
In Vitro and In Vivo Studies of a Verapamil-Containing Gastroretentive Solid Foam Capsule

 ,
,  ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Experimental Animals
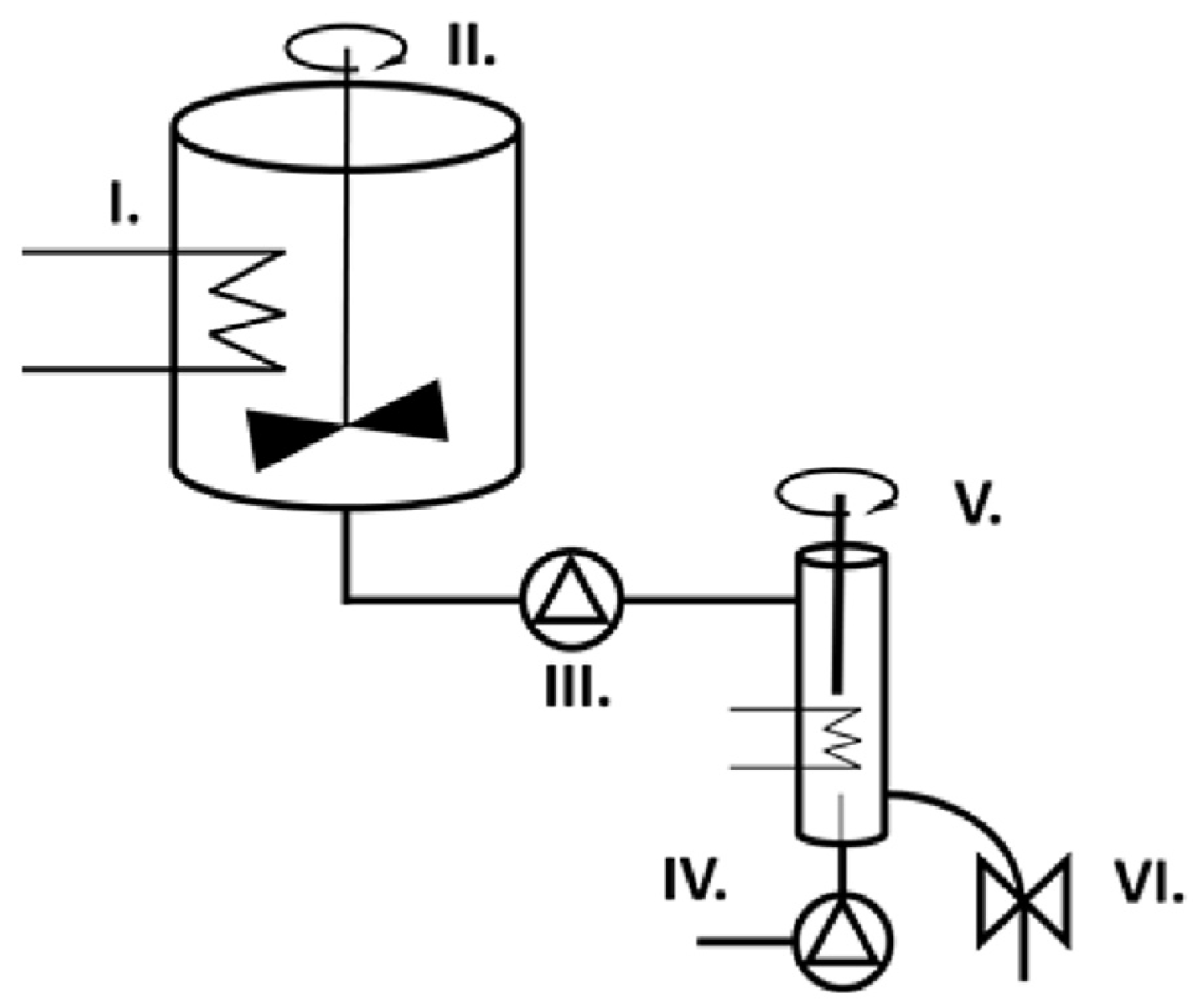
2.2. Preparation of Solid Foam Capsules
2.3. Determination of the Densities of the Samples
2.4. SEM Analysis
2.5. Dissolution Test
2.6. Mathematical Analysis of the Drug Release Profiles
2.7. Validation of Production
2.8. Microtomography and Size Distribution of Foam Cells
2.9. Texture Analysis
2.10. Water Uptake and Matrix Erosion Studies
2.11. Floating Strength Determination
2.12. Dissolution Test after Long-Term and Accelerated Storage Conditions
2.13. In Vivo Pharmacokinetic Study
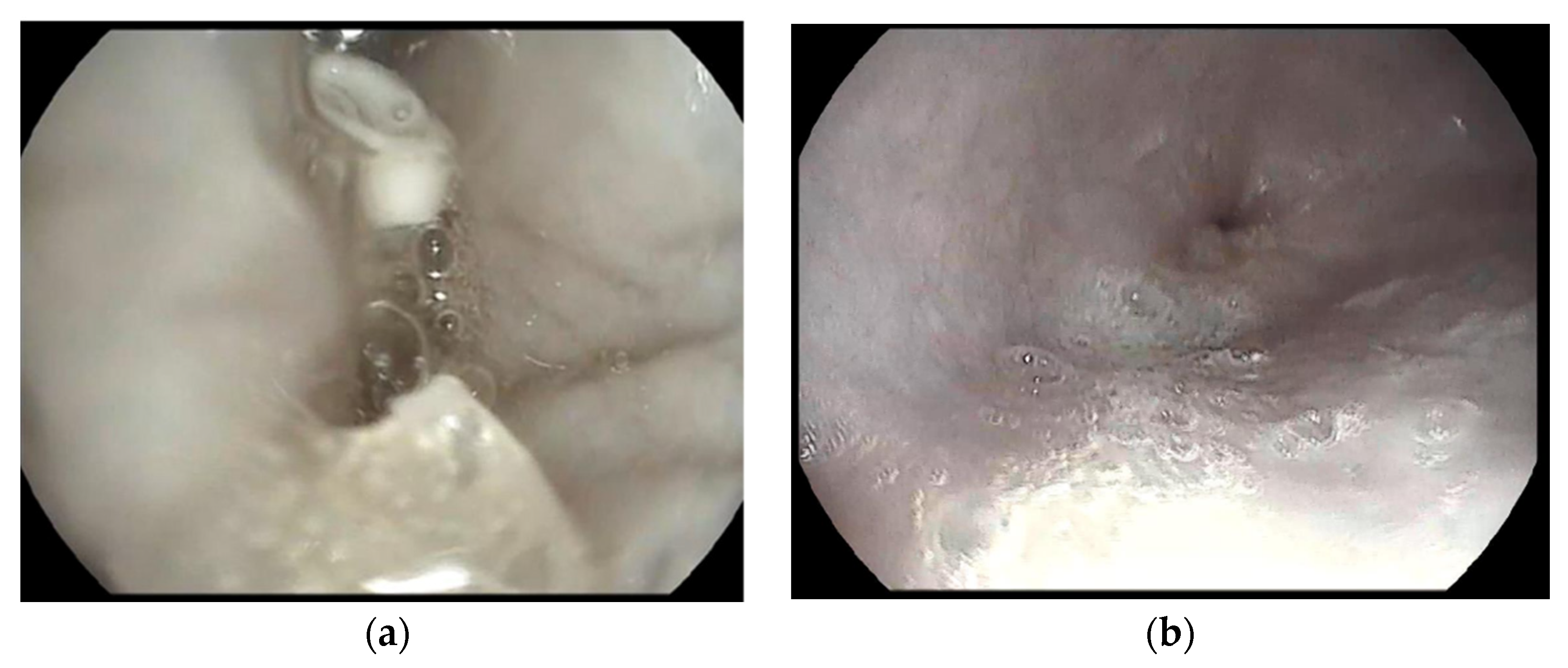
2.14. Gastroscopy
2.15. Quantitative Determination of Verapamilin Plasma
2.16. Statistical Analysis
3. Results
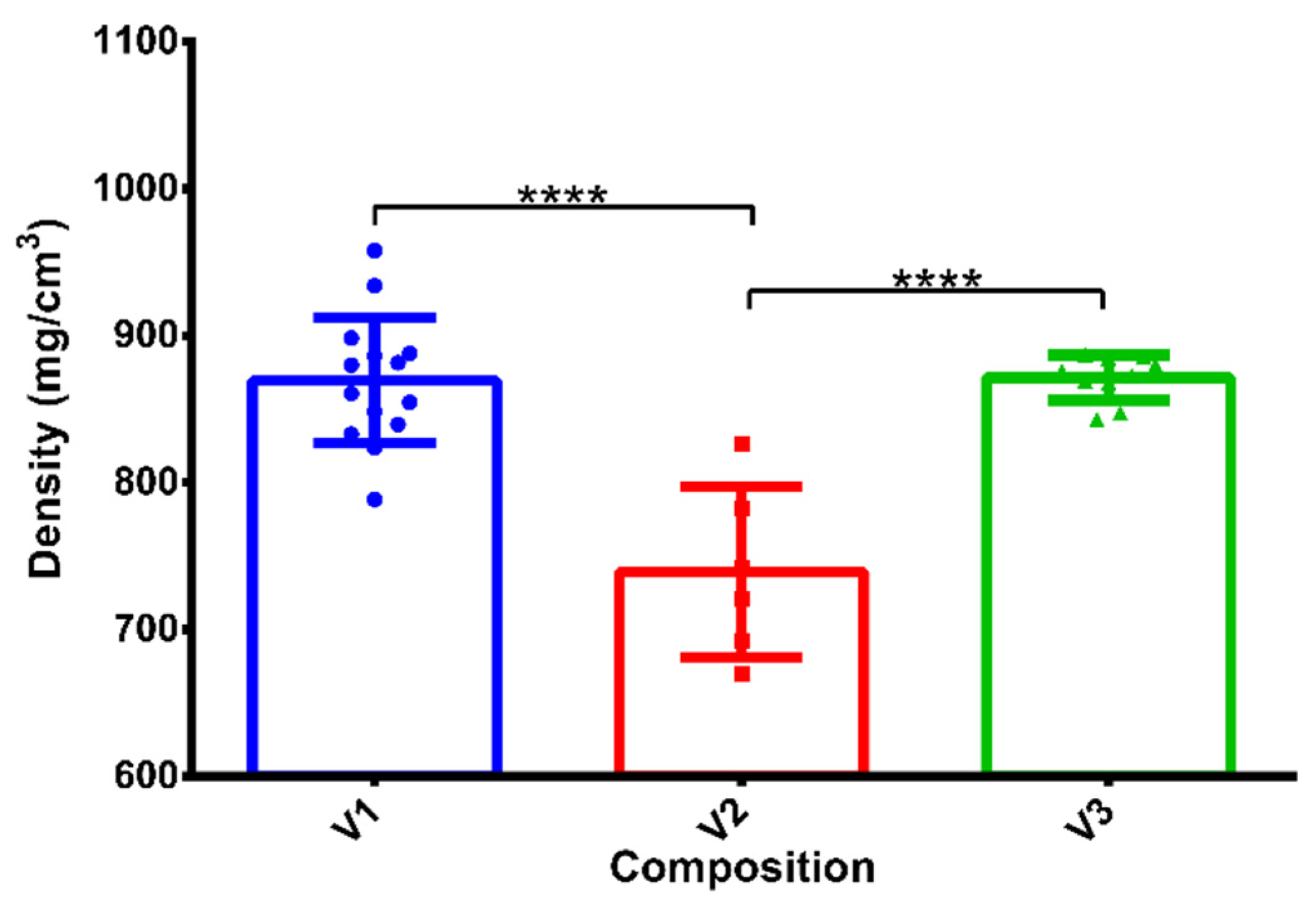
3.1. Density of Compositions
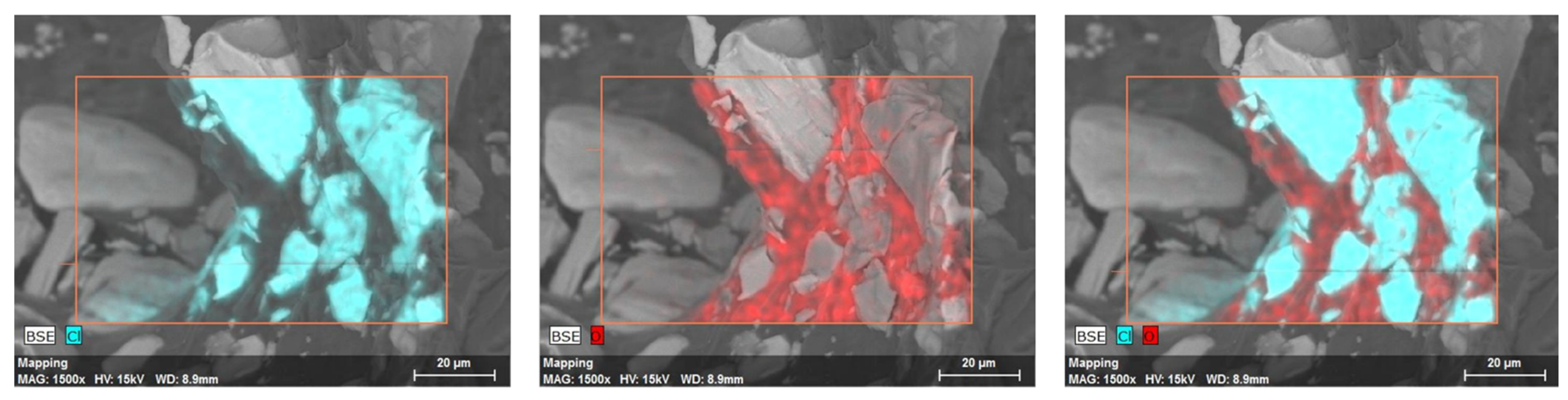
3.2. SEM Analysis
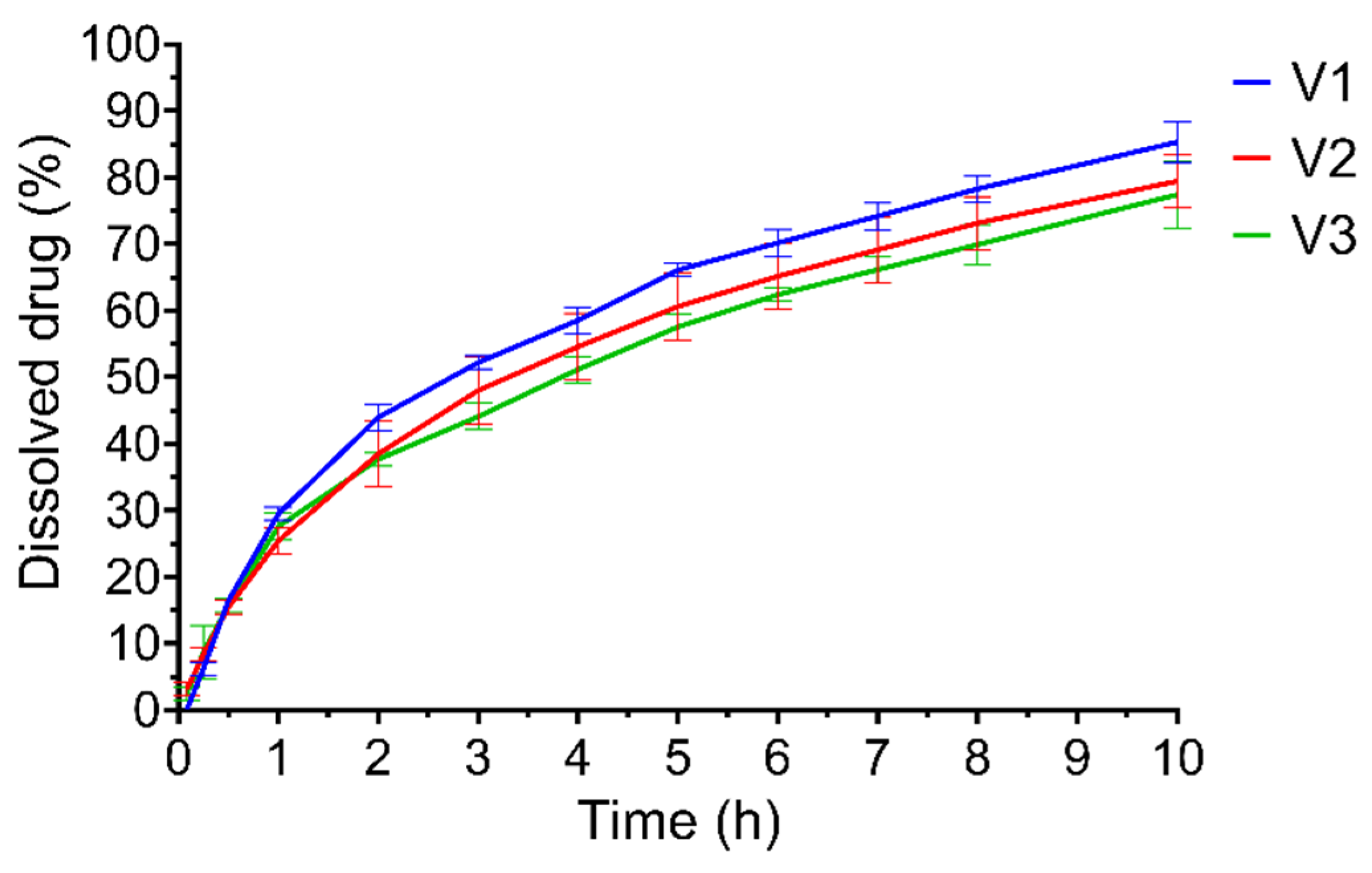
3.3. Dissolution Test
3.4. Validation of Production
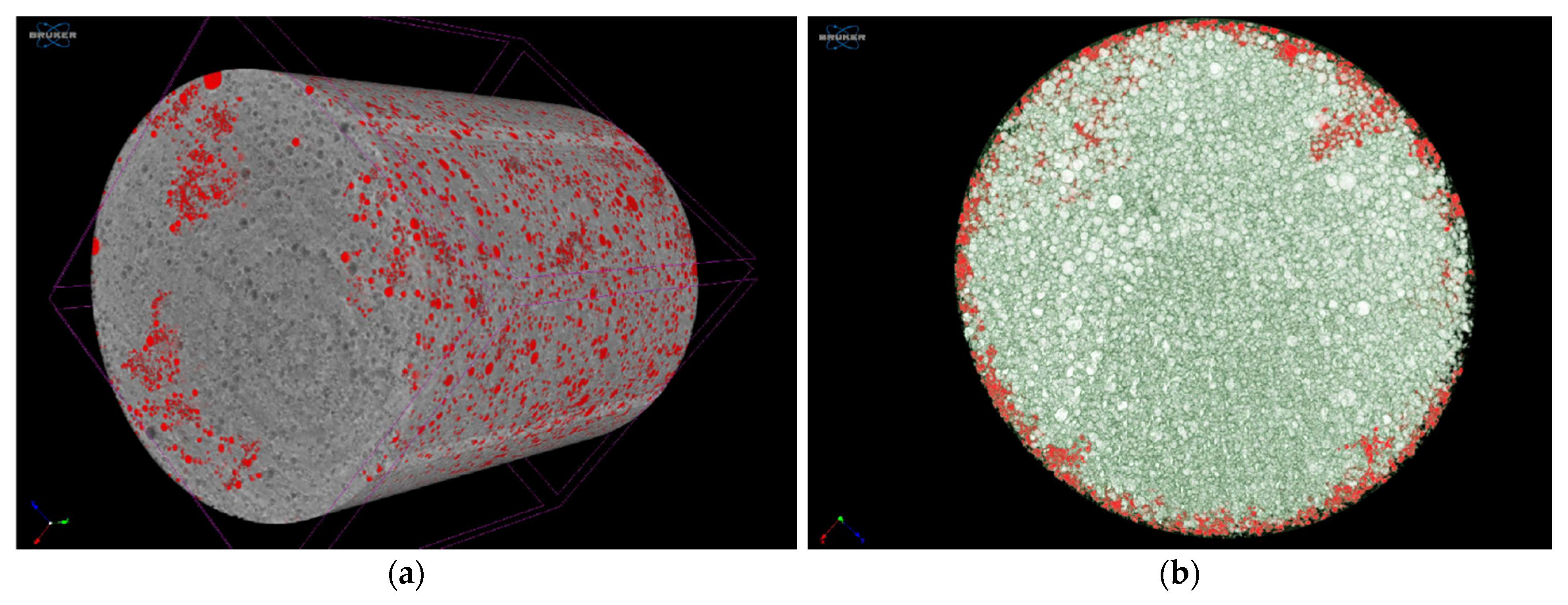
3.5. Microtomography
3.6. Texture Analysis
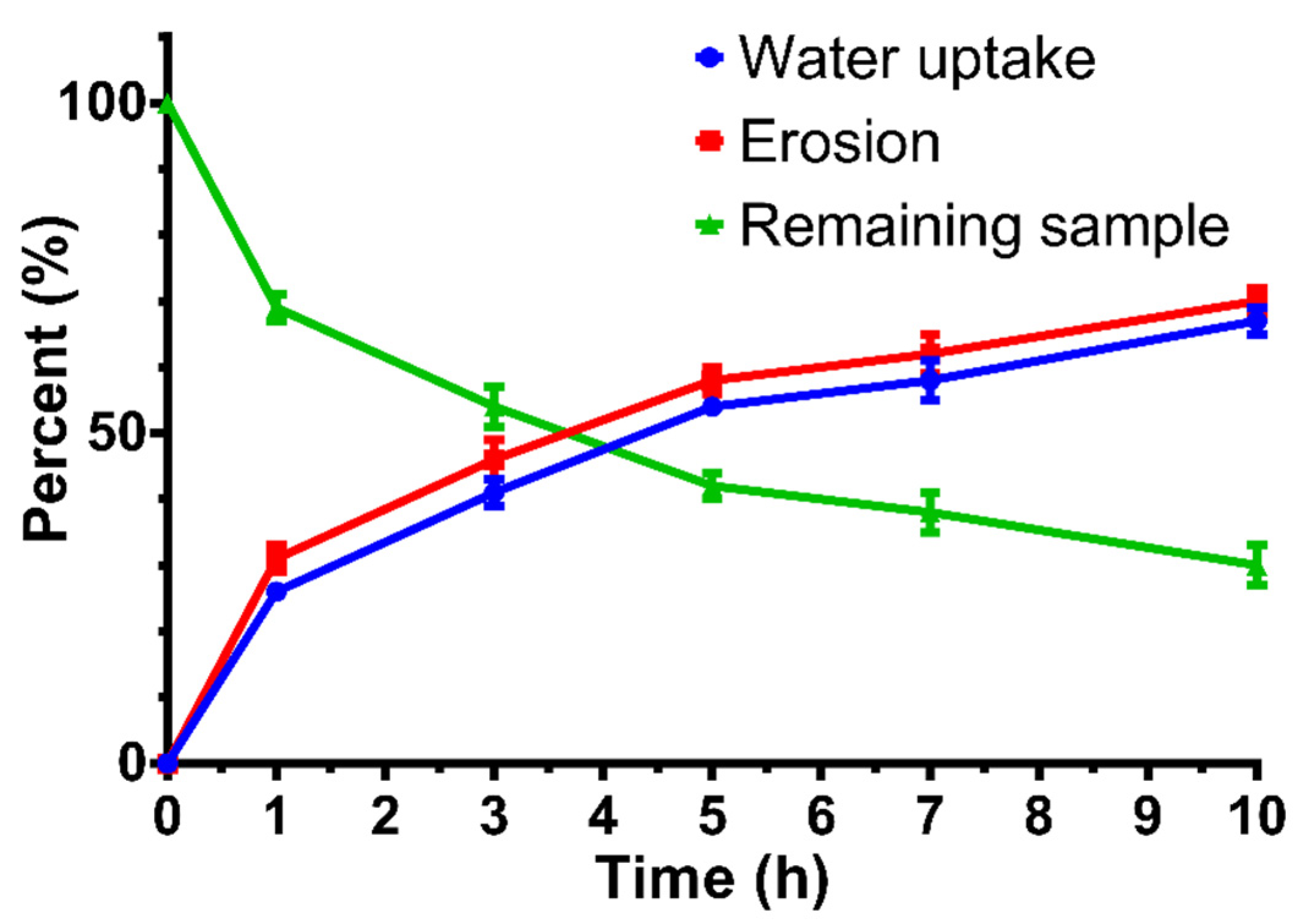
3.7. Water Uptake and Matrix Erosion Studies
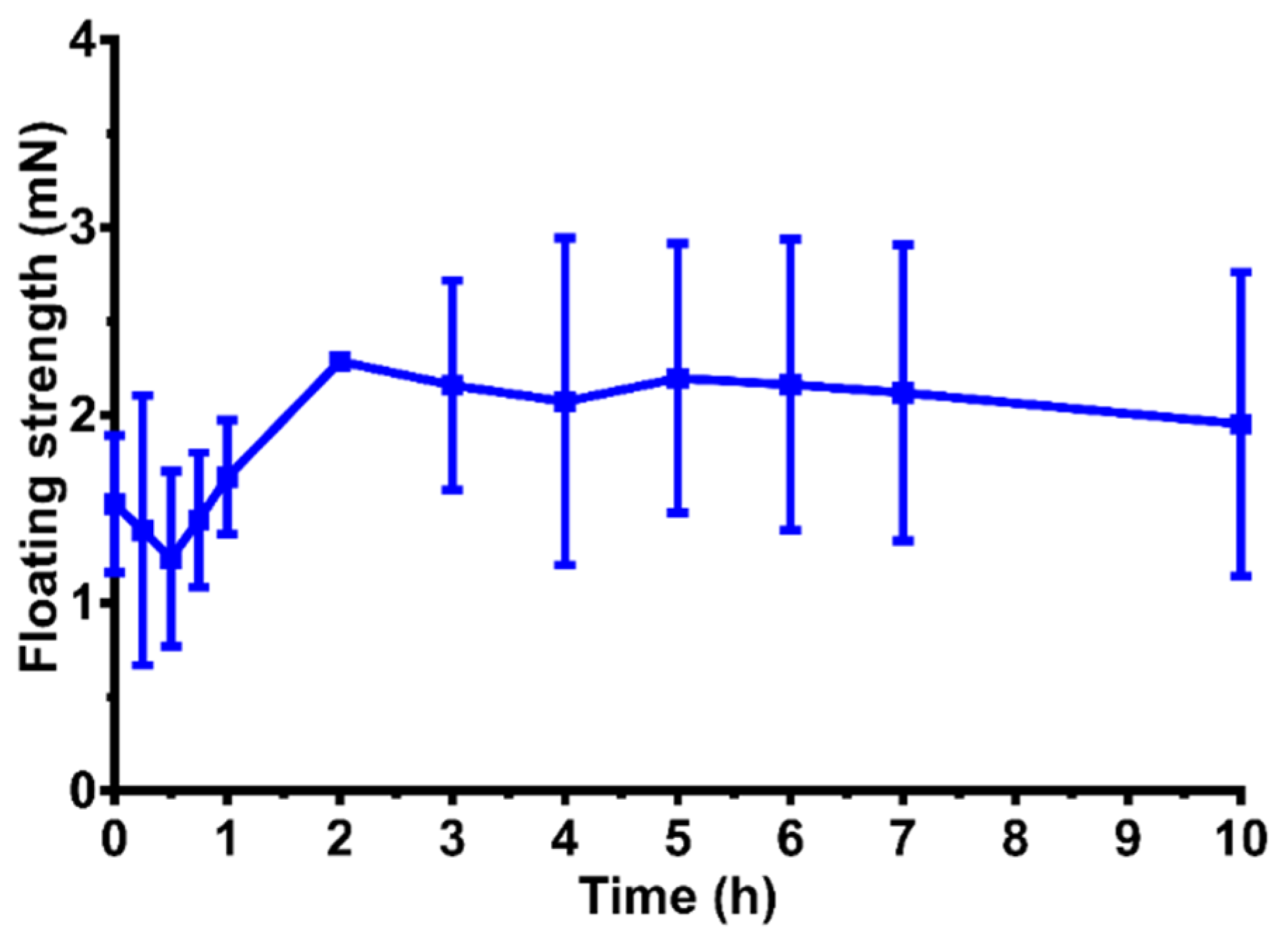
3.8. Floating Strength Determination
3.9. Dissolution Test after Long-Term and Accelerated Storage Conditions
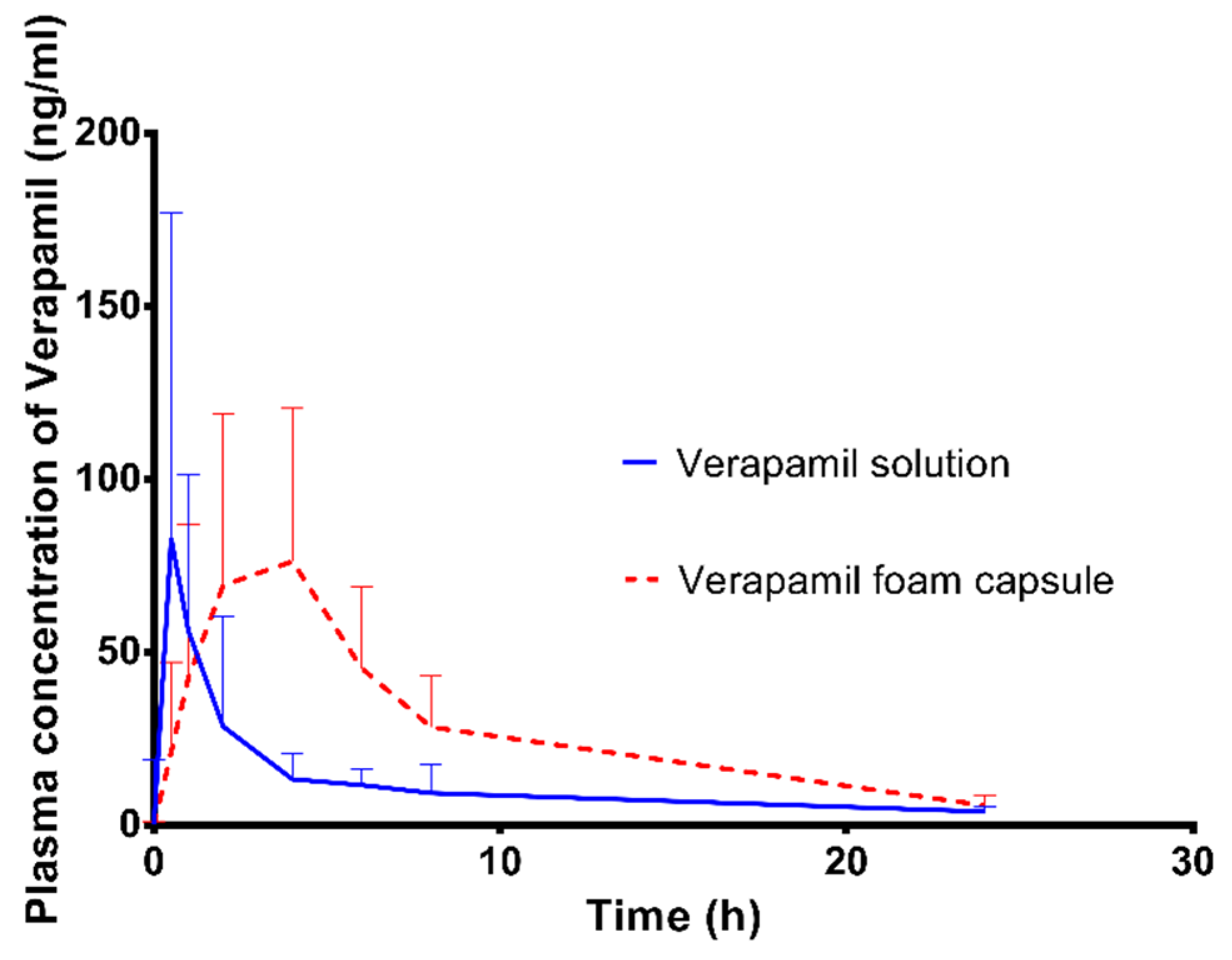
