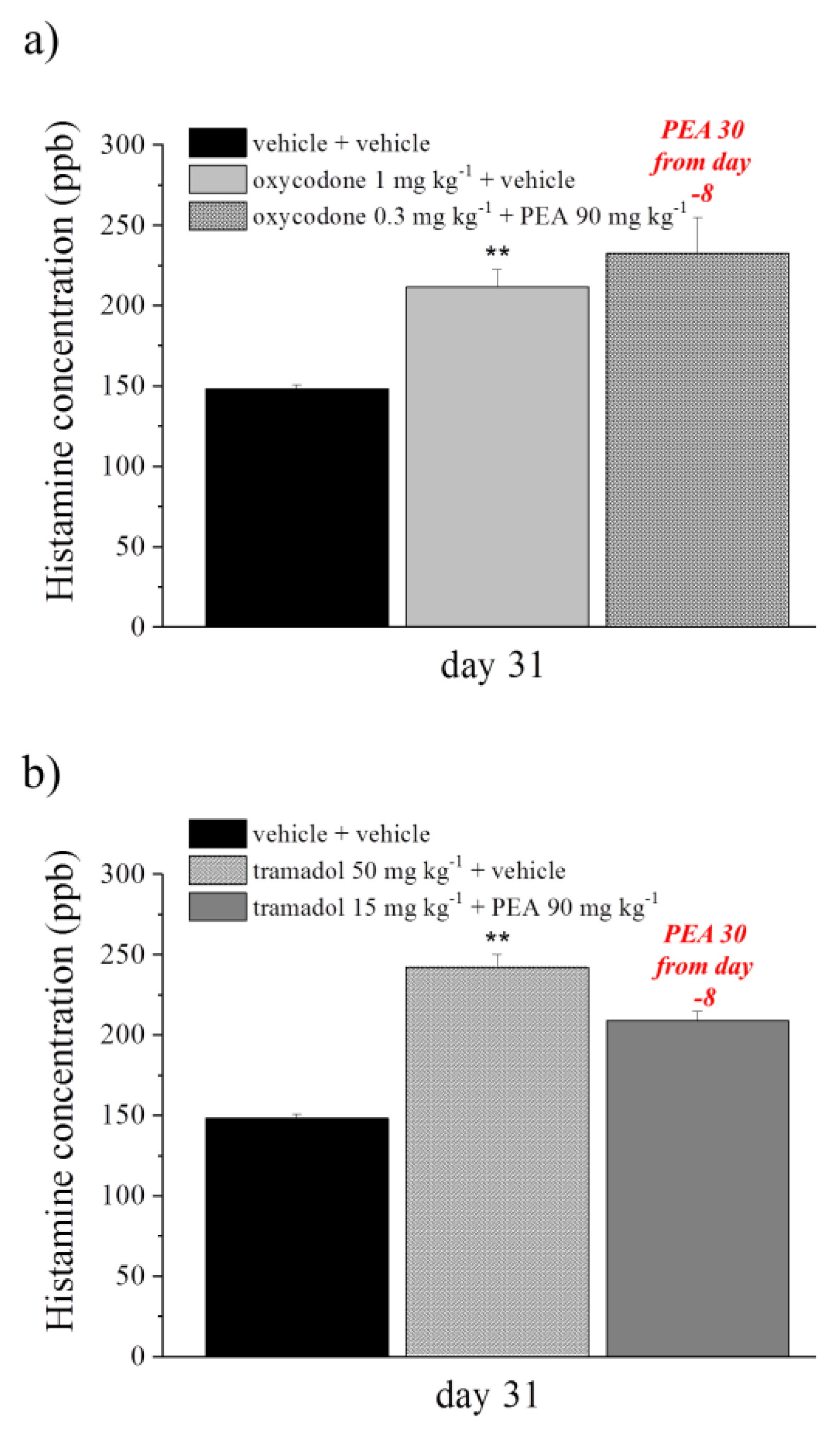
2.8. Histamine Dosage by HPLC-MS/MS
- Solvent A: 10% CH3CN 5 mM HCOOH 15 mM HCOONH4;
- Solvent B: 90% CH3CN 15 mM HCOOH 5 mM HCOONH4;
- Solvent C (used for sample loading): 90% CH3CN 17.5 mM HCOOH 2.5 mM HCOONH4.
