
Lipid Nanoparticle Delivery Systems to Enable mRNA-Based Therapeutics

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Lipid Based Delivery Systems
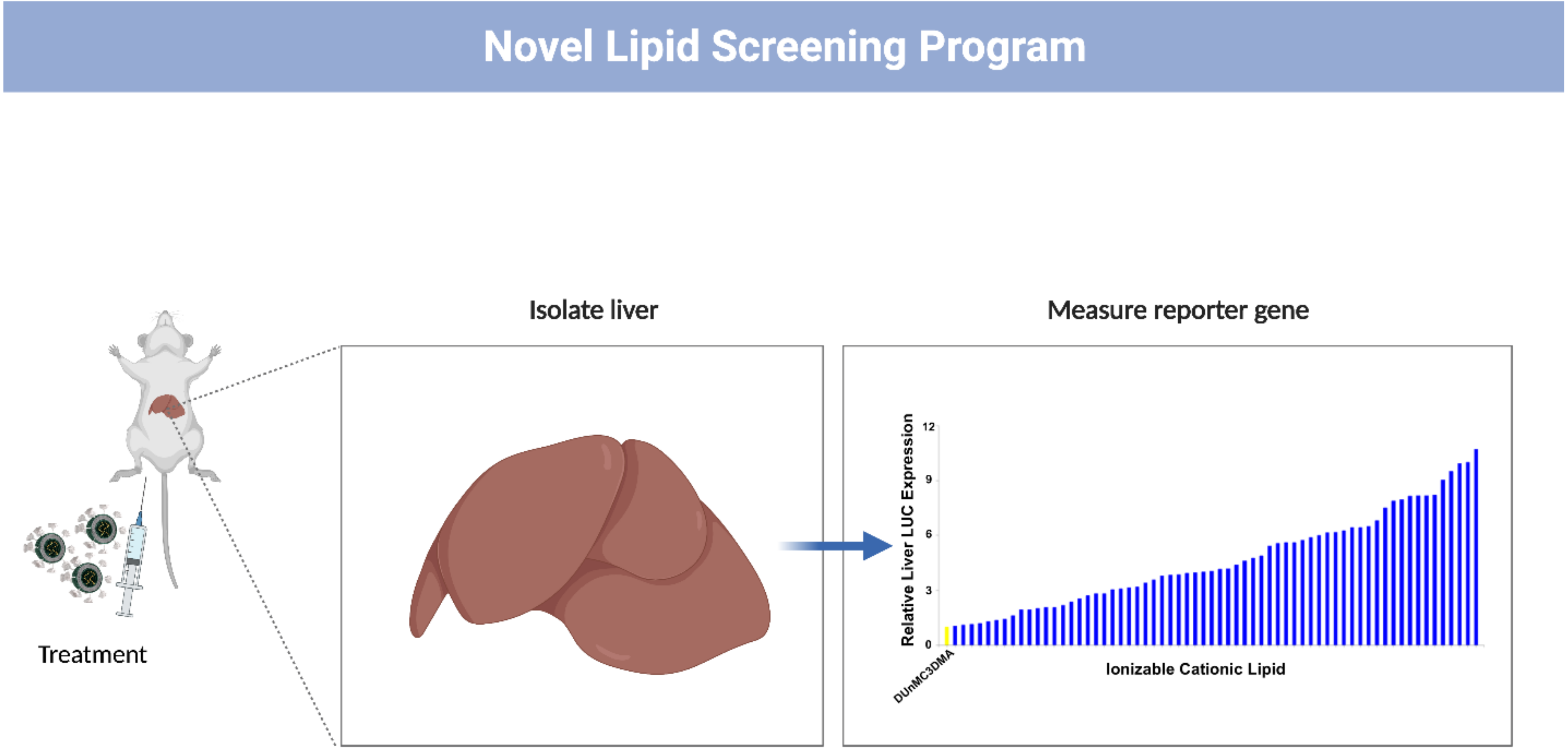
3. siRNA-LNP Development
4. Clinical siRNA-LNP Experiences
5. mRNA-LNP Development
5.1. Protein/Enzyme Replacement Therapy
5.2. Passive Immunization
5.3. Prophylactic Vaccines
5.3.1. Influenza
5.3.2. Malaria Vaccine
5.3.3. HIV Vaccine
5.3.4. Herpes Simplex Virus
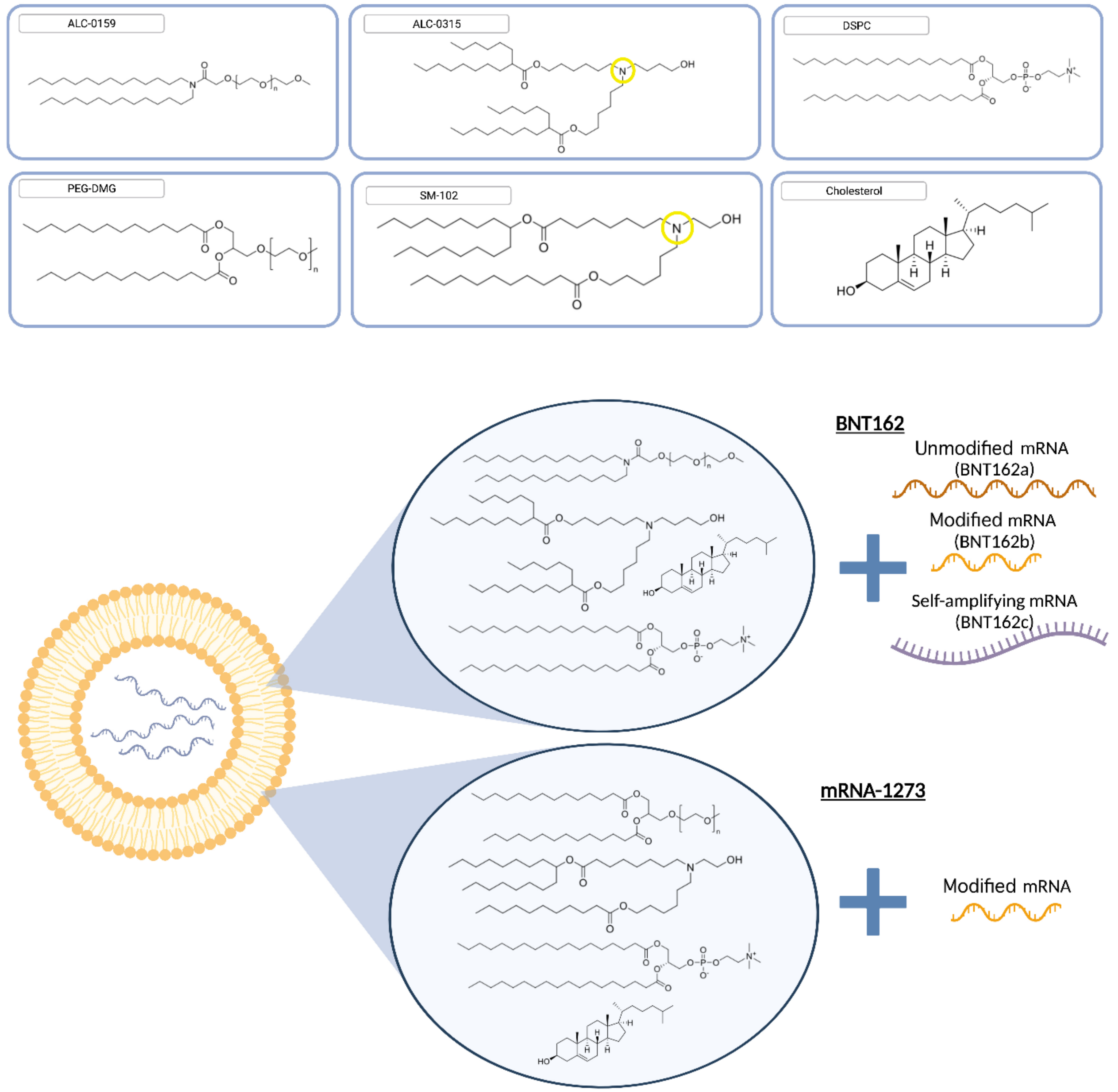
6. From Concept to Reality: Approval of the First mRNA-LNP Vaccine
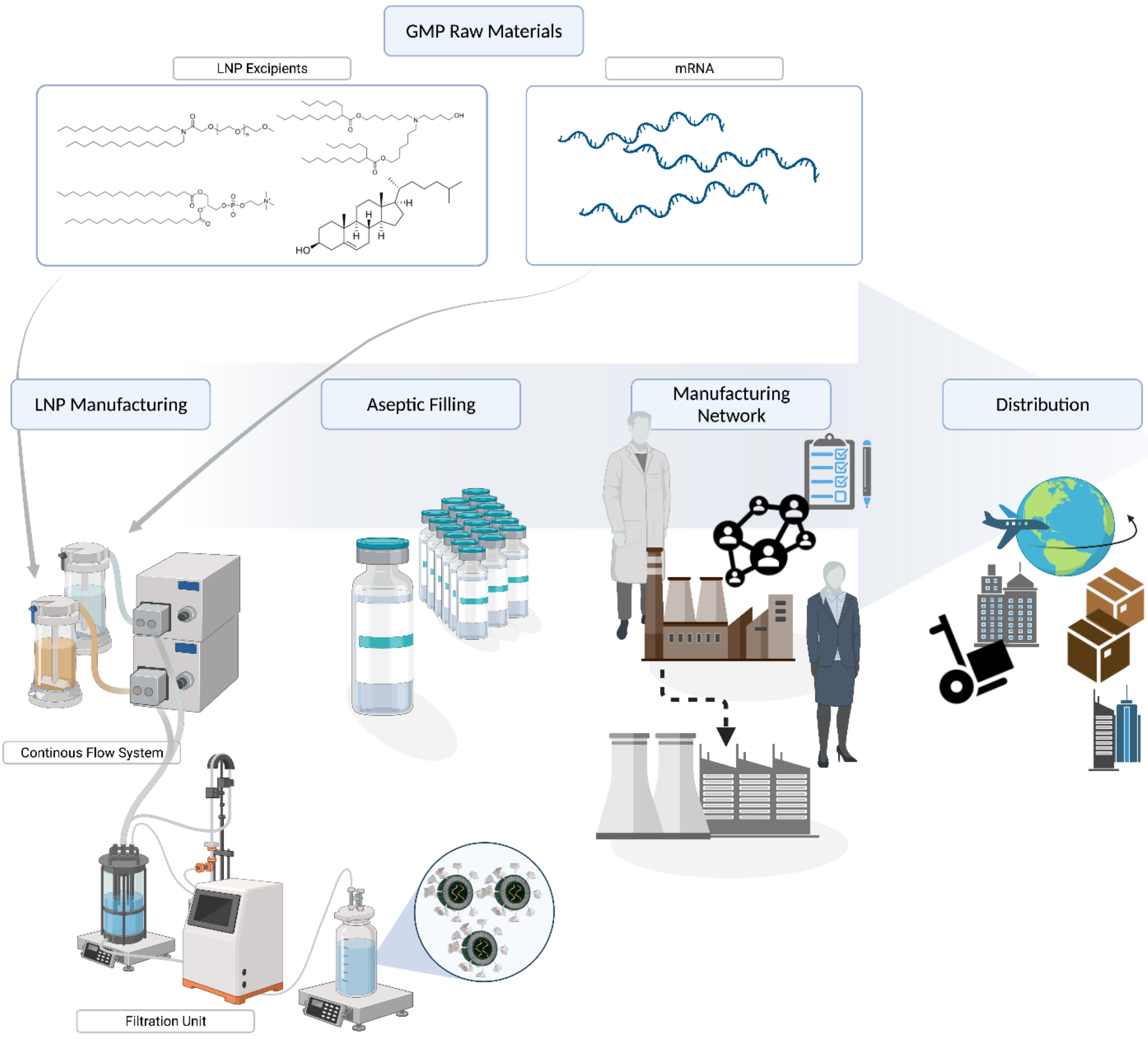
7. Lessons of Sourcing Material and Manufacturing Leading to Commercialization Scales
8. Next Generation COVID-19 mRNA-Vaccines
9. Breakthrough and Challenges of mRNA-LNP
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Htun, Y.M.; Thiha, K.; Aung, A.; Aung, N.M.; Oo, T.W.; Win, P.S.; Sint, N.H.; Naing, K.M.; Min, A.K.; Tun, K.M.; et al. Assessment of depressive symptoms in patients with COVID-19 during the second wave of epidemic in Myanmar: A cross-sectional single-center study. PLoS ONE 2021, 16, e0252189. [Google Scholar] [CrossRef] [PubMed]
- Bate, J.; Pham, P.T.; Borelli, J.L. Be My Safe Haven: Parent–Child Relationships and Emotional Health During COVID. J. Pediatr. Psychol. 2021, 46, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Xia, H.; Zou, J.; Weaver, S.C.; Swanson, K.A.; Cai, H.; Cutler, M.; Cooper, D.; Muik, A.; et al. BNT162b2-elicited neutralization of B.1.617 and other SARS-CoV-2 variants. Nature 2021, 596, 273–275. [Google Scholar] [CrossRef]
- Sahin, U.; Muik, A.; Vogler, I.; Derhovanessian, E.; Kranz, L.M.; Vormehr, M.; Quandt, J.; Bidmon, N.; Ulges, A.; Baum, A.; et al. BNT162b2 vaccine induces neutralizing antibodies and poly-specific T cells in humans. Nature 2021, 595, 572–577. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Xia, H.; Zhang, X.; Zou, J.; Fontes-Garfias, C.R.; Weaver, S.C.; Swanson, K.A.; Cai, H.; Sarkar, R.; et al. BNT162b2-Elicited Neutralization against New SARS-CoV-2 Spike Variants. N. Engl. J. Med. 2021, 385, 472–474. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Xia, H.; Zhang, X.; Fontes-Garfias, C.R.; Swanson, K.A.; Cai, H.; Sarkar, R.; Chen, W.; Cutler, M.; et al. Neutralizing Activity of BNT162b2-Elicited Serum. N. Engl. J. Med. 2021, 384, 1466–1468. [Google Scholar] [CrossRef]
- Wu, K.; Werner, A.P.; Moliva, J.I.; Koch, M.; Choi, A.; Stewart-Jones, G.B.E.; Bennett, H.; Boyoglu-Barnum, S.; Shi, W.; Graham, B.S.; et al. mRNA-1273 vaccine induces neutralizing antibodies against spike mutants from global SARS-CoV-2 variants. bioRxiv 2021. [Google Scholar] [CrossRef]
- Cobb, M. Who discovered messenger RNA? Curr. Biol. 2015, 25, R526–R532. [Google Scholar] [CrossRef] [Green Version]
- Weissman, D.; Ni, H.; Scales, D.; Dude, A.; Capodici, J.; McGibney, K.; Abdool, A.; Isaacs, S.N.; Cannon, G.; Karikó, K. HIV Gag mRNA Transfection of Dendritic Cells (DC) Delivers Encoded Antigen to MHC Class I and II Molecules, Causes DC Maturation, and Induces a Potent Human In Vitro Primary Immune Response. J. Immunol. 2000, 165, 4710–4717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capodici, J.; Karikó, K.; Weissman, D. Inhibition of HIV-1 infection by small interfering RNA-mediated RNA interference. J. Immunol. 2002, 169, 5196–5201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karikó, K.; Bhuyan, P.; Capodici, J.; Ni, H.; Lubinski, J.; Friedman, H.; Weissman, D. Exogenous siRNA Mediates Sequence-Independent Gene Suppression by Signaling through Toll-Like Receptor. Cells Tissues Organs 2004, 177, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Bhuyan, P.; Capodici, J.; Weissman, D. Small Interfering RNAs Mediate Sequence-Independent Gene Suppression and Induce Immune Activation by Signaling through Toll-Like Receptor. J. Immunol. 2004, 172, 6545–6549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karikó, K.; Ni, H.; Capodici, J.; Lamphier, M.; Weissman, D. mRNA Is an Endogenous Ligand for Toll-like Receptor. J. Biol. Chem. 2004, 279, 12542–12550. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Weissman, D.; Welsh, F.A. Inhibition of Toll-like Receptor and Cytokine Signaling—A Unifying Theme in Ischemic Tolerance. J. Cereb. Blood Flow Metab. 2004, 24, 1288–1304. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immun. 2005, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Weissman, D. Naturally occurring nucleoside modifications suppress the immunostimulatory activity of RNA: Implication for therapeutic RNA development. Curr. Opin. Drug Discov. Dev. 2007, 10, 523–532. [Google Scholar]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine into mRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Anderson, B.R.; Muramatsu, H.; Nallagatla, S.R.; Bevilacqua, P.C.; Sansing, L.H.; Weissman, D.; Karikó, K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 2010, 38, 5884–5892. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karikó, K.; Muramatsu, H.; Keller, J.M.; Weissman, D. Increased Erythropoiesis in Mice Injected with Submicrogram Quantities of Pseudouridine-containing mRNA Encoding Erythropoietin. Mol. Ther. 2012, 20, 948–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardi, N.; Muramatsu, H.; Weissman, D.; Karikó, K. In Vitro Transcription of Long RNA Containing Modified Nucleosides. Vaccine Des. 2012, 969, 29–42. [Google Scholar] [CrossRef]