3.10. In Vivo Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tewabe, A.; Abate, A.; Tamrie, M.; Seyfu, A.; Abdela, S.E. Targeted Drug Delivery—From Magic Bullet to Nanomedicine: Principles, Challenges, and Future Perspectives. J. Multidiscip. Healthc. 2021, 14, 1711–1724. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, J.; Thapa, P.; Maharjan, R.; Jeong, S.H. Current State and Future Perspectives on Gastroretentive Drug Delivery Systems. Pharmaceutics 2019, 11, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasvári, G.; Haimhoffer, Á.; Horváth, L.; Budai, I.; Trencsényi, G.; Béresová, M.; Dobó-Nagy, C.; Váradi, J.; Bácskay, I.; Ujhelyi, Z.; et al. Development and Characterisation of Gastroretentive Solid Dosage Form Based on Melt Foaming. AAPS PharmSciTech 2019, 20, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Dalavi, V.V.; Patil, J.S. Gastroretentive drug delivery system of an antiretroviral agent. Int. J. PharmTech Res. 2009, 1, 1678–1684. [Google Scholar]
- Haimhoffer, Á.; Fenyvesi, F.; Lekli, I.; Béresová, M.; Bak, I.; Czagány, M.; Vasvári, G.; Bácskay, I.; Tóth, J.; Budai, I. Preparation of Acyclovir-Containing Solid Foam by Ultrasonic Batch Technology. Pharmaceutics 2021, 13, 1571. [Google Scholar] [CrossRef]
- Singh, B.; Garg, B.; Chaturvedi, S.C.; Arora, S.; Mandsaurwale, R.; Kapil, R.; Singh, B. Formulation development of gastroretentive tablets of lamivudine using the floating-bioadhesive potential of optimized polymer blends. J. Pharm. Pharmacol. 2012, 64, 654–669. [Google Scholar] [CrossRef]