2.9. Statistical Analysis
3. Results
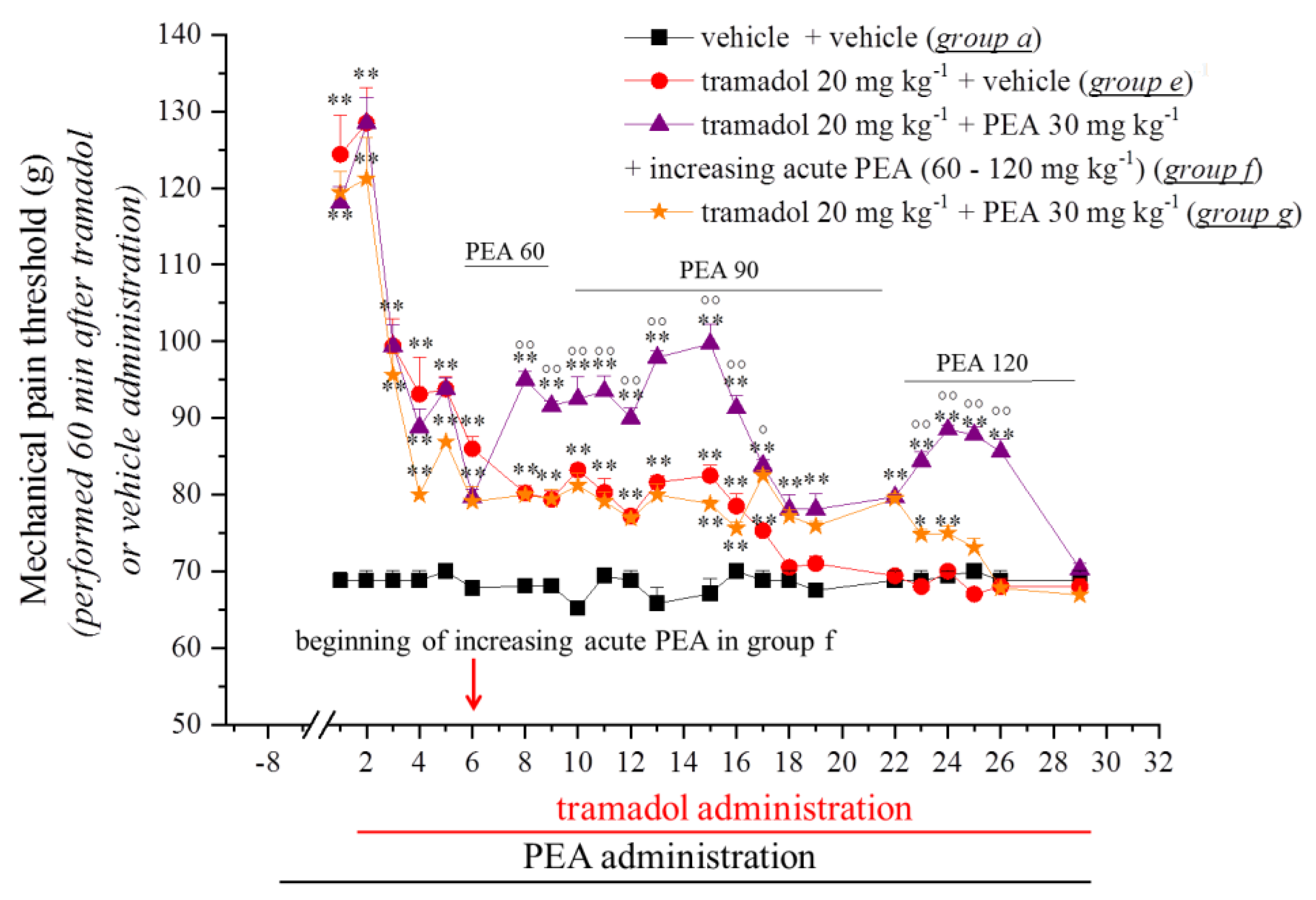
3.1. PEA Modulates the Onset of Tramadol and Oxycodone Antinociception and Tolerance



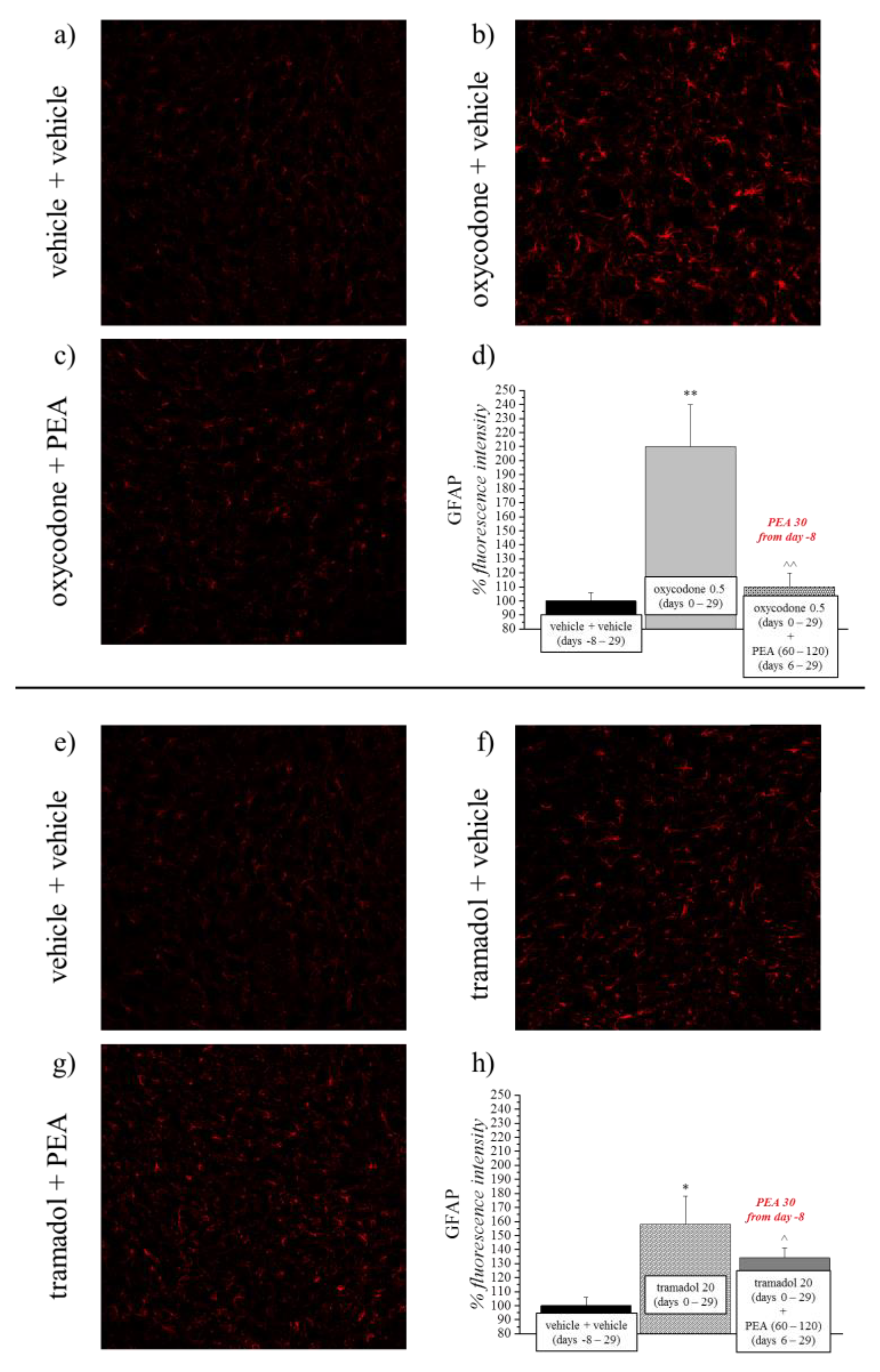
3.2. PEA Reduces the Astrocyte Activation Evoked by Opioids
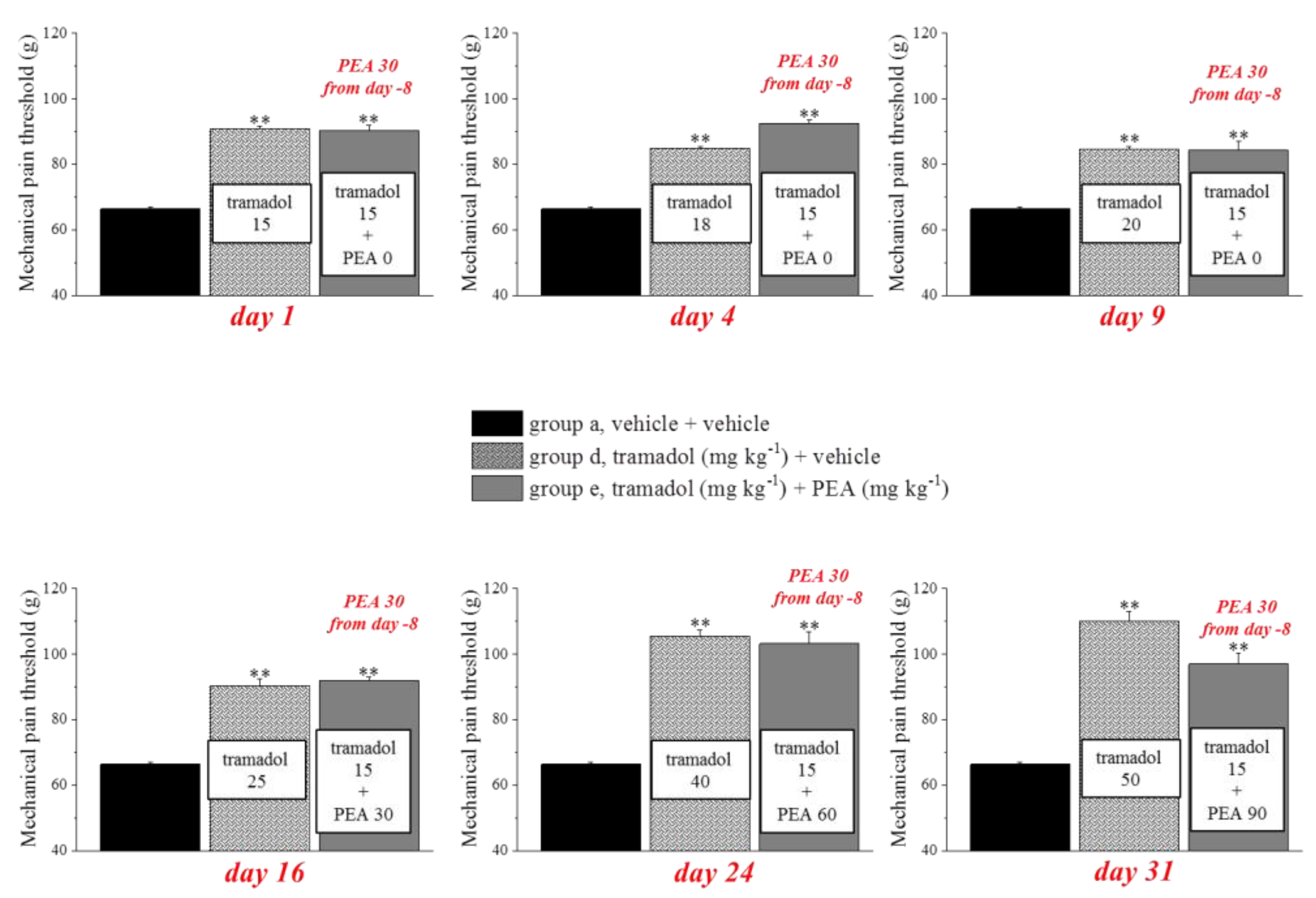
3.3. Different Combinations of PEA with Tramadol or Oxycodone Modulate Opioids’ Analgesia
3.4. Effect of PEA Treatment on Ex Vivo Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fishman, S.M. Recognizing Pain Management as a Human Right: A First Step. Anesth. Analg. 2007, 105, 8–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Pain Summit of the In Declaration of Montréal: Declaration That Access to Pain Management Is a Fundamental Human Right. J. Pain Palliat. Care Pharmacother. 2011, 25, 29–31. [CrossRef] [PubMed]
- Lohman, D.; Schleifer, R.; Amon, J.J. Access to pain treatment as a human right. BMC Med. 2010, 8, 8. [Google Scholar] [CrossRef]
- Volkow, N.D.; Collins, F.S. The Role of Science in Addressing the Opioid Crisi. N. Engl. J. Med. 2017, 377, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Els, C.; Jackson, T.D.; Kunyk, D.; Lappi, V.G.; Sonnenberg, B.; Hagtvedt, R.; Sharma, S.; Kolahdooz, F.; Straube, S. Adverse events associated with medium-and long-term use of opioids for chronic non-cancer pain: An overview of Cochrane Reviews. Cochrane Database Syst. Rev. 2017, 2018. [Google Scholar] [CrossRef]
- Benyamin, R.; Trescot, A.M.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; Glaser, S.E.; Vallejo, R. Opioid complications and side effects. Pain Physician 2008, 11, S105–S120. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Giusti, P. Glia and Mast Cells as Targets for Palmitoylethanolamide, an Anti-inflammatory and Neuroprotective Lipid Mediator. Mol. Neurobiol. 2013, 48, 340–352. [Google Scholar] [CrossRef]
- Di Cesare Mannelli, L.; Corti, F.; Micheli, L.; Zanardelli, M.; Ghelardini, C. Delay of Morphine Tolerance by Palmitoylethanolamide. Biomed Res. Int. 2015, 2015, 894732. [Google Scholar] [CrossRef] [Green Version]
- Di Cesare Mannelli, L.; Micheli, L.; Lucarini, E.; Ghelardini, C. Ultramicronized N-Palmitoylethanolamine Supplementation for Long-Lasting, Low-Dosed Morphine Antinociception. Front. Pharmacol. 2018, 9, 473. [Google Scholar] [CrossRef] [Green Version]
- Mika, J.; Wawrzczak-Bargiela, A.; Osikowicz, M.; Makuch, W.; Przewlocka, B. Attenuation of morphine tolerance by minocycline and pentoxifylline in naive and neuropathic mice. Brain. Behav. Immun. 2009, 23, 75–84. [Google Scholar] [CrossRef]