- Weissman, D.; Pardi, N.; Muramatsu, H.; Karikó, K. HPLC Purification of In Vitro Transcribed Long RNA. Methods Mol. Biol. 2012, 969, 43–54. [Google Scholar] [CrossRef]
- Grudzien-Nogalska, E.; Stepinski, J.; Jemielity, J.; Zuberek, J.; Stolarski, R.; Rhoads, R.E.; Darzynkiewicz, E. Synthesis of Anti-Reverse Cap Analogs (ARCAs) and their Applications in mRNA Translation and Stability. Methods Enzymol. 2007, 431, 203–227. [Google Scholar] [CrossRef] [PubMed]
- Jemielity, J.; Stepinski, J.; Jaremko, M.; Haber, D.; Stolarski, R.; Rhoads, R.E.; Darzynkiewicz, E. Synthesis of Novel mRNA 5′ Cap-Analogues: Dinucleoside P1, P3-Tri-, P1, P4-Tetra-, and P1, P5-Pentaphosphates. Nucleosides Nucleotides Nucleic Acids 2003, 22, 691–694. [Google Scholar] [CrossRef]
- Stepinski, J.; Waddell, C.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl(3’-O-methyl)GpppG and 7-methyl (3’-deoxy)GpppG. RNA 2001, 7, 1486–1495. [Google Scholar]
- Laczko, D.; Hogan, M.J.; Toulmin, S.A.; Hicks, P.; Lederer, K.; Gaudette, B.T.; Castano, D.; Amanat, F.; Muramatsu, H.; Oguin, T.H., III; et al. A Single Immunization with Nucleoside-Modified mRNA Vaccines Elicits Strong Cellular and Humoral Immune Responses against SARS-CoV-2 in Mice. Immunity 2020, 53, 724–732.e7. [Google Scholar] [CrossRef]
- Pardi, N.; LaBranche, C.C.; Ferrari, G.; Cain, D.W.; Tombácz, I.; Parks, R.J.; Muramatsu, H.; Mui, B.L.; Tam, Y.K.; Karikó, K.; et al. Characterization of HIV-1 Nucleoside-Modified mRNA Vaccines in Rabbits and Rhesus Macaques. Mol. Ther.-Nucleic Acids 2019, 15, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Parkhouse, K.; Kirkpatrick, E.; McMahon, M.; Zost, S.; Mui, B.L.; Tam, Y.K.; Karikó, K.; Barbosa, C.J.; Madden, T.D.; et al. Nucleoside-modified mRNA immunization elicits influenza virus hemagglutinin stalk-specific antibodies. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Secreto, A.J.; Shan, X.; Debonera, F.; Glover, J.; Yi, Y.; Muramatsu, H.; Ni, H.; Mui, B.L.; Tam, Y.K.; et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 2017, 8, 14630. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phua, K.K.; Leong, K.W.; Nair, S.K. Transfection efficiency and transgene expression kinetics of mRNA delivered in naked and nanoparticle format. J. Control. Release 2013, 166, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, J.J.; Palmer, L.; Ossanlou, M.; MacLachlan, I.; Graham, R.W.; Zhang, Y.P.; Hope, M.J.; Scherrer, P.; Cullis, P.R. Stabilized plasmid-lipid particles: Construction and characterization. Gene Ther. 1999, 6, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Saravolac, E.G.; Ludkovski, O.; Skirrow, R.; Ossanlou, M.; Zhang, Y.P.; Giesbrecht, C.; Thompson, J.; Thomas, S.; Stark, H.; Cullis, P.R.; et al. Encapsulation of Plasmid DNA in Stabilized Plasmid–Lipid Particles Composed of Different Cationic Lipid Concentration for Optimal Transfection Activity. J. Drug Target. 2000, 7, 423–437. [Google Scholar] [CrossRef]
- Mok, K.W.; Lam, A.M.; Cullis, P.R. Stabilized plasmid-lipid particles: Factors influencing plasmid entrapment and transfection properties. Biochim. et Biophys. Acta (BBA)-Biomembr. 1999, 1419, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.P.; Sekirov, L.; Saravolac, E.G.; Wheeler, J.J.; Tardi, P.; Clow, K.; Leng, E.; Sun, R.; Cullis, P.R.; Scherrer, P. Stabilized plasmid-lipid particles for regional gene therapy: Formulation and transfection properties. Gene Ther. 1999, 6, 1438–1447. [Google Scholar] [CrossRef] [Green Version]
- Semple, S.C.; Klimuk, S.K.; Harasym, T.O.; Dos Santos, N.; Ansell, S.M.; Wong, K.F.; Maurer, N.; Stark, H.; Cullis, P.R.; Hope, M.J.; et al. Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: Formation of novel small multilamellar vesicle structures. Biochim. et Biophys. Acta (BBA)-Biomembr. 2001, 1510, 152–166. [Google Scholar] [CrossRef] [Green Version]
- Semple, S.C.; Klimuk, S.K.; Harasym, T.O.; Hope, M.J. Lipid-based formulations of antisense oligonucleotides for systemic delivery applications. Methods Enzymol. 2000, 313, 322–341. [Google Scholar] [CrossRef] [PubMed]
- Maurer, N.; Wong, K.F.; Stark, H.; Louie, L.; McIntosh, D.; Wong, T.; Scherrer, P.; Semple, S.C.; Cullis, P.R. Spontaneous Entrapment of Polynucleotides upon Electrostatic Interaction with Ethanol-Destabilized Cationic Liposomes. Biophys. J. 2001, 80, 2310–2326. [Google Scholar] [CrossRef] [Green Version]
- Zelphati, O.; Nguyen, C.; Ferrari, M.; Felgner, J.; Tsai, Y.; Felgner, P.L. Stable and monodisperse lipoplex formulations for gene delivery. Gene Ther. 1998, 5, 1272–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahn, A.; Vreeland, W.N.; Gaitan, A.M.; Locascio, L.E. Controlled Vesicle Self-Assembly in Microfluidic Channels with Hydrodynamic Focusing. J. Am. Chem. Soc. 2004, 126, 2674–2675. [Google Scholar] [CrossRef]
- Sun, Y.; Li, X.; Düzgüneşν, N.; Takaoka, Y.; Ohi, S.; Hirota, S. The Shape Parameter of Liposomes and DNA-Lipid Complexes Determined by Viscometry Utilizing Small Sample Volumes. Biophys. J. 2003, 85, 1223–1232. [Google Scholar] [CrossRef] [Green Version]