- Porwal, A.; Dwivedi, H.; Pathak, K. Decades of research in drug targeting using gastroretentive drug delivery systems for antihypertensive therapy. Braz. J. Pharm. Sci. 2017, 53, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Elkheshen, S.A.; Yassin, A.E.B.; Alsuwayeh, S.; Alkhaled, F.A. In vitro and in vivo evaluation of floating controlled release dosage forms of verapamil hydrochloride. Pharm. Ind. 2004, 66, 1364–1372. [Google Scholar]
- Sawicki, W. Pharmacokinetics of verapamil and norverapamil from controlled release floating pellets in humans. Eur. J. Pharm. Biopharm. 2002, 53, 29–35. [Google Scholar] [CrossRef]
- Lopes, C.M.; Bettencourt, C.; Rossi, A.; Buttini, F.; Barata, P. Overview on gastroretentive drug delivery systems for improving drug bioavailability. Int. J. Pharm. 2016, 510, 144–158. [Google Scholar] [CrossRef]
- Louis, M.M.; Badawy, A.A.; Nessem, D.I.; Nevine, S.A.E. Drotaverine hydrochloride gastroretentive floating mini-tablets: Formulation, in-vitro and in-vivo evaluation. J. Drug Deliv. Sci. Technol. 2020, 57, 101733–101741. [Google Scholar] [CrossRef]
- Raval, J.A.; Patel, M.M. Formulation and characterization of gastroretentive discs containing famotidine. Braz. Arch. Biol. Technol. 2011, 54, 293–300. [Google Scholar] [CrossRef]