- Mika, J. Modulation of microglia can attenuate neuropathic pain symptoms and enhance morphine effectiveness. Pharmacol. Rep. 2008, 60, 297–307. [Google Scholar] [PubMed]
- Skaper, S.D.; Facci, L.; Barbierato, M.; Zusso, M.; Bruschetta, G.; Impellizzeri, D.; Cuzzocrea, S.; Giusti, P. N-Palmitoylethanolamine and Neuroinflammation: A Novel Therapeutic Strategy of Resolution. Mol. Neurobiol. 2015, 52, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; D’Agostino, G.; Pacini, A.; Russo, R.; Zanardelli, M.; Ghelardini, C.; Calignano, A. Palmitoylethanolamide Is a Disease-Modifying Agent in Peripheral Neuropathy: Pain Relief and Neuroprotection Share a PPAR-Alpha-Mediated Mechanism. Mediators Inflamm. 2013, 2013, 328797. [Google Scholar] [CrossRef] [PubMed]
- Della Rocca, G.; Gamba, D. Chronic Pain in Dogs and Cats: Is There Place for Dietary Intervention with Micro-Palmitoylethanolamide? Animals 2021, 11, 952. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.M.; Vandevoorde, S.; Diependaele, G.; Govaerts, S.J.; Robert, A.R. Anticonvulsant Activity of N-Palmitoylethanolamide, a Putative Endocannabinoid, in Mice. Epilepsia 2002, 42, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Agostino, G.; Russo, R.; Avagliano, C.; Cristiano, C.; Meli, R.; Calignano, A. Palmitoylethanolamide Protects Against the Amyloid-β25-35-Induced Learning and Memory Impairment in Mice, an Experimental Model of Alzheimer Disease. Neuropsychopharmacology 2012, 37, 1784–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, E.; Impellizzeri, D.; Mazzon, E.; Paterniti, I.; Cuzzocrea, S. Neuroprotective Activities of Palmitoylethanolamide in an Animal Model of Parkinson’s Disease. PLoS ONE 2012, 7, e41880. [Google Scholar] [CrossRef] [Green Version]
- Skaper, S.D.; Giusti, P.; Facci, L. Microglia and mast cells: Two tracks on the road to neuroinflammation. FASEB J. 2012, 26, 3103–3117. [Google Scholar] [CrossRef]
- Petrosino, S.; Schiano Moriello, A. Palmitoylethanolamide: A Nutritional Approach to Keep Neuroinflammation within Physiological Boundaries-A Systematic Review. Int. J. Mol. Sci. 2020, 21, 9526. [Google Scholar] [CrossRef]
- Mazzari, S.; Canella, R.; Petrelli, L.; Marcolongo, G.; Leon, A. N-(2-Hydroxyethyl)hexadecanamide is orally active in reducing edema formation and inflammatory hyperalgesia by down-modulating mast cell activation. Eur. J. Pharmacol. 1996, 300, 227–236. [Google Scholar] [CrossRef]