- Jeffs, L.B.; Palmer, L.R.; Ambegia, E.G.; Giesbrecht, C.; Ewanick, S.; MacLachlan, I. A Scalable, Extrusion-Free Method for Efficient Liposomal Encapsulation of Plasmid DNA. Pharm. Res. 2005, 22, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Belliveau, N.; Huft, J.; Lin, P.J.; Chen, S.; Leung, A.K.; Leaver, T.J.; Wild, A.W.; Lee, J.B.; Taylor, R.J.; Tam, Y.K.; et al. Microfluidic Synthesis of Highly Potent Limit-size Lipid Nanoparticles for In Vivo Delivery of siRNA. Mol. Ther.-Nucleic Acids 2012, 1, e37. [Google Scholar] [CrossRef]
- Leonetti, C.; Biroccio, A.; Benassi, B.; Stringaro, A.; Stoppacciaro, A.; Semple, S.C.; Zupi, G. Encapsulation of c-myc antisense oligodeoxynucleotides in lipid particles improves antitumoral efficacy in vivo in a human melanoma line. Cancer Gene Ther. 2001, 8, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Mui, B.; Raney, S.G.; Semple, S.C.; Hope, M.J. Immune stimulation by a CpG-containing oligodeoxynucleotide is enhanced when encapsulated and delivered in lipid particles. J. Pharmacol. Exp. Ther. 2001, 298, 1185–1192. [Google Scholar]
- Zimmermann, T.S.; Lee, A.C.H.; Akinc, A.; Bramlage, B.; Bumcrot, D.; Fedoruk, M.N.; Harborth, J.; Heyes, J.A.; Jeffs, L.B.; John, M.; et al. RNAi-mediated gene silencing in non-human primates. Nature 2006, 441, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the Potency of siRNA Lipid Nanoparticles for Hepatic Gene Silencing In Vivo**. Angew. Chem. Int. Ed. 2012, 51, 8529–8533. [Google Scholar] [CrossRef] [PubMed]
- Alabi, C.A.; Love, K.T.; Sahay, G.; Yin, H.; Luly, K.M.; Langer, R.; Anderson, D.G. Multiparametric approach for the evaluation of lipid nanoparticles for siRNA delivery. Proc. Natl. Acad. Sci. USA 2013, 110, 12881–12886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.J.; Tam, Y.Y.C.; Hafez, I.; Sandhu, A.; Chen, S.; Ciufolini, M.A.; Nabi, I.R.; Cullis, P.R. Influence of cationic lipid composition on uptake and intracellular processing of lipid nanoparticle formulations of siRNA. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 233–246. [Google Scholar] [CrossRef]
- Hafez, I.M.; Cullis, P.R. Roles of lipid polymorphism in intracellular delivery. Adv. Drug Deliv. Rev. 2001, 47, 139–148. [Google Scholar] [CrossRef]
- Hafez, I.M.; Maurer, N.; Cullis, P.R. On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther. 2001, 8, 1188–1196. [Google Scholar] [CrossRef] [Green Version]
- Van Dyke, R.W. Acidification of Lysosomes and Endosomes. Subcell. Biochem. 1996, 27, 331–360. [Google Scholar] [CrossRef]
- Mui, B.L.; Tam, Y.K.; Jayaraman, M.; Ansell, S.M.; Du, X.; Tam, Y.Y.C.; Lin, P.J.; Chen, S.; Narayanannair, J.K.; Rajeev, K.G.; et al. Influence of Polyethylene Glycol Lipid Desorption Rates on Pharmacokinetics and Pharmacodynamics of siRNA Lipid Nanoparticles. Mol. Ther.-Nucleic Acids 2013, 2, e139. [Google Scholar] [CrossRef]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted Delivery of RNAi Therapeutics with Endogenous and Exogenous Ligand-Based Mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Kumar, V.; Qin, J.; Jiang, Y.; Duncan, R.G.; Brigham, B.; Fishman, S.; Nair, J.K.; Akinc, A.; Barros, S.A.; Kasperkovitz, P.V. Shielding of Lipid Nanoparticles for siRNA Delivery: Impact on Physicochemical Properties, Cytokine Induction, and Efficacy. Mol. Ther.-Nucleic Acids 2014, 3, e210. [Google Scholar] [CrossRef] [PubMed]
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Racie, T.; et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 1864–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlman, J.E.; Kauffman, K.J.; Langer, R.; Anderson, D.G. Nanotechnology for In vivo Targeted siRNA Delivery. Adv. Genet. 2014, 88, 37–69. [Google Scholar] [CrossRef] [PubMed]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R.; et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Shapiro, G.I.; Lorusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-Humans Trial of an RNA Interference Therapeutic Targeting VEGF and KSP in Cancer Patients with Liver Involvement. Cancer Discov. 2013, 3, 406–418. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, G.; Ozpolat, B.; Coleman, R.L.; Sood, A.K.; Lopez-Berestein, G. Preclinical and clinical development of siRNA-based therapeutics. Adv. Drug Deliv. Rev. 2015, 87, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef]
- Fitzgerald, K.; Frank-Kamenetsky, M.; Shulga-Morskaya, S.; Liebow, A.; Bettencourt, B.R.; Sutherland, J.E.; Hutabarat, R.M.; Clausen, V.A.; Karsten, V.; Cehelsky, J.; et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: A randomised, single-blind, placebo-controlled, phase 1 trial. Lancet 2014, 383, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Thess, A.; Grund, S.; Mui, B.L.; Hope, M.J.; Baumhof, P.; Fotin-Mleczek, M.; Schlake, T. Sequence-engineered mRNA Without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Mol. Ther. 2015, 23, 1456–1464. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.; Polydefkis, M.; González-Duarte, A.; Wixner, J.; Kristen, A.V.; Schmidt, H.H.; Berk, J.L.; López, I.A.L.; Dispenzieri, A.; Quan, D.; et al. Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. Lancet Neurol. 2020, 20, 49–59. [Google Scholar] [CrossRef]