- Rouge, N.; Buri, P.; Doelker, E. Drug absorption sites in the gastrointestinal tract and dosage forms for site-specific delivery. Int. J. Pharm. 1996, 136, 117–139. [Google Scholar] [CrossRef]
- Yassin, A.E.B. Chitosan Beads as a New Gastroretentive System of Verapamil. Sci. Pharm. 2006, 74, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Bhukya, N.; Dhurke, R. Novel gastroretentive mucoadhesive pulsatile tablet for Verapamil hydrochloride. J. Chem. Pharm. Res. 2016, 8, 1190–1198. [Google Scholar]
- Mali, K.K.; Dhawale, S.C. Design and optimization of modified tamarind gum-based floating-bioadhesive tablets of verapamil hydrochloride. Asian J. Pharm. 2016, 10, 239–250. [Google Scholar]
- Klausner, E.A.; Lavy, E.; Friedman, M.; Hoffman, A. Expandable gastroretentive dosage forms. J. Control. Release 2003, 90, 143–162. [Google Scholar] [CrossRef]
- Davis, S.S.; Stockwell, A.F.; Taylor, M.J.; Hardy, J.G.; Whalley, D.R.; Wilson, C.G.; Bechgaard, H.; Christensen, F.N. The effect of density on the gastric emptying of single- and multiple-unit dosage forms. Pharm. Res. 1986, 3, 208–213. [Google Scholar] [CrossRef]
- Streubel, A.; Siepmann, J.; Bodmeier, R. Floating microparticles based on low density foam powder. Int. J. Pharm. 2002, 241, 279–292. [Google Scholar] [CrossRef]
- Shinde, S.; Zadbuke, N.; Padalkar, A.; Shahi, S. Formulation developement and evaluation of floating matrix tablet of Verapamil HCl. Asian J. Pharm. 2013, 7, 27. [Google Scholar] [CrossRef]