- Petrosino, S.; Cordaro, M.; Verde, R.; Schiano Moriello, A.; Marcolongo, G.; Schievano, C.; Siracusa, R.; Piscitelli, F.; Peritore, A.F.; Crupi, R.; et al. Oral Ultramicronized Palmitoylethanolamide: Plasma and Tissue Levels and Spinal Anti-hyperalgesic Effect. Front. Pharmacol. 2018, 9, 249. [Google Scholar] [CrossRef] [PubMed]
- Leighton, G.E.; Rodriguez, R.E.; Hill, R.G.; Hughes, J. Kappa-Opioid agonists produce antinociception after i.v. and i.c.v. but not intrathecal administration in the rat. Br. J. Pharmacol. 1988, 93, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Castellano, B.; González, B.; Jensen, M.B.; Pedersen, E.B.; Finsen, B.R.; Zimmer, J. A double staining technique for simultaneous demonstration of astrocytes and microglia in brain sections and astroglial cell cultures. J. Histochem. Cytochem. 1991, 39, 561–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cesare Mannelli, L.; Pacini, A.; Corti, F.; Boccella, S.; Luongo, L.; Esposito, E.; Cuzzocrea, S.; Maione, S.; Calignano, A.; Ghelardini, C. Antineuropathic Profile of N-Palmitoylethanolamine in a Rat Model of Oxaliplatin-Induced Neurotoxicity. PLoS ONE 2015, 10, e0128080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cesare Mannelli, L.; Pacini, A.; Bonaccini, L.; Zanardelli, M.; Mello, T.; Ghelardini, C. Morphologic Features and Glial Activation in Rat Oxaliplatin-Dependent Neuropathic Pain. J. Pain 2013, 14, 1585–1600. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; Pacini, A.; Micheli, L.; Tani, A.; Zanardelli, M.; Ghelardini, C. Glial role in oxaliplatin-induced neuropathic pain. Exp. Neurol. 2014, 261, 22–33. [Google Scholar] [CrossRef]
- Woolf, C.J.; Bennett, G.J.; Doherty, M.; Dubner, R.; Kidd, B.; Koltzenburg, M.; Lipton, R.; Loeser, J.D.; Payne, R.; Torebjork, E. Towards a mechanism-based classification of pain? Pain 1998, 77, 227–229. [Google Scholar] [CrossRef]
- Prescott, S.A.; Ratté, S. Chapter 23–Somatosensation and Pain. In Conn’s Translational Neuroscience; Conn, P.M., Ed.; Academic Press: San Diego, CA, USA, 2017; pp. 517–539. ISBN 978-0-12-802381-5. [Google Scholar]
- Woolf, C.J.; Mannion, R.J. Neuropathic pain: Aetiology, symptoms, mechanisms, and management. Lancet 1999, 353, 1959–1964. [Google Scholar] [CrossRef]
- Paul, A.K.; Smith, C.M.; Rahmatullah, M.; Nissapatorn, V.; Wilairatana, P.; Spetea, M.; Gueven, N.; Dietis, N. Opioid Analgesia and Opioid-Induced Adverse Effects: A Review. Pharmaceuticals 2021, 14, 1091. [Google Scholar] [CrossRef]
- Fishbain, D.A.; Cole, B.; Lewis, J.; Rosomoff, H.L.; Rosomoff, R.S. What percentage of chronic nonmalignant pain patients exposed to chronic opioid analgesic therapy develop abuse/addiction and/or aberrant drug-related behaviors? A structured evidence-based review. Pain Med. 2008, 9, 444–459. [Google Scholar] [CrossRef] [Green Version]