- Coelho, T.; Adams, D.; Conceição, I.; Waddington-Cruz, M.; Schmidt, H.H.; Buades, J.; Campistol, J.; Berk, J.L.; Polydefkis, M.; Wang, J.J.; et al. A phase II, open-label, extension study of long-term patisiran treatment in patients with hereditary transthyretin-mediated (hATTR) amyloidosis. Orphanet J. Rare Dis. 2020, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Obici, L.; Berk, J.L.; Gonzalez-Duarte, A.; Coelho, T.; Gillmore, J.; Schmidt, H.H.; Schilling, M.; Yamashita, T.; Labeyrie, C.; Brannagan, T.H., 3rd; et al. Quality of life outcomes in APOLLO, the phase 3 trial of the RNAi therapeutic patisiran in patients with hereditary transthyretin-mediated amyloidosis. Amyloid 2020, 27, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Duarte, A.; Berk, J.L.; Quan, D.; Mauermann, M.L.; Schmidt, H.H.; Polydefkis, M.; Waddington-Cruz, M.; Ueda, M.; Conceição, I.M.; Kristen, A.V.; et al. Analysis of autonomic outcomes in APOLLO, a phase III trial of the RNAi therapeutic patisiran in patients with hereditary transthyretin-mediated amyloidosis. J. Neurol. 2019, 267, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Adams, D.; Suhr, O.B.; Dyck, P.J.; Litchy, W.J.; Leahy, R.G.; Chen, J.; Gollob, J.; Coelho, T. Trial design and rationale for APOLLO, a Phase 3, placebo-controlled study of patisiran in patients with hereditary ATTR amyloidosis with polyneuropathy. BMC Neurol. 2017, 17, 1–12. [Google Scholar] [CrossRef]
- Suhr, O.B.; Coelho, T.; Buades, J.; Pouget, J.; Conceicao, I.; Berk, J.; Schmidt, H.; Waddington-Cruz, M.; Campistol, J.M.; Bettencourt, B.R.; et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: A phase II multi-dose study. Orphanet J. Rare Dis. 2015, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Maier, M.A.; Jayaraman, M.; Matsuda, S.; Liu, J.; Barros, S.; Querbes, W.; Tam, Y.K.; Ansell, S.M.; Kumar, V.; Qin, J.; et al. Biodegradable Lipids Enabling Rapidly Eliminated Lipid Nanoparticles for Systemic Delivery of RNAi Therapeutics. Mol. Ther. 2013, 21, 1570–1578. [Google Scholar] [CrossRef] [Green Version]
- Conway, A.; Mendel, M.; Kim, K.; McGovern, K.; Boyko, A.; Zhang, L.; Miller, J.C.; DeKelver, R.C.; Paschon, D.E.; Mui, B.L.; et al. Non-viral Delivery of Zinc Finger Nuclease mRNA Enables Highly Efficient In Vivo Genome Editing of Multiple Therapeutic Gene Targets. Mol. Ther. 2019, 27, 866–877. [Google Scholar] [CrossRef] [Green Version]
- Thran, M.; Mukherjee, J.; Pönisch, M.; Fiedler, K.; Thess, A.; Mui, B.L.; Hope, M.J.; Tam, Y.K.; Horscroft, N.; Heidenreich, R.; et al. mRNA mediates passive vaccination against infectious agents, toxins, and tumors. EMBO Mol. Med. 2017, 9, 1434–1447. [Google Scholar] [CrossRef]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef]
- Mallory, K.L.; Taylor, J.A.; Zou, X.; Waghela, I.N.; Schneider, C.G.; Sibilo, M.Q.; Punde, N.M.; Perazzo, L.C.; Savransky, T.; Sedegah, M.; et al. Messenger RNA expressing PfCSP induces functional, protective immune responses against malaria in mice. NPJ Vaccines 2021, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Weissman, D.; Alameh, M.-G.; de Silva, T.; Collini, P.; Hornsby, H.; Brown, R.; LaBranche, C.C.; Edwards, R.J.; Sutherland, L.; Santra, S.; et al. D614G Spike Mutation Increases SARS CoV-2 Susceptibility to Neutralization. Cell Host Microbe 2020, 29, 23–31.e4. [Google Scholar] [CrossRef] [PubMed]
- McKay, P.F.; Hu, K.; Blakney, A.K.; Samnuan, K.; Brown, J.C.; Penn, R.; Zhou, J.; Bouton, C.R.; Rogers, P.; Polra, K.; et al. Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nat. Commun. 2020, 11, 3523. [Google Scholar] [CrossRef]
- Rothgangl, T.; Dennis, M.K.; Lin PJ, C.; Oka, R.; Witzigmann, D.; Villiger, L.; Qi, W.; Hruzova, M.; Kissling, L.; Lenggenhager, D.; et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat. Biotechnol. 2021, 39, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Villiger, L.; Rothgangl, T.; Witzigmann, D.; Oka, R.; Lin, P.J.C.; Qi, W.; Janjuha, S.; Berk, C.; Ringnalda, F.; Beattie, M.B.; et al. In vivo cytidine base editing of hepatocytes without detectable off-target mutations in RNA and DNA. Nat. Biomed. Eng. 2021, 5, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.O.; Pardi, N.; Parks, R.; Santra, S.; Mu, Z.; Sutherland, L.; Scearce, R.; Barr, M.; Eaton, A.; Hernandez, G.; et al. Lipid nanoparticle encapsulated nucleoside-modified mRNA vaccines elicit polyfunctional HIV-1 antibodies comparable to proteins in nonhuman primates. NPJ Vaccines 2021, 6, 50. [Google Scholar] [CrossRef] [PubMed]
- Frenck, R.W., Jr.; Klein, N.P.; Kitchin, N.; Gurtman, A.; Absalon, J.; Lockhart, S.; Perez, J.L.; Walter, E.B.; Senders, S.; Bailey, R.; et al. Safety, Immunogenicity, and Efficacy of the BNT162b2 Covid-19 Vaccine in Adolescents. New Engl. J. Med. 2021, 385, 239–250. [Google Scholar] [CrossRef]
- Walsh, E.E.; Frenck, R.W., Jr.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based COVID-19 Vaccine Candidates. N. Engl. J. Med. 2020, 383, 2439–2450. [Google Scholar] [CrossRef]