- Patel, A.; Modasiya, M.; Shah, D.; Patel, V. Development and in vivo floating behavior of verapamil HCL intragastric floating tablets. AAPS PharmSciTech 2009, 10, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousry, C.; Amin, M.M.; Elshafeey, A.H.; El Gazayerly, O.N. Ultrahigh verapamil-loaded controlled release polymeric beads using superamphiphobic substrate: D-optimal statistical design, in vitro and in vivo performance. Drug Deliv. 2018, 25, 1448–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chikhalikar, S.S.; Wakade, R.B. Floating drug delivery system—An approach to oral controlled drug delivery. Int. J. PharmTech Res. 2012, 4, 1812–1826. [Google Scholar]
- Haimhoffer, Á.; Vasvári, G.; Trencsényi, G.; Béresová, M.; Budai, I.; Czomba, Z.; Rusznyák, Á.; Váradi, J.; Bácskay, I.; Ujhelyi, Z.; et al. Process Optimization for the Continuous Production of a Gastroretentive Dosage Form Based on Melt Foaming. AAPS PharmSciTech 2021, 22, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Benet, L.Z. Predicting Drug Disposition via Application of BCS: Transport/Absorption/ Elimination Interplay and Development of a Biopharmaceutics Drug Disposition Classification System. Pharm. Res. 2005, 22, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Vogelpoel, H.; Welink, J.; Amidon, G.L.; Junginger, H.E.; Midha, K.K.; Möller, H.; Olling, M.; Shah, V.P.; Barends, D.M. Biowaiver monographs for immediate release solid oral dosage forms based on biopharmaceutics classification system (BCS) literature data: Verapamil hydrochloride, propranolol hydrochloride, and atenolol. J. Pharm. Sci. 2004, 93, 1945–1956. [Google Scholar] [CrossRef] [PubMed]