- Paul, A.K.; Gueven, N.; Dietis, N. Profiling the Effects of Repetitive Morphine Administration on Motor Behavior in Rats. Molecules 2021, 26, 4355. [Google Scholar] [CrossRef] [PubMed]
- Buntin-Mushock, C.; Phillip, L.; Moriyama, K.; Palmer, P.P. Age-Dependent Opioid Escalation in Chronic Pain Patients. Anesth. Analg. 2005, 100, 1740–1745. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.K.; Gueven, N.; Dietis, N. Morphine dosing strategy plays a key role in the generation and duration of the produced antinociceptive tolerance. Neuropharmacology 2017, 121, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.S.; Kosek, P.S.; Staats, P.; Fisher, R.; Schultz, D.M.; Leong, M. Phase II, open-label, multicenter study of combined intrathecal morphine and ziconotide: Addition of ziconotide in patients receiving intrathecal morphine for severe chronic pain. Pain Med. 2008, 9, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirooie, S.; Esmaeili, J.; Sureda, A.; Esmaeili, N.; Mirzaee Saffari, P.; Yousefi-Manesh, H.; Dehpour, A.R. Evaluation of the effects of metformin administration on morphine tolerance in mice. Neurosci. Lett. 2020, 716, 134638. [Google Scholar] [CrossRef] [PubMed]
- Inturrisi, C.E. Clinical Pharmacology of Opioids for Pain. Clin. J. Pain 2002, 18, S3–S13. [Google Scholar] [CrossRef]
- Gugliandolo, E.; Peritore, A.F.; Piras, C.; Cuzzocrea, S.; Crupi, R. Palmitoylethanolamide and Related ALIAmides: Prohomeostatic Lipid Compounds for Animal Health and Wellbeing. Vet. Sci. 2020, 7, 78. [Google Scholar] [CrossRef]
- Epps, D.E.; Schmid, P.C.; Natarajan, V.; Schmid, H.H.O. N-acylethanolamine accumulation in infarcted myocardium. Biochem. Biophys. Res. Commun. 1979, 90, 628–633. [Google Scholar] [CrossRef]
- Alhouayek, M.; Muccioli, G.G. Harnessing the anti-inflammatory potential of palmitoylethanolamide. Drug Discov. Today 2014, 19, 1632–1639. [Google Scholar] [CrossRef]
- Balvers, M.G.J.; Verhoeckx, K.C.M.; Meijerink, J.; Wortelboer, H.M.; Witkamp, R.F. Measurement of palmitoylethanolamide and other N-acylethanolamines during physiological and pathological conditions. CNS Neurol. Disord. Drug Targets 2013, 12, 23–33. [Google Scholar] [CrossRef]