- Sabnis, S.; Kumarasinghe, E.S.; Salerno, T.; Mihai, C.; Ketova, T.; Senn, J.J.; Lynn, A.; Bulychev, A.; McFadyen, I.; Chan, J.; et al. A Novel Amino Lipid Series for mRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-human Primates. Mol. Ther. 2018, 26, 1509–1519. [Google Scholar] [CrossRef] [Green Version]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther. Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauffman, K.J.; Dorkin, J.R.; Yang, J.H.; Heartlein, M.W.; DeRosa, F.; Mir, F.F.; Fenton, O.S.; Anderson, D.G. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Dorkin, J.R.; Wang, W.; Chang, P.H.; Webber, M.J.; Tang, B.C.; Yang, J.; Abutbul-Ionita, I.; Danino, D.; DeRosa, F.; et al. Poly(glycoamidoamine) Brushes Formulated Nanomaterials for Systemic siRNA and mRNA Delivery in Vivo. Nano Lett. 2016, 16, 842–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenton, O.S.; Kauffman, K.J.; McClellan, R.L.; Appel, E.A.; Dorkin, J.R.; Tibbitt, M.W.; Heartlein, M.W.; DeRosa, F.; Langer, R.; Anderson, D.G. Bioinspired Alkenyl Amino Alcohol Ionizable Lipid Materials for Highly Potent In Vivo mRNA Delivery. Adv. Mater. 2016, 28, 2939–2943. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Tonnu, N.; Tachikawa, K.; Limphong, P.; Vega, J.B.; Karmali, P.P.; Chivukula, P.; Verma, I.M. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E1941–E1950. [Google Scholar] [CrossRef] [Green Version]
- Prieve, M.G.; Harvie, P.; Monahan, S.D.; Roy, D.; Li, A.G.; Blevins, T.L.; Paschal, A.E.; Waldheim, M.; Bell, E.C.; Galperin, A.; et al. Targeted mRNA Therapy for Ornithine Transcarbamylase Deficiency. Mol. Ther. 2018, 26, 801–813. [Google Scholar] [CrossRef]
- DeRosa, F.; Smith, L.; Shen, Y.; Huang, Y.; Pan, J.; Xie, H.; Yahalom, B.; Heartlein, M.W. Improved Efficacy in a Fabry Disease Model Using a Systemic mRNA Liver Depot System as Compared to Enzyme Replacement Therapy. Mol. Ther. 2019, 27, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Cao, J.; Huang, P.; An, P.; Badlani, D.; Vaid, K.A.; Zhao, S.; Wang, D.Q.; Zhuo, J.; Yin, L.; et al. Synthetic human ABCB4 mRNA therapy rescues severe liver disease phenotype in a BALB/c.Abcb4 (-/-) mouse model of PFIC3. J. Hepatol. 2020, 74, 1416–1428. [Google Scholar] [CrossRef]
- Cao, J.; An, D.; Galduroz, M.; Zhuo, J.; Liang, S.; Eybye, M.; Frassetto, A.; Kuroda, E.; Funahashi, A.; Santana, J.; et al. mRNA Therapy Improves Metabolic and Behavioral Abnormalities in a Murine Model of Citrin Deficiency. Mol. Ther. 2019, 27, 1242–1251. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Tran, D.M.; Cavedon, A.; Cai, X.; Rajendran, R.; Lyle, M.J.; Martini, P.G.; Miao, C.H. Treatment of Hemophilia A Using Factor VIII Messenger RNA Lipid Nanoparticles. Mol. Ther.-Nucleic Acids 2020, 20, 534–544. [Google Scholar] [CrossRef]
- Parra-Guillen, Z.P.; Fontanellas, A.; Jiang, L.; Jericó, D.; Martini, P.; Vera-Yunca, D.; Hard, M.; Guey, L.T.; Troconiz, I.F. Disease pharmacokinetic–pharmacodynamic modelling in acute intermittent porphyria to support the development of mRNA -based therapies. J. Cereb. Blood Flow Metab. 2020, 177, 3168–3182. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Yin, L.; Theisen, M.; Zhuo, J.; Siddiqui, S.; Levy, B.; Presnyak, V.; Frassetto, A.; Milton, J.; Salerno, T.; et al. Systemic mRNA Therapy for the Treatment of Fabry Disease: Preclinical Studies in Wild-Type Mice, Fabry Mouse Model, and Wild-Type Non-human Primates. Am. J. Hum. Genet. 2019, 104, 625–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakrishnan, B.; An, D.; Nguyen, V.; DeAntonis, C.; Martini, P.G.V.; Lai, K. Novel mRNA-Based Therapy Reduces Toxic Galactose Metabolites and Overcomes Galactose Sensitivity in a Mouse Model of Classic Galactosemia. Mol. Ther. 2020, 28, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Karadagi, A.; Cavedon, A.G.; Zemack, H.; Nowak, G.; Eybye, M.E.; Zhu, X.; Guadagnin, E.; White, R.A.; Rice, L.M.; Frassetto, A.L.; et al. Systemic modified messenger RNA for replacement therapy in alpha 1-antitrypsin deficiency. Sci. Rep. 2020, 10, 7052. [Google Scholar] [CrossRef]
- Truong, B.; Allegri, G.; Liu, X.-B.; Burke, K.E.; Zhu, X.; Cederbaum, S.D.; Häberle, J.; Martini, P.G.V.; Lipshutz, G.S. Lipid nanoparticle-targeted mRNA therapy as a treatment for the inherited metabolic liver disorder arginase deficiency. Proc. Natl. Acad. Sci. USA 2019, 116, 21150–21159. [Google Scholar] [CrossRef]
- Kose, N.; Fox, J.M.; Sapparapu, G.; Bombardi, R.; Tennekoon, R.N.; de Silva, A.D.; Elbashir, S.M.; Theisen, M.A.; Humphris-Narayanan, E.; Ciaramella, G.; et al. A lipid-encapsulated mRNA encoding a potently neutralizing human monoclonal antibody protects against chikungunya infection. Sci. Immunol. 2019, 4, eaaw6647. [Google Scholar] [CrossRef]
- Jiang, L.; Park, J.-S.; Yin, L.; Laureano, R.; Jacquinet, E.; Yang, J.; Liang, S.; Frassetto, A.; Zhuo, J.; Yan, X.; et al. Dual mRNA therapy restores metabolic function in long-term studies in mice with propionic acidemia. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- An, D.; Schneller, J.L.; Frassetto, A.; Liang, S.; Zhu, X.; Park, J.-S.; Theisen, M.; Hong, S.-J.; Zhou, J.; Rajendran, R.; et al. Systemic Messenger RNA Therapy as a Treatment for Methylmalonic Acidemia. Cell Rep. 2017, 21, 3548–3558. [Google Scholar] [CrossRef] [Green Version]
- Rybakova, Y.; Kowalski, P.; Huang, Y.; Gonzalez, J.; Heartlein, M.W.; DeRosa, F.; Delcassian, D.; Anderson, D.G. mRNA Delivery for Therapeutic Anti-HER2 Antibody Expression In Vivo. Mol. Ther. 2019, 27, 1415–1423. [Google Scholar] [CrossRef]
- Richner, J.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.; Julander, J.G.; Tang, W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell 2017, 169, 176. [Google Scholar] [CrossRef] [Green Version]