- Kirsten, R.; Nelson, K.; Kirsten, D.; Heintz, B. Clinical Pharmacokinetics of Vasodilators. Clin. Pharmacokinet. 1998, 34, 457–482. [Google Scholar] [CrossRef]
- Stegemann, S.; Bornem, C. Hard Gelatin Capsules Today—And Tomorrow; Capsugel Library: Greenwood, SC, USA, 2002; pp. 1–23. [Google Scholar]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm.-Drug Res. 2010, 67, 217–223. [Google Scholar]
- Samaha, D.; Shehayeb, R.; Kyriacos, S. Modeling and comparison of dissolution profiles of diltiazem modified-release formulations. Dissolution Technol. 2009, 16, 41–46. [Google Scholar] [CrossRef]
- Simons, F.J.; Wagner, K.G. Modeling, design and manufacture of innovative floating gastroretentive drug delivery systems based on hot-melt extruded tubes. Eur. J. Pharm. Biopharm. 2019, 137, 196–208. [Google Scholar] [CrossRef]
- Chytil, L.; Štrauch, B.; Cvačka, J.; Marešová, V.; Widimský, J.; Holaj, R.; Slanař, O. Determination of doxazosin and verapamil in human serum by fast LC–MS/MS: Application to document non-compliance of patients. J. Chromatogr. B 2010, 878, 3167–3173. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V. Das Quantitation and Stability of Verapamil Hydrochloride using High-Performance Liquid Chromatography. Drug Dev. Ind. Pharm. 1985, 11, 1497–1506. [Google Scholar] [CrossRef]
