
In Vitro Methodologies for Evaluating Colon-Targeted Pharmaceutical Products and Industry Perspectives for Their Applications

 ,
,  , and
, and 
Abstract
:1. Introduction
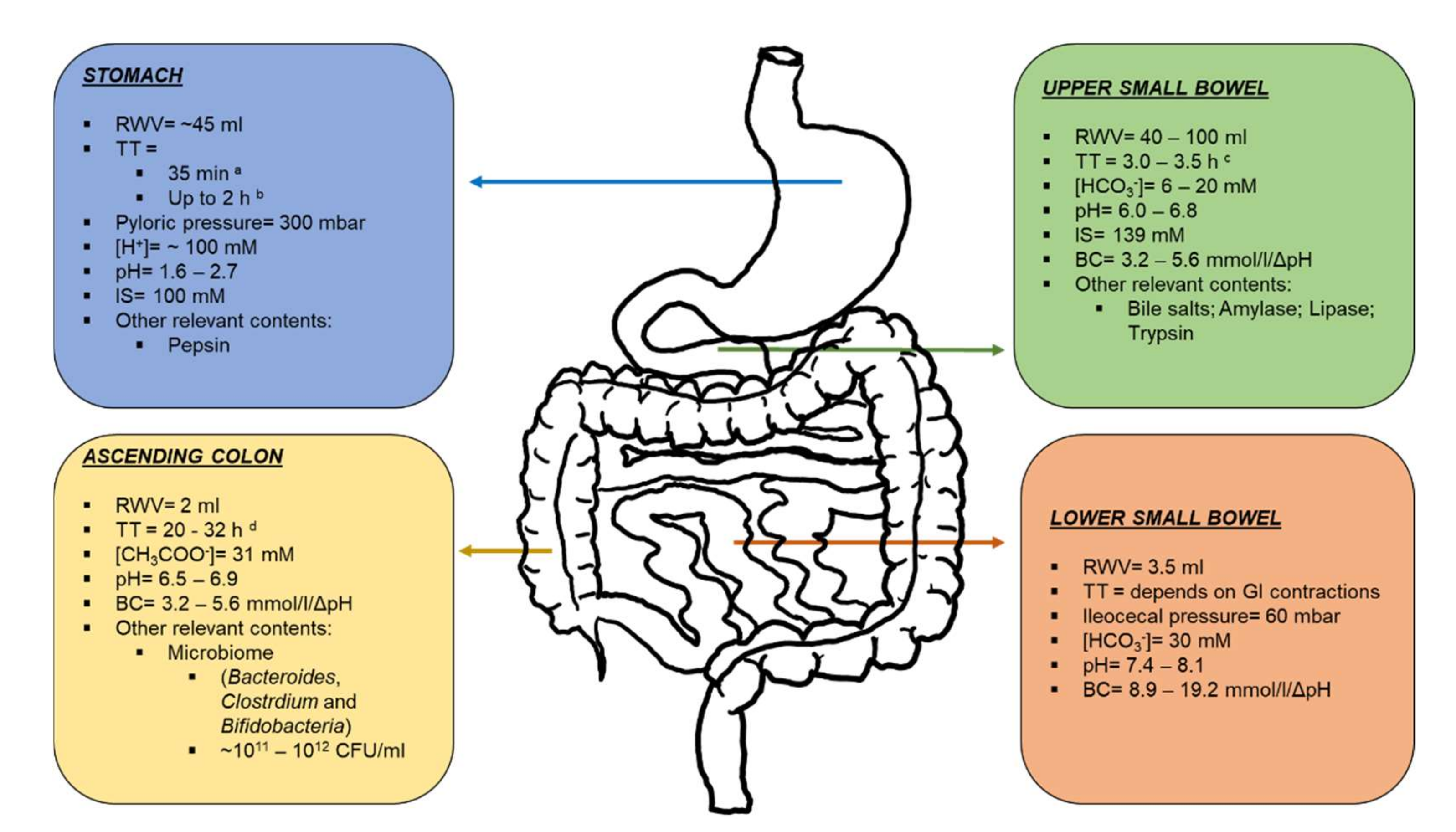
2. Gastrointestinal Physiology: “Bypassing the Upper Gastrointestinal Tract and Targeting Distal Release”
2.1. Stomach and Upper Small Intestine
2.2. Ileum, Caecum and Colon
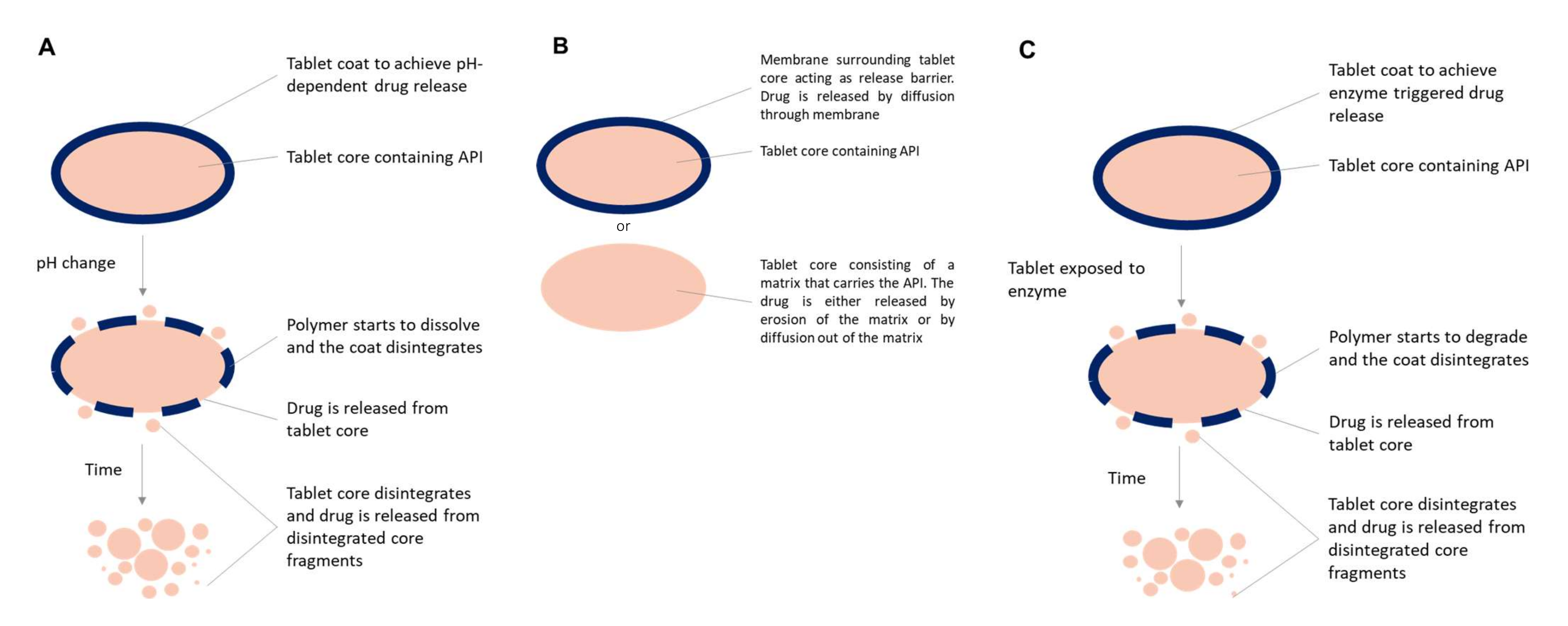
3. Formulation Approaches
3.1. pH-Dependent Systems
3.2. Time-Controlled Systems
3.3. Enzymatic Triggered Release Systems
4. In Vitro Methodologies for pH-Dependent Colonic Delivery Products
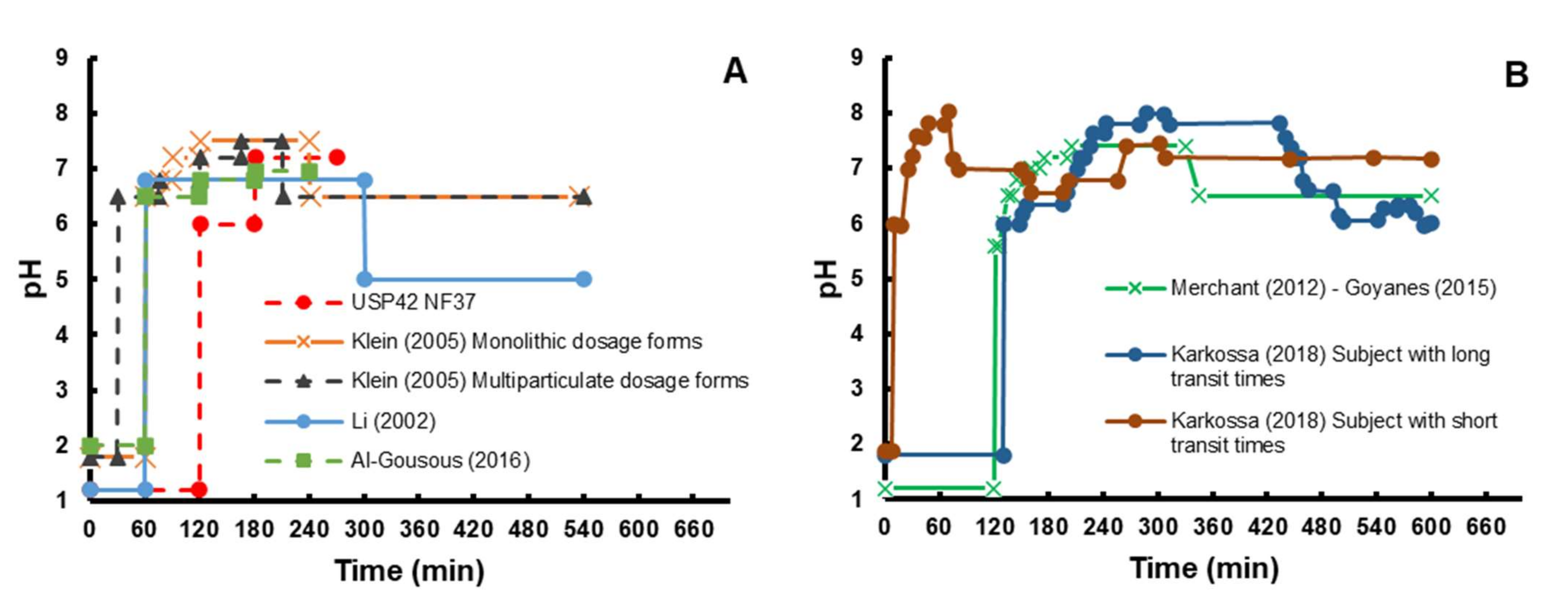
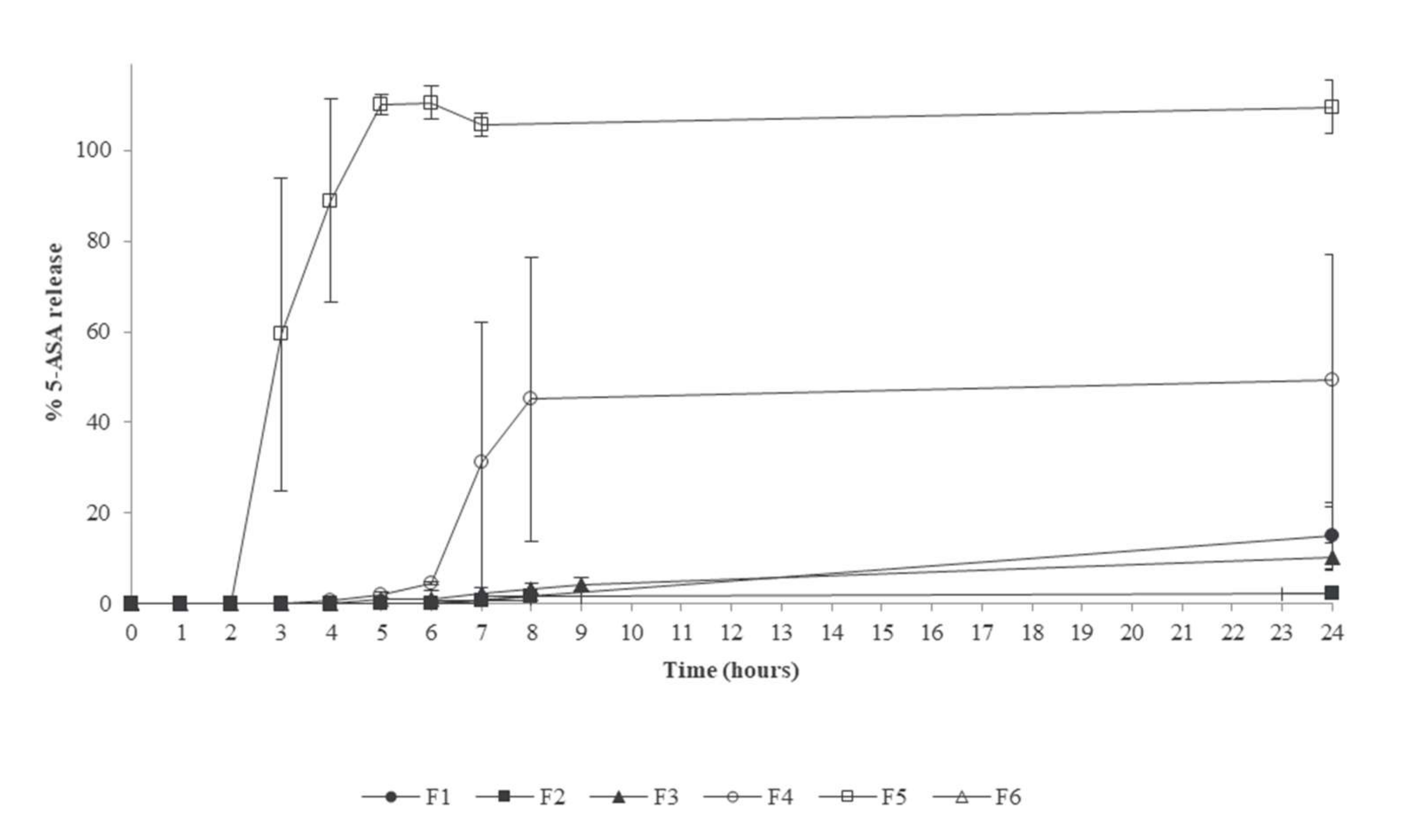
4.1. Pharmacopeial-Based Methods
4.2. Methods Based on Biorelevant Buffers