- Wollner, C.J.; Richner, M.; Hassert, M.A.; Pinto, A.K.; Brien, J.D.; Richner, J.M. A Dengue Virus Serotype 1 mRNA-LNP Vaccine Elicits Protective Immune Responses. J. Virol. 2021, 95, e02482-20. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.S.; Jenks, J.A.; Pardi, N.; Goodwin, M.; Roark, H.; Edwards, W.; McLellan, J.S.; Pollara, J.; Weissman, D.; Permar, S.R. Human Cytomegalovirus Glycoprotein B Nucleoside-Modified mRNA Vaccine Elicits Antibody Responses with Greater Durability and Breadth than MF59-Adjuvanted gB Protein Immunization. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Huang, E.; Yuzhakov, O.; Ramanathan, P.; Ciaramella, G.; Bukreyev, A. Modified mRNA-Based Vaccines Elicit Robust Immune Responses and Protect Guinea Pigs from Ebola Virus Disease. J. Infect. Dis. 2018, 217, 451–455. [Google Scholar] [CrossRef] [PubMed]
- VanBlargan, L.A.; Himansu, S.; Foreman, B.M.; Ebel, G.D.; Pierson, T.C.; Diamond, M.S. An mRNA Vaccine Protects Mice against Multiple Tick-Transmitted Flavivirus Infections. Cell Rep. 2018, 25, 3382–3392.e3. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, Q.-T.; Choi, Y.-K. Targeting Antigens for Universal Influenza Vaccine Development. Viruses 2021, 13, 973. [Google Scholar] [CrossRef]
- Magini, D.; Giovani, C.; Mangiavacchi, S.; Maccari, S.; Cecchi, R.; Ulmer, J.B.; De Gregorio, E.; Geall, A.J.; Brazzoli, M.; Bertholet, S. Self-Amplifying mRNA Vaccines Expressing Multiple Conserved Influenza Antigens Confer Protection against Homologous and Heterosubtypic Viral Challenge. PLoS ONE 2016, 11, e0161193. [Google Scholar] [CrossRef]
- Hekele, A.; Bertholet, S.; Archer, J.; Gibson, D.G.; Palladino, G.; Brito, L.A.; Otten, G.R.; Brazzoli, M.; Buccato, S.; Bonci, A.; et al. Rapidly produced SAM((R)) vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microbes Infect. 2013, 2, e52. [Google Scholar] [CrossRef]
- Feldman, R.A.; Fuhr, R.; Smolenov, I.; Ribeiro, A.; Panther, L.; Watson, M.; Senn, J.J.; Smith, M.; Almarsson, Ö.; Pujar, H.S.; et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 2019, 37, 3326–3334. [Google Scholar] [CrossRef]
- Lindgren, G.; Ols, S.; Liang, F.; Thompson, E.A.; Lin, A.; Hellgren, F.; Bahl, K.; John, S.; Yuzhakov, O.; Hassett, K.J.; et al. Induction of Robust B Cell Responses after Influenza mRNA Vaccination Is Accompanied by Circulating Hemagglutinin-Specific ICOS+ PD-1+ CXCR3+ T Follicular Helper Cells. Front. Immunol. 2017, 8, 1539. [Google Scholar] [CrossRef] [Green Version]
- Bahl, K.; Senn, J.J.; Yuzhakov, O.; Bulychev, A.; Brito, L.A.; Hassett, K.J.; Laska, M.E.; Smith, M.; Almarsson, Örn; Thompson, J.; et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol. Ther. 2017, 25, 1316–1327. [Google Scholar] [CrossRef] [Green Version]
- Freyn, A.W.; da Silva, J.R.; Rosado, V.C.; Bliss, C.M.; Pine, M.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Ferreira, L.C.D.S.; Weissman, D.; et al. A Multi-Targeting, Nucleoside-Modified mRNA Influenza Virus Vaccine Provides Broad Protection in Mice. Mol. Ther. 2020, 28, 1569–1584. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Qi, Y.; Wang, M.; Yu, N.; Nan, F.; Zhang, H.; Tian, M.; Li, C.; Lu, H.; Jin, N. mRNA Vaccines Encoding the HA Protein of Influenza a H1N1 Virus Delivered by Cationic Lipid Nanoparticles Induce Protective Immune Responses in Mice. Vaccines 2020, 8, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Haas, H.; et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2017, 26, 446–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rts, S.C.T.P. Clinical Trials Partnership. Efficacy and safety of the RTS, S/AS01 malaria vaccine during 18 months after vaccination: A phase 3 randomized, controlled trial in children and young infants at 11 African sites. PLoS Med. 2014, 11, e1001685. [Google Scholar]
- Young, W.C.; Carpp, L.N.; Chaudhury, S.; Regules, J.A.; Bergmann-Leitner, E.S.; Ockenhouse, C.; Wille-Reece, U.; de Camp, A.C.; Hughes, E.; Mahoney, C.; et al. Comprehensive Data Integration Approach to Assess Immune Responses and Correlates of RTS, S/AS01-Mediated Protection From Malaria Infection in Controlled Human Malaria Infection Trials. Front Big Data 2021, 4, 672460. [Google Scholar] [CrossRef]
- Jongo, S.A.; Church, L.W.P.; Mtoro, A.T.; Chakravarty, S.; Ruben, A.J.; Ii, P.A.S.; Kassim, K.R.; Mpina, M.; Tumbo, A.-M.; Milando, F.A.; et al. Safety and Differential Antibody and T-Cell Responses to the Plasmodium falciparum Sporozoite Malaria Vaccine, PfSPZ Vaccine, by Age in Tanzanian Adults, Adolescents, Children, and Infants. Am. J. Trop. Med. Hyg. 2019, 100, 1433–1444. [Google Scholar] [CrossRef]
- Draper, S.J.; Sack, B.K.; King, C.R.; Nielsen, C.M.; Rayner, J.C.; Higgins, M.; Long, C.A.; Seder, R.A. Malaria Vaccines: Recent Advances and New Horizons. Cell Host Microbe 2018, 24, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Richie, T.L.; Billingsley, P.F.; Sim, B.K.; James, E.R.; Chakravarty, S.; Epstein, J.E.; Lyke, K.E.; Mordmuller, B.; Alonso, P.; Duffy, P.E.; et al. Progress with Plasmodium falciparum sporozoite (PfSPZ)-based malaria vaccines. Vaccine 2015, 33, 7452–7461. [Google Scholar] [CrossRef]
- Ledford, H. Malaria vaccine wows and seeds COVID-19 vaccine effort. Nat. Biotechnol. 2021, 39, 786–787. [Google Scholar] [CrossRef]
- Raj, D.K.; Mohapatra, A.D.; Jnawali, A.; Zuromski, J.; Jha, A.; Cham-Kpu, G.; Sherman, B.; Rudlaff, R.M.; Nixon, C.E.; Hilton, N.; et al. Anti-PfGARP activates programmed cell death of parasites and reduces severe malaria. Nature 2020, 582, 104–108. [Google Scholar] [CrossRef]