- Esposito, E.; Cuzzocrea, S. Palmitoylethanolamide in homeostatic and traumatic central nervous system injuries. CNS Neurol. Disord. Drug Targets 2013, 12, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.S. Effect of diet on tissue levels of palmitoylethanolamide. CNS Neurol. Disord. Drug Targets 2013, 12, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Rinne, P.; Guillamat-Prats, R.; Rami, M.; Bindila, L.; Ring, L.; Lyytikäinen, L.-P.; Raitoharju, E.; Oksala, N.; Lehtimäki, T.; Weber, C.; et al. Palmitoylethanolamide Promotes a Proresolving Macrophage Phenotype and Attenuates Atherosclerotic Plaque Formation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2562–2575. [Google Scholar] [CrossRef]
- Roviezzo, F.; Rossi, A.; Caiazzo, E.; Orlando, P.; Riemma, M.A.; Iacono, V.M.; Guarino, A.; Ialenti, A.; Cicala, C.; Peritore, A.; et al. Palmitoylethanolamide Supplementation during Sensitization Prevents Airway Allergic Symptoms in the Mouse. Front. Pharmacol. 2017, 8, 857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solorzano, C.; Zhu, C.; Battista, N.; Astarita, G.; Lodola, A.; Rivara, S.; Mor, M.; Russo, R.; Maccarrone, M.; Antonietti, F.; et al. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc. Natl. Acad. Sci. USA 2009, 106, 20966–20971. [Google Scholar] [CrossRef] [Green Version]
- Skaper, S.D.; Facci, L.; Giusti, P. Mast cells, glia and neuroinflammation: Partners in crime? Immunology 2014, 141, 314–327. [Google Scholar] [CrossRef]
- Silvasti, M.; Rosenberg, P.; Seppälä, T.; Svartling, N.; Pitkänen, M. Comparison of analgesic efficacy of oxycodone and morphine in postoperative intravenous patient-controlled analgesia. Acta Anaesthesiol. Scand. 1998, 42, 576–580. [Google Scholar] [CrossRef]
- Kalso, E.; Vainio, A.; Mattila, M.J.; Rosenberg, P.H.; Seppαlä, T. Morphine and Oxycodone in the Management of Cancer Pain: Plasma Levels Determined by Chemical and Radioreceptor Assays. Pharmacol. Toxicol. 1990, 67, 322–328. [Google Scholar] [CrossRef]
- Pöyhiä, R. Opioids in anaesthesia: A questionnaire survey in Finland. Eur. J. Anaesthesiol. 1994, 11, 221–230. [Google Scholar]
- Nuuttnen, L.S.; Wuolijoki, E.; Pentikäinen, I.T. Diclofenac and oxycodone in treatment of postoperative pain: A double-blind trial. Acta Anaesthesiol. Scand. 1986, 30, 620–624. [Google Scholar] [CrossRef]
- Lehmann, K.A.; Kratzenberg, U.; Schroeder-Bark, B.; Horrichs-Haermeyer, G. Postoperative Patient-Controlled Analgesia with Tramadol: Analgesic Efficacy and Minimum Effective Concentrations. Clin. J. Pain 1990, 6, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Luongo, L.; Guida, F.; Boccella, S.; Bellini, G.; Gatta, L.; Rossi, F.; de Novellis, V.; Maione, S. Palmitoylethanolamide reduces formalin-induced neuropathic-like behaviour through spinal glial/microglial phenotypical changes in mice. CNS Neurol. Disord. Drug Targets 2013, 12, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Caprioli, A.; Coccurello, R.; Rapino, C.; Di Serio, S.; Di Tommaso, M.; Vertechy, M.; Vacca, V.; Battista, N.; Pavone, F.; Maccarrone, M.; et al. The Novel Reversible Fatty Acid Amide Hydrolase Inhibitor ST4070 Increases Endocannabinoid Brain Levels and Counteracts Neuropathic Pain in Different Animal Models. J. Pharmacol. Exp. Ther. 2012, 342, 188–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganley, O.H.; Robinson, H.J. Antianaphylactic and antiserotonin activity of a compound obtained from egg yolk, peanut oil, and soybean lecithin. J. Allergy 1959, 30, 415–419. [Google Scholar] [CrossRef]
- Beggiato, S.; Tomasini, M.C.; Ferraro, L. Palmitoylethanolamide (PEA) as a Potential Therapeutic Agent in Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 821. [Google Scholar] [CrossRef] [Green Version]
- Levi-Montalcini, R.; Skaper, S.D.; Dal Toso, R.; Petrelli, L.; Leon, A. Nerve growth factor: From neurotrophin to neurokine. Trends Neurosci. 1996, 19, 514–520. [Google Scholar] [CrossRef]
- Aloe, L.; Leon, A.; Levi-Montalcini, R. A proposed autacoid mechanism controlling mastocyte behaviour. Agents Actions 1993, 39, C145–C147. [Google Scholar] [CrossRef]
- Cantarella, G.; Scollo, M.; Lempereur, L.; Saccani-Jotti, G.; Basile, F.; Bernardini, R. Endocannabinoids inhibit release of nerve growth factor by inflammation-activated mast cells. Biochem. Pharmacol. 2011, 82, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The Nuclear Receptor Peroxisome Proliferator-Activated Receptor-α Mediates the Anti-Inflammatory Actions of Palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef]
- Cordaro, M.; Cuzzocrea, S.; Crupi, R. An Update of Palmitoylethanolamide and Luteolin Effects in Preclinical and Clinical Studies of Neuroinflammatory Events. Antioxidants 2020, 9, 216. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.-S.V.; Barrett, D.A.; Randall, M.D. “Entourage” effects of N-palmitoylethanolamide and N-oleoylethanolamide on vasorelaxation to anandamide occur through TRPV1 receptors. Br. J. Pharmacol. 2008, 155, 837–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.; Naito, T.; Sato, H.; Hiraide, T.; Yamada, Y.; Kawakami, J. Impact of CYP genotype and inflammatory markers on the plasma concentrations of tramadol and its demethylated metabolites and drug tolerability in cancer patients. Eur. J. Clin. Pharmacol. 2018, 74, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Eidson, L.N.; Murphy, A.Z. Blockade of Toll-Like Receptor 4 Attenuates Morphine Tolerance and Facilitates the Pain Relieving Properties of Morphine. J. Neurosci. 2013, 33, 15952–15963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavendra, V.; Rutkowski, M.D.; DeLeo, J.A. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J. Neurosci. 2002, 22, 9980–9989. [Google Scholar] [CrossRef] [Green Version]