4.2.1. Bicarbonate Buffer
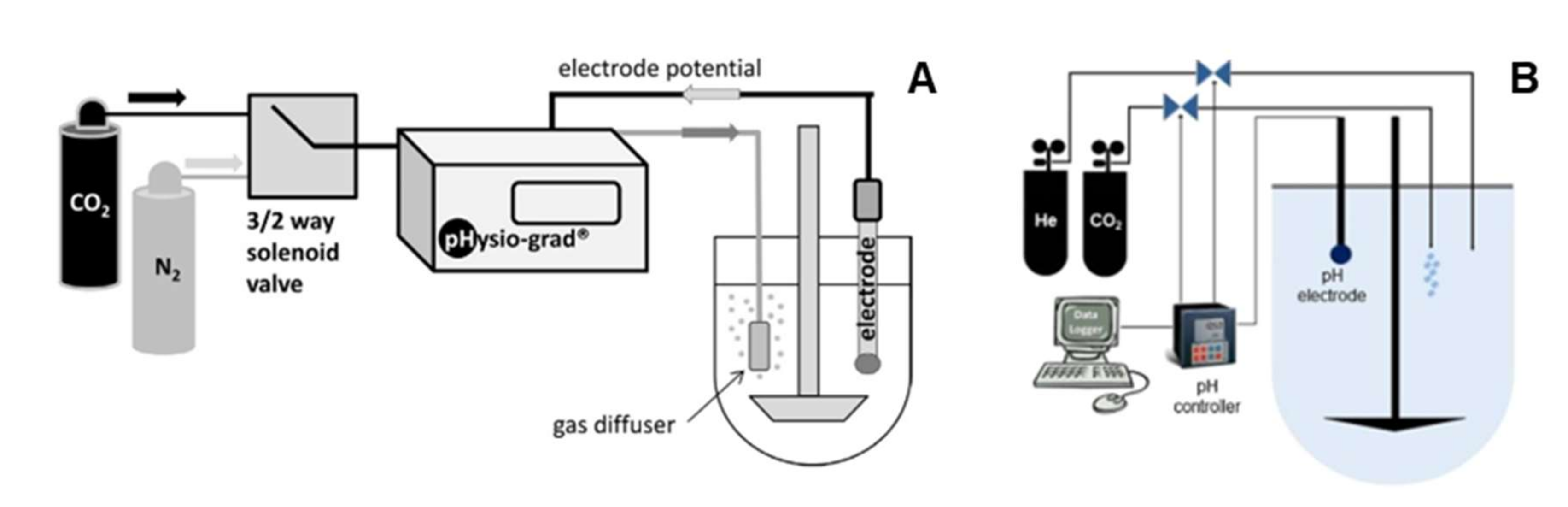
4.2.2. Manual Methods to Stabilize Bicarbonate pH
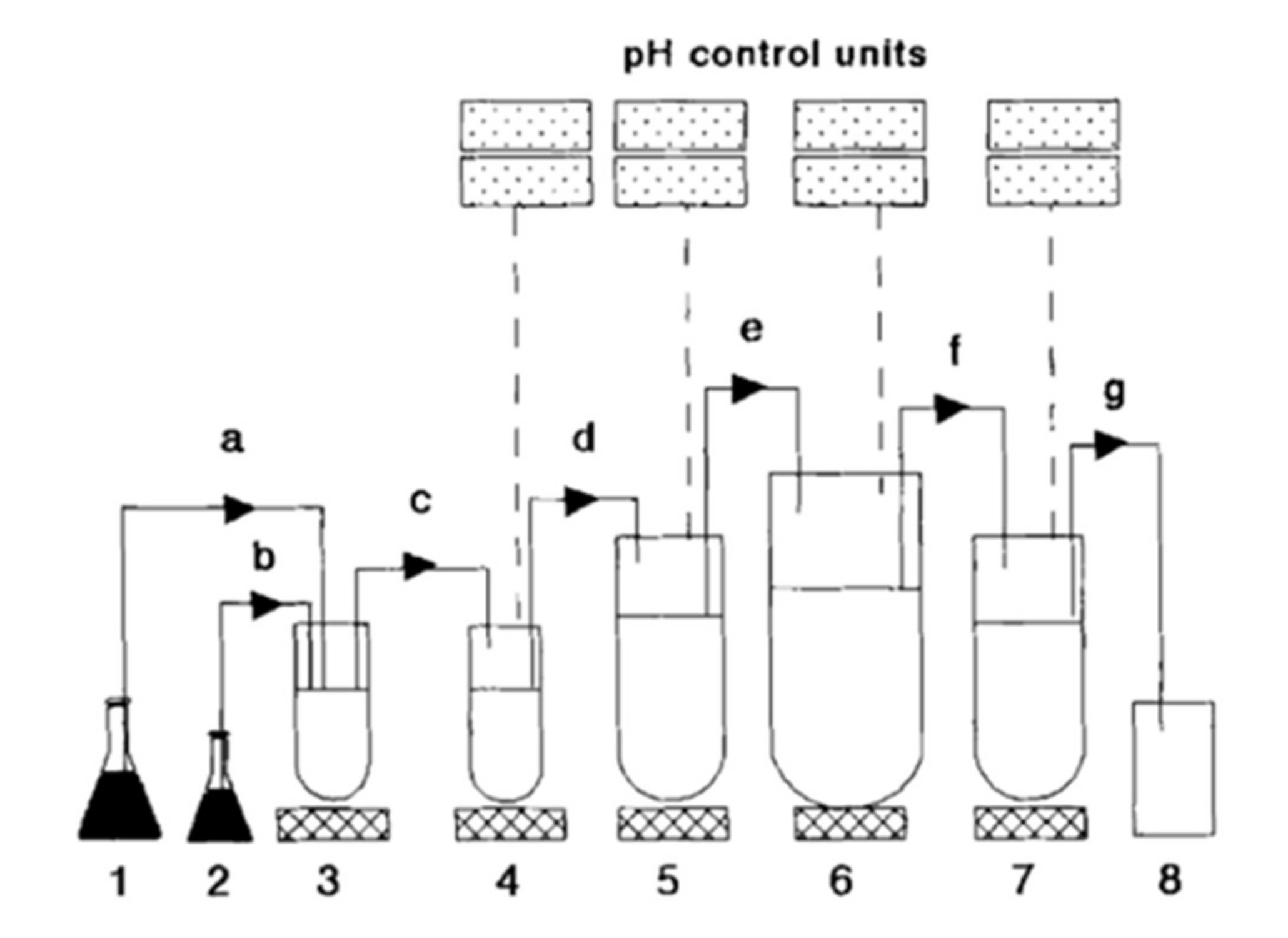
4.2.3. Automated Methods to Stabilize Bicarbonate pH
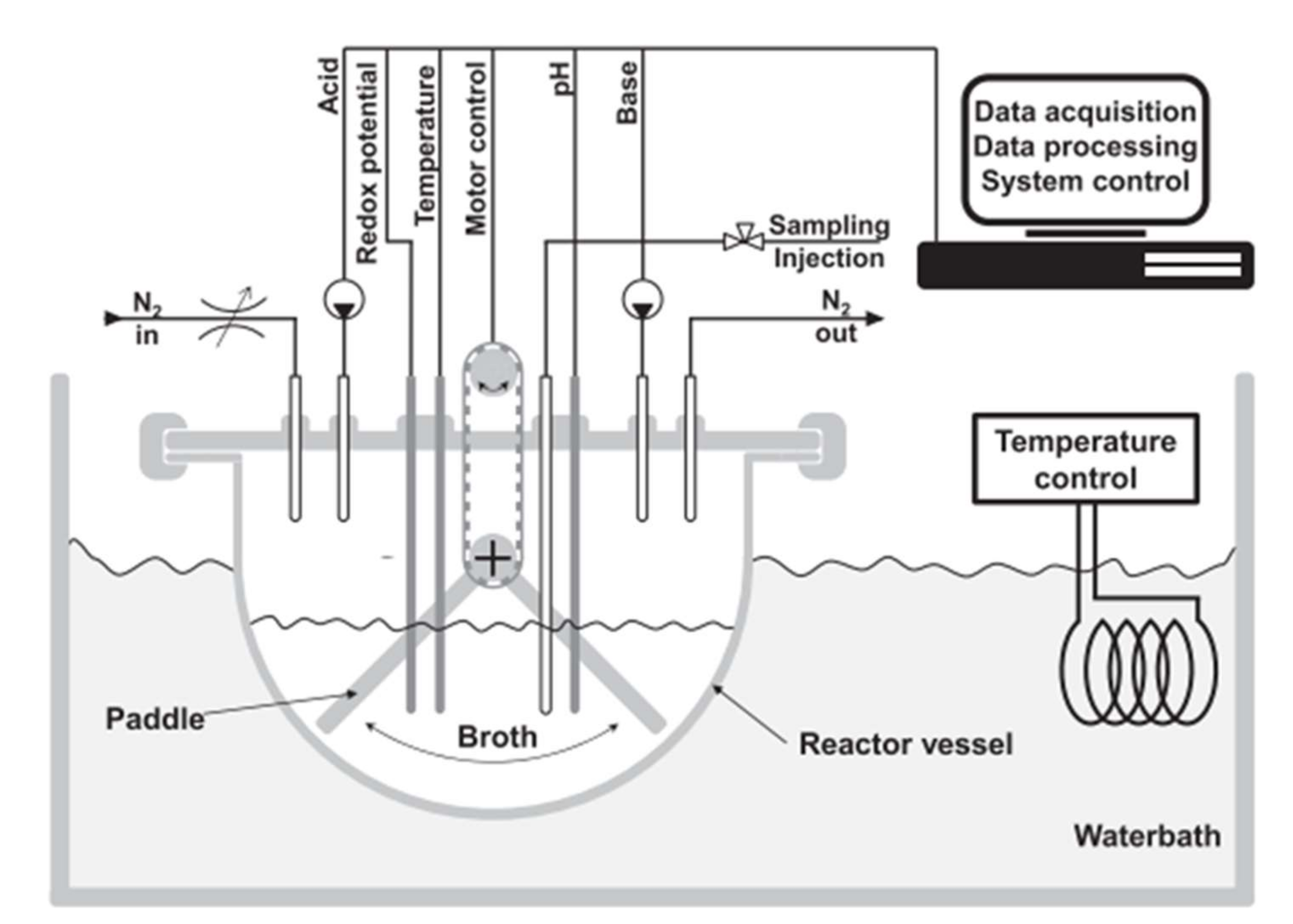
4.2.4. Bicarbonate-Free Bio-Relevant Methods
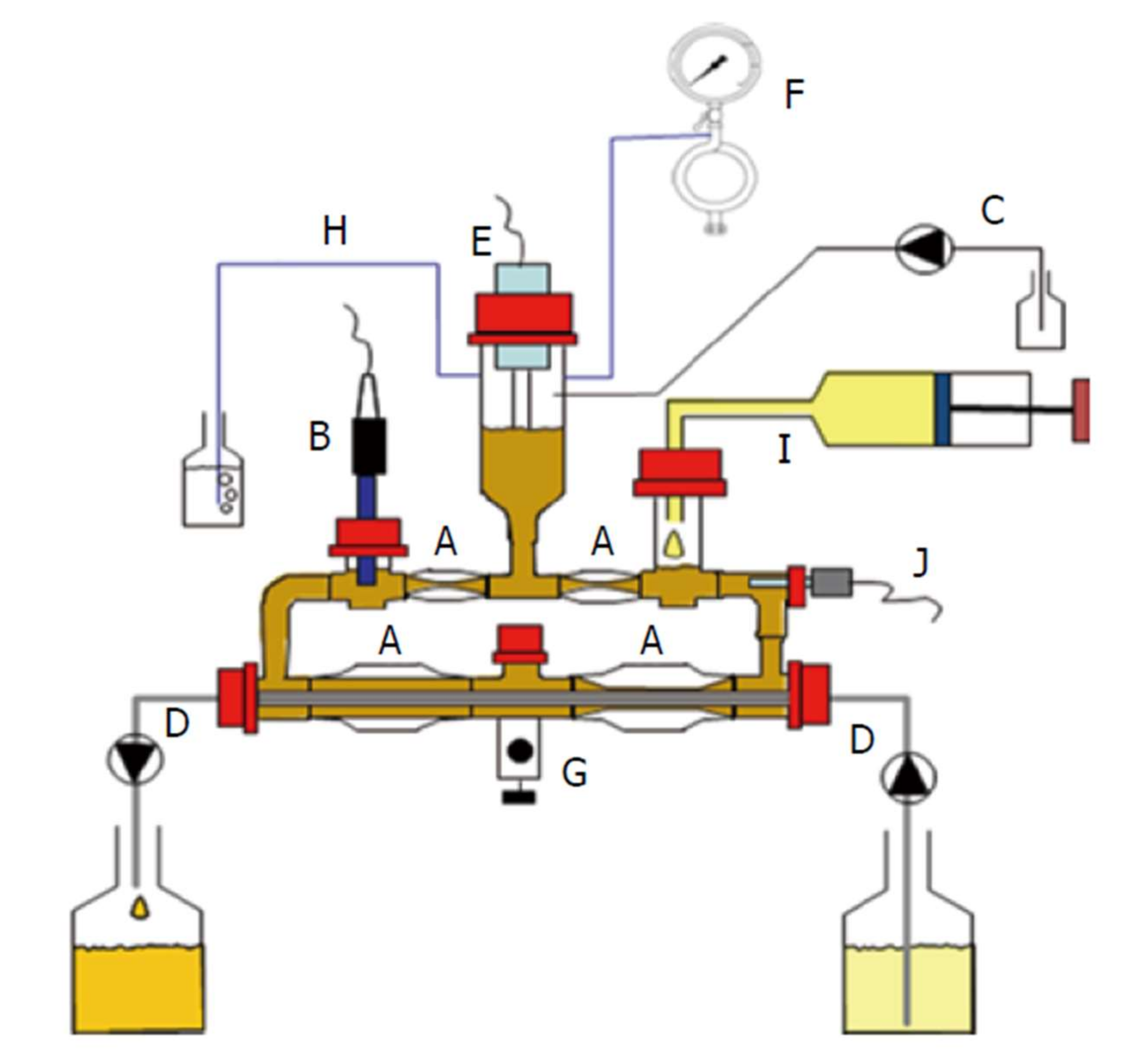
5. In Vitro Methodologies for Time-Controlled Release Products
6. In Vitro Methodologies for Enzymatic-Triggered Drug-Release Systems
7. Future Perspectives on In Vitro Testing for Locally-Acting Colon-Targeting Drug Products
7.1. Methods for Formulation Development Purposes
7.1.1. pH-Dependent Systems
7.1.2. Time-Controlled Systems
7.1.3. Enzymatic Triggered Systems
7.2. Methods for Quality Control Purposes
7.2.1. pH-Dependent Systems