- Fischer, W.; Giorgi, E.E.; Chakraborty, S.; Nguyen, K.; Bhattacharya, T.; Theiler, J.; Goloboff, P.A.; Yoon, H.; Abfalterer, W.; Foley, B.T.; et al. HIV-1 and SARS-CoV-2: Patterns in the evolution of two pandemic pathogens. Cell Host Microbe 2021, 29, 1093–1110. [Google Scholar] [CrossRef] [PubMed]
- Blakney, A.K.; McKay, P.F.; Yus, B.I.; Aldon, Y.; Shattock, R.J. Inside out: Optimization of lipid nanoparticle formulations for exterior complexation and in vivo delivery of saRNA. Gene Ther. 2019, 26, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Moyo, N.; Wee, E.; Korber, B.; Bahl, K.; Falcone, S.; Himansu, S.; Wong, A.; Dey, A.; Feinberg, M.; Hanke, T. Tetravalent Immunogen Assembled from Conserved Regions of HIV-1 and Delivered as mRNA Demonstrates Potent Preclinical T-Cell Immunogenicity and Breadth. Vaccines 2020, 8, 360. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Hook, L.M.; Pardi, N.; Wang, F.; Myles, A.; Cancro, M.P.; Cohen, G.H.; Weissman, D.; Friedman, H.M. Nucleoside-modified mRNA encoding HSV-2 glycoproteins C, D, and E prevents clinical and subclinical genital herpes. Sci. Immunol. 2019, 4, eaaw7083. [Google Scholar] [CrossRef]
- Egan, K.P.; Hook, L.M.; Naughton, A.; Pardi, N.; Awasthi, S.; Cohen, G.H.; Weissman, D.; Friedman, H.M. An HSV-2 nucleoside-modified mRNA genital herpes vaccine containing glycoproteins gC, gD, and gE protects mice against HSV-1 genital lesions and latent infection. PLOS Pathog. 2020, 16, e1008795. [Google Scholar] [CrossRef]
- LaTourette, P.C., 2nd; Awasthi, S.; Desmond, A.; Pardi, N.; Cohen, G.H.; Weissman, D.; Friedman, M.H. Protection against herpes simplex virus type 2 infection in a neonatal murine model using a trivalent nucleoside-modified mRNA in lipid nanoparticle vaccine. Vaccine 2020, 38, 7409–7413. [Google Scholar] [CrossRef]
- Aldrich, C.; Leroux-Roels, I.; Huang, K.B.; Bica, M.A.; Loeliger, E.; Schoenborn-Kellenberger, O.; Walz, L.; Leroux-Roels, G.; von Sonnenburg, F.; Oostvogels, L. Proof-of-concept of a low-dose unmodified mRNA-based rabies vaccine formulated with lipid nanoparticles in human volunteers: A phase 1 trial. Vaccine 2021, 39, 1310–1318. [Google Scholar] [CrossRef]
- Walsh, E.E.; Frenck, R.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. RNA-Based COVID-19 Vaccine BNT162b2 Selected for a Pivotal Efficacy Study. medRxiv 2020. [Google Scholar]
- Vogel, A.B.; Kanevsky, I.; Che, Y.; Swanson, K.A.; Muik, A.; Vormehr, M.; Kranz, L.M.; Walzer, K.C.; Hein, S.; Guler, A.; et al. BNT162b vaccines protect rhesus macaques from SARS-CoV-2. Nature 2021, 592, 283–289. [Google Scholar] [CrossRef]
- Ji, R.R.; Qu, Y.; Zhu, H.; Yang, Y.; Vogel, A.B.; Sahin, U.; Qin, C.; Hui, A. BNT162b2 Vaccine Encoding the SARS-CoV-2 P2 S Protects Transgenic hACE2 Mice against COVID-19. Vaccines 2021, 9, 324. [Google Scholar] [CrossRef]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schafer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.S.; Flynn, B.; Foulds, K.E.; Francica, J.R.; Boyoglu-Barnum, S.; Werner, A.P.; Flach, B.; O’Connell, S.; Bock, K.W.; Minai, M.; et al. Evaluation of the mRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. N. Engl. J. Med. 2020, 383, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- Challener, C.A. Meeting the Demand for Lipid Excipients. BioPharm Int. 2021, 34, 18–20. [Google Scholar]
- Schoenitz, M.; Grundemann, L.; Augustin, W.; Scholl, S. Fouling in microstructured devices: A review. Chem. Commun. 2015, 51, 8213–8228. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, R. When microfluidic devices go bad. How does fouling occur in microfluidic devices, and what can be done about it? Anal. Chem. 2005, 77, 429A–432A. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauch, S.; Roth, N.; Schwendt, K.; Fotin-Mleczek, M.; Mueller, S.O.; Petsch, B. mRNA-based SARS-CoV-2 vaccine candidate CVnCoV induces high levels of virus-neutralising antibodies and mediates protection in rodents. NPJ Vaccines 2021, 6, 57. [Google Scholar] [CrossRef]
- Kremsner, P.G.; Mann, P.; Kroidl, A.; Leroux-Roels, I.; Schindler, C.; Gabor, J.J.; Schunk, M.; Leroux-Roels, G.; Bosch, J.J.; Fendel, R.; et al. Safety and immunogenicity of an mRNA-lipid nanoparticle vaccine candidate against SARS-CoV-2 A phase 1 randomized clinical trial. Wien Klin Wochenschr. 2021, 133, 931–941. [Google Scholar] [CrossRef]
- Kremsner, P.; Mann, P.; Bosch, J.; Fendel, R.; Gabor, J.J.; Kreidenweiss, A.; Kroidl, A.; Leroux-Roels, I.; Leroux-Roels, G.; Schindler, C.; et al. Phase 1 Assessment of the Safety and Immunogenicity of an mRNA- Lipid Nanoparticle Vaccine Candidate Against SARS-CoV-2 in Human Volunteers. medRxiv 2020. [Google Scholar]
- Kremsner, P.G.; Guerrero, R.A.A.; Arana-Arri, E.; Martinez, G.J.A.; Bonten, M.; Chandler, R.; Corral, G.; de Block, E.J.L.; Ecker, L.; Gabor, J.J.; et al. Efficacy and Safety of the CVnCoV SARS-CoV-2 mRNA Vaccine Candidate in Ten Countries in Europe and Latin America (HERALD): A Randomised, Observer-Blinded, Placebo-Controlled, Phase 2b/3 Trial. Lancet Infect Dis. 2021. [Google Scholar] [CrossRef]
- Anderson, E.J.; Rouphael, N.G.; Widge, A.T.; Jackson, L.A.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Safety and Immunogenicity of SARS-CoV-2 mRNA-1273 Vaccine in Older Adults. N. Engl. J. Med. 2020, 383, 2427–2438. [Google Scholar] [CrossRef]