- Gattani, S.G.; Londhe, S.; Chalikwar, S.S.; Amrutkar, J.R. Formulation and evaluation of verapamil hydrochloride transmucosal drug delivery system. Eur. J. Parenter. Pharm. Sci. 2010, 15, 125–129. [Google Scholar]
- Lunio, R.; Sawicki, W.; Skoczen, P.; Walentynowicz, O.; Kubasik-Juraniec, J. Compressibility of Gastroretentive Pellets Coated with Eudragit NE Using a Single-Stroke and a Rotary Tablet Press. Pharm. Dev. Technol. 2008, 13, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Binks, B.P.; Vishal, B. Particle-stabilized oil foams. Adv. Colloid Interface Sci. 2021, 291, 102404. [Google Scholar] [CrossRef]
- Fameau, A.-L.; Fujii, S. Stimuli-responsive liquid foams: From design to applications. Curr. Opin. Colloid Interface Sci. 2020, 50, 101380–101398. [Google Scholar] [CrossRef]
- Murakami, R.; Bismarck, A. Particle-Stabilized Materials: Dry Oils and (Polymerized) Non-Aqueous Foams. Adv. Funct. Mater. 2010, 20, 732–737. [Google Scholar] [CrossRef]
- Khan, A.D.; Meenakshi, B. Formulation and Evaluation of Floating beads of Verapamil hydrochloride. Int. J. PharmTech Res. 2011, 3, 1537–1546. [Google Scholar]
- FDA. Guidance for Industry; Process Validation: General Principles and Practices. Available online: https://www.fda.gov/files/drugs/published/Process-Validation--General-Principles-and-Practices.pdf (accessed on 5 December 2021).