7.2.2. Time-Controlled Systems
7.2.3. Enzymatic Triggered Systems
8. Considerations for In Vivo Methods
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- CDER/FDA. Guidance for Industry Bioavailability and Bioequivalence Studies for Orally Administered Drug Products—General Guidance for Industry Bioavailability and Bioequivalence. FDA Guid. 2002, 1–24. [Google Scholar]
- Lemmens, G.; Van Camp, A.; Kourula, S.; Vanuytsel, T.; Augustijns, P. Drug Disposition in the Lower Gastrointestinal Tract: Targeting and Monitoring. Pharmaceutics 2021, 13, 161. [Google Scholar] [CrossRef]
- Vinarov, Z.; Abdallah, M.; Agundez, J.A.G.; Allegaert, K.; Basit, A.W.; Braeckmans, M.; Ceulemans, J.; Corsetti, M.; Griffin, B.T.; Grimm, M.; et al. Impact of Gastrointestinal Tract Variability on Oral Drug Absorption and Pharmacokinetics: An UNGAP Review. Eur. J. Pharm. Sci. 2021, 162, 105812. [Google Scholar] [CrossRef] [PubMed]
- Koziolek, M.; Grimm, M.; Becker, D.; Iordanov, V.; Zou, H.; Shimizu, J.; Wanke, C.; Garbacz, G.; Weitschies, W. Investigation of PH and Temperature Profiles in the GI Tract of Fasted Human Subjects Using the Intellicap® System. J. Pharm. Sci. 2015, 104, 2855–2863. [Google Scholar] [CrossRef] [PubMed]
- Dressman, J.B.; Berardi, R.R.; Dermentzoglou, L.C.; Russell, T.L.; Schmaltz, S.P.; Barnett, J.L.; Jarvenpaa, K.M. Upper Gastrointestinal (GI) PH in Young, Healthy Men and Women. Pharm. Res. Off. J. Am. Assoc. Pharm. Sci. 1990, 7, 756–761. [Google Scholar] [CrossRef]