- van Doremalen, N.; Fischer, R.J.; Schulz, J.E.; Holbrook, M.G.; Smith, B.J.; Lovaglio, J.; Petsch, B.; Munster, V.J. Immunogenicity of Low-Dose Prime-Boost Vaccination of mRNA Vaccine CV07050101 in Non-Human Primates. Viruses 2021, 13, 1645. [Google Scholar] [CrossRef] [PubMed]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef]
- Gebre, M.S.; Rauch, S.; Roth, N.; Yu, J.; Chandrashekar, A.; Mercado, N.B.; He, X.; Liu, J.; McMahan, K.; Martinot, A.; et al. Optimization of non-coding regions for a non-modified mRNA COVID-19 vaccine. Nature 2021, 601, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Corleis, B.; Rauch, S.; Roth, N.; Mühe, J.; Halwe, N.J.; Ulrich, L.; Fricke, C.; Schön, J.; Kraft, A.; et al. CVnCoV and CV2CoV protect human ACE2 transgenic mice from ancestral B BavPat1 and emerging B.1.351 SARS-CoV-2. Nat. Commun. 2021, 12, 4048. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Sirk, S.J.; Shui, S.-L.; Liu, J. Genome-Editing Technologies: Principles and Applications. Cold Spring Harb. Perspect. Biol. 2016, 8, a023754. [Google Scholar] [CrossRef] [Green Version]
- Sago, C.D.; Lokugamage, M.; Paunovska, K.; Vanover, D.A.; Monaco, C.M.; Shah, N.N.; Castro, M.G.; Anderson, S.E.; Rudoltz, T.G.; Lando, G.N.; et al. High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc. Natl. Acad. Sci. USA 2018, 115, E9944–E9952. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, A.D.S.; Paunovska, K.; Cristian, M.A.; Dahlman, J.E. Treating Cystic Fibrosis with mRNA and CRISPR. Hum. Gene Ther. 2020, 31, 940–955. [Google Scholar] [CrossRef]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; Van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef] [Green Version]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. New Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef]
- Song, C.-Q.; Jiang, T.; Richter, M.; Rhym, L.H.; Koblan, L.; Zafra, M.P.; Schatoff, E.M.; Doman, J.L.; Cao, Y.; Dow, L.E.; et al. Adenine base editing in an adult mouse model of tyrosinaemia. Nat. Biomed. Eng. 2020, 4, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Henderson, J.M.; Coote, K.; Cheng, Y.; Valley, H.C.; Zhang, X.-O.; Wang, Q.; Rhym, L.H.; Cao, Y.; Newby, G.A.; et al. Chemical modifications of adenine base editor mRNA and guide RNA expand its application scope. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Lee, J.B.; Zhang, K.; Tam, Y.Y.C.; Tam, Y.K.; Belliveau, N.M.; Sung, V.Y.; Lin, P.J.; LeBlanc, E.; Ciufolini, M.A.; Rennie, P.S.; et al. Lipid nanoparticle siRNA systems for silencing the androgen receptor in human prostate cancer in vivo. Int. J. Cancer 2012, 131, E781–E790. [Google Scholar] [CrossRef] [PubMed]
- Tam, Y.Y.C.; Chen, S.; Zaifman, J.; Tam, Y.K.; Lin, P.J.; Ansell, S.; Roberge, M.; Ciufolini, M.A.; Cullis, P.R. Small molecule ligands for enhanced intracellular delivery of lipid nanoparticle formulations of siRNA. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Parhiz, H.; Shuvaev, V.V.; Pardi, N.; Khoshnejad, M.; Kiseleva, R.Y.; Brenner, J.S.; Uhler, T.; Tuyishime, S.; Mui, B.L.; Tam, Y.K.; et al. PECAM-1 directed re-targeting of exogenous mRNA providing two orders of magnitude enhancement of vascular delivery and expression in lungs independent of apolipoprotein E-mediated uptake. J. Control. Release 2018, 291, 106–115. [Google Scholar] [CrossRef]
- Marcos-Contreras, O.A.; Greineder, C.F.; Kiseleva, R.Y.; Parhiz, H.; Walsh, L.R.; Zuluaga-Ramirez, V.; Myerson, J.W.; Hood, E.D.; Villa, C.H.; Tombacz, I.; et al. Selective targeting of nanomedicine to inflamed cerebral vasculature to enhance the blood–brain barrier. Proc. Natl. Acad. Sci. USA 2020, 117, 3405–3414. [Google Scholar] [CrossRef]
- Tombacz, I.; Laczko, D.; Shahnawaz, H.; Muramatsu, H.; Natesan, A.; Yadegari, A.; Papp, T.E.; Alameh, M.G.; Shuvaev, V.; Mui, B.L.; et al. Highly efficient CD4+ T cell targeting and genetic recombination using engineered CD4+ cell-homing mRNA-LNP. Mol Ther. 2021, 29, 3293–3304. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jeong, M.; Hur, S.; Cho, Y.; Park, J.; Jung, H.; Seo, Y.; Woo, H.A.; Nam, K.T.; Lee, K.; et al. Engineered ionizable lipid nanoparticles for targeted delivery of RNA therapeutics into different types of cells in the liver. Sci. Adv. 2021, 7, eabf4398. [Google Scholar] [CrossRef]
- Gan, Z.; Lokugamage, M.P.; Hatit, M.Z.C.; Loughrey, D.; Paunovska, K.; Sato, M.; Cristian, A.; Dahlman, J.E. Nanoparticles containing constrained phospholipids deliver mRNA to liver immune cells in vivo without targeting ligands. Bioeng. Transl. Med. 2020, 5, 10161. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semple, S.C.; Leone, R.; Barbosa, C.J.; Tam, Y.K.; Lin, P.J.C. Lipid Nanoparticle Delivery Systems to Enable mRNA-Based Therapeutics. Pharmaceutics 2022, 14, 398. https://doi.org/10.3390/pharmaceutics14020398
Semple SC, Leone R, Barbosa CJ, Tam YK, Lin PJC. Lipid Nanoparticle Delivery Systems to Enable mRNA-Based Therapeutics. Pharmaceutics. 2022; 14(2):398. https://doi.org/10.3390/pharmaceutics14020398
Chicago/Turabian StyleSemple, Sean C., Robert Leone, Christopher J. Barbosa, Ying K. Tam, and Paulo J. C. Lin. 2022. "Lipid Nanoparticle Delivery Systems to Enable mRNA-Based Therapeutics" Pharmaceutics 14, no. 2: 398. https://doi.org/10.3390/pharmaceutics14020398