- Narang, A.S.; Sheverev, V.A.; Stepaniuk, V.; Badawy, S.; Stevens, T.; Macias, K.; Wolf, A.; Pandey, P.; Bindra, D.; Varia, S. Real-Time Assessment of Granule Densification in High Shear Wet Granulation and Application to Scale-up of a Placebo and a Brivanib Alaninate Formulation. J. Pharm. Sci. 2015, 104, 1019–1034. [Google Scholar] [CrossRef]
- Qiu, Y.; Duan, J.Z. In Vitro/In Vivo Correlations. In Developing Solid Oral Dosage Forms; Elsevier: Amsterdam, The Netherlands, 2017; pp. 415–452. ISBN 9780128024478. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition 1 | PEG 4000 | Stearic Acid | Verapamil-HCl |
|---|---|---|---|
| V1 | 75.0% | 10.0% | 15.0% |
| V2 | 72.5% | 12.5% | 15.0% |
| V3 | 70.0% | 15.0% | 15.0% |
| Model | Equations [29,30] | Graph | |
|---|---|---|---|
| Zero-order | (2) | The graph of the drug-dissolved fraction vs. time is linear. | |
| First-order | (3) | Linear graph of the released amount of drug (expressed by decimal logarithm) vs. time. | |
| Korsmeyer-Peppas model | (4) | Straight line graph of released drug vs. the square root of time. | |
| Correlation Coefficient (R2) | |||
|---|---|---|---|
| Composition | Zero 1 | First 1 | Korsmeyer–Peppas 1 |
| V1 | 0.7467 | 0.9740 | 0.9371 |
| V2 | 0.7572 | 0.9646 | 0.9589 |
| V3 | 0.7511 | 0.9881 | 0.9619 |
| V2 Composition 1 | |||
|---|---|---|---|
| Validation Properties | Batch I | Batch II | Batch III |
| Average weight of batch ± SD (mg) | 885.9 ± 24.8 | 898.8 ± 36.5 | 894.5 ± 18.15 |
| Maximum absolute deviation from average weight (all batches %) 3 | 5.0 | 8.8 | 5.5 |
| Average API content of batch ± SD (mg) | 106.9 ± 4.0 | 117.2 ± 8.5 | 122.1 ± 5.2 |
| Maximum absolute deviation from average API content of all batches (%) 3 | 11.9 | 10.7 | 11.4 |
| Difference factor 2 | 4.19 | 4.95 | 2.60 |
| Similarity factor 2 | 99.98 | 99.98 | 99.99 |
| Composition | Verapamil Solution | Verapamil Foam Capsule | Analysis of Variance 1 |
|---|---|---|---|
| Cmax (ng/mL) | 90.24 ± 89.02 | 101.20 ± 46.15 | NS |
| Tmax (h) | 1.00 ± 0.54 | 2.67 ± 1.51 | IS |
| AUC (0–24 h) | 254.5 ± 175.4 | 682.4 ± 297.1 | IS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haimhoffer, Á.; Vasvári, G.; Budai, I.; Béresová, M.; Deák, Á.; Németh, N.; Váradi, J.; Sinka, D.; Bácskay, I.; Vecsernyés, M.; et al. In Vitro and In Vivo Studies of a Verapamil-Containing Gastroretentive Solid Foam Capsule. Pharmaceutics 2022, 14, 350. https://doi.org/10.3390/pharmaceutics14020350
Haimhoffer Á, Vasvári G, Budai I, Béresová M, Deák Á, Németh N, Váradi J, Sinka D, Bácskay I, Vecsernyés M, et al. In Vitro and In Vivo Studies of a Verapamil-Containing Gastroretentive Solid Foam Capsule. Pharmaceutics. 2022; 14(2):350. https://doi.org/10.3390/pharmaceutics14020350
Chicago/Turabian StyleHaimhoffer, Ádám, Gábor Vasvári, István Budai, Monika Béresová, Ádám Deák, Norbert Németh, Judit Váradi, Dávid Sinka, Ildikó Bácskay, Miklós Vecsernyés, and et al. 2022. "In Vitro and In Vivo Studies of a Verapamil-Containing Gastroretentive Solid Foam Capsule" Pharmaceutics 14, no. 2: 350. https://doi.org/10.3390/pharmaceutics14020350