- Karamanolis, G.; Theofanidou, I.; Yiasemidou, M.; Giannoulis, E.; Triantafyllou, K.; Ladas, S.D. A Glass of Water Immediately Increases Gastric PH in Healthy Subjects. Dig. Dis. Sci. 2008, 53, 3128–3132. [Google Scholar] [CrossRef]
- Lindahl, A.; Ungell, A.L.; Knutson, L.; Lennernäs, H. Characterization of Fluids from the Stomach and Proximal Jejunum in Men and Women. Pharm. Res. 1997, 14, 497–502. [Google Scholar] [CrossRef]
- Kalantzi, L.; Goumas, K.; Kalioras, V.; Abrahamsson, B.; Dressman, J.B.; Reppas, C. Characterization of the Human Upper Gastrointestinal Contents under Conditions Simulating Bioavailability/Bioequivalence Studies. Pharm. Res. 2006, 23, 165–176. [Google Scholar] [CrossRef]
- Grimm, M.; Koziolek, M.; Kühn, J.P.; Weitschies, W. Interindividual and Intraindividual Variability of Fasted State Gastric Fluid Volume and Gastric Emptying of Water. Eur. J. Pharm. Biopharm. 2018, 127, 309–317. [Google Scholar] [CrossRef]
- Mudie, D.M.; Murray, K.; Hoad, C.L.; Pritchard, S.E.; Garnett, M.C.; Amidon, G.L.; Gowland, P.A.; Spiller, R.C.; Amidon, G.E.; Marciani, L. Quantification of Gastrointestinal Liquid Volumes and Distribution Following a 240 ML Dose of Water in the Fasted State. Mol. Pharm. 2014, 11, 3039–3047. [Google Scholar] [CrossRef]
- Schiller, C.; Fröhlich, C.P.; Giessmann, T.; Siegmund, W.; Mönnikes, H.; Hosten, N.; Weitschies, W. Intestinal Fluid Volumes and Transit of Dosage Forms as Assessed by Magnetic Resonance Imaging. Aliment. Pharmacol. Ther. 2005, 22, 971–979. [Google Scholar] [CrossRef]