- DeLeo, J.A.; Tanga, F.Y.; Tawfik, V.L. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2004, 10, 40–52. [Google Scholar] [CrossRef]
- Congiu, M.; Micheli, L.; Santoni, M.; Sagheddu, C.; Muntoni, A.L.; Makriyannis, A.; Malamas, M.S.; Ghelardini, C.; Di Cesare Mannelli, L.; Pistis, M. N-Acylethanolamine Acid Amidase Inhibition Potentiates Morphine Analgesia and Delays the Development of Tolerance. Neurotherapeutics 2021, 18, 2722–2736. [Google Scholar] [CrossRef]
- Machelska, H.; Celik, M.Ö. Opioid Receptors in Immune and Glial Cells-Implications for Pain Control. Front. Immunol. 2020, 11, 300. [Google Scholar] [CrossRef] [Green Version]
- Skaper, S.D. Mast Cell-Glia Dialogue in Chronic Pain and Neuropathic Pain: Blood-Brain Barrier Implications. CNS Neurol. Disord. Drug Targets 2016, 15, 1072–1078. [Google Scholar] [CrossRef]
- Green, D.P.; Limjunyawong, N.; Gour, N.; Pundir, P.; Dong, X. A Mast-Cell-Specific Receptor Mediates Neurogenic Inflammation and Pain. Neuron 2019, 101, 412–420.e3. [Google Scholar] [CrossRef] [Green Version]
- McGrath, J.C.; Lilley, E. Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. Br. J. Pharmacol. 2015, 172, 3189–3193. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micheli, L.; Lucarini, E.; Toti, A.; Ferrara, V.; Ciampi, C.; Parisio, C.; Bartolucci, G.; Di Cesare Mannelli, L.; Ghelardini, C. Effects of Ultramicronized N-Palmitoylethanolamine Supplementation on Tramadol and Oxycodone Analgesia and Tolerance Prevention. Pharmaceutics 2022, 14, 403. https://doi.org/10.3390/pharmaceutics14020403
Micheli L, Lucarini E, Toti A, Ferrara V, Ciampi C, Parisio C, Bartolucci G, Di Cesare Mannelli L, Ghelardini C. Effects of Ultramicronized N-Palmitoylethanolamine Supplementation on Tramadol and Oxycodone Analgesia and Tolerance Prevention. Pharmaceutics. 2022; 14(2):403. https://doi.org/10.3390/pharmaceutics14020403
Chicago/Turabian StyleMicheli, Laura, Elena Lucarini, Alessandra Toti, Valentina Ferrara, Clara Ciampi, Carmen Parisio, Gianluca Bartolucci, Lorenzo Di Cesare Mannelli, and Carla Ghelardini. 2022. "Effects of Ultramicronized N-Palmitoylethanolamine Supplementation on Tramadol and Oxycodone Analgesia and Tolerance Prevention" Pharmaceutics 14, no. 2: 403. https://doi.org/10.3390/pharmaceutics14020403