- Koziolek, M.; Grimm, M.; Schneider, F.; Jedamzik, P.; Sager, M.; Kühn, J.P.; Siegmund, W.; Weitschies, W. Navigating the Human Gastrointestinal Tract for Oral Drug Delivery: Uncharted Waters and New Frontiers. Adv. Drug Deliv. Rev. 2016, 101, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Abuhelwa, A.Y.; Foster, D.J.R.; Upton, R.N. A Quantitative Review and Meta-Models of the Variability and Factors Affecting Oral Drug Absorption—Part II: Gastrointestinal Transit Time. AAPS J. 2016, 18, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K. Biopharmaceutics Modeling and Simulations: Theory, Practice, Methods, and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar] [CrossRef]
- Helander, H.F.; Fändriks, L. Surface Area of the Digestive Tract-Revisited. Scand. J. Gastroenterol. 2014, 49, 681–689. [Google Scholar] [CrossRef] [PubMed]
- McConnell, E.L.; Fadda, H.M.; Basit, A.W. Gut Instincts: Explorations in Intestinal Physiology and Drug Delivery. Int. J. Pharm. 2008, 364, 213–226. [Google Scholar] [CrossRef]
- Davis, S.S.; Hardy, J.G.; Fara, J.W. Transit of Pharmaceutical Dosage Forms through the Small Intestine. Gut 1986, 27, 886–892. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Siva, S.; Wo, J.M.; Fadda, H.M. Assessment of Small Intestinal Transit Times in Ulcerative Colitis and Crohn’s Disease Patients with Different Disease Activity Using Video Capsule Endoscopy. AAPS PharmSciTech 2017, 18, 404–409. [Google Scholar] [CrossRef]
- Weitschies, W.; Kosch, O.; Mönnikes, H.; Trahms, L. Magnetic Marker Monitoring: An Application of Biomagnetic Measurement Instrumentation and Principles for the Determination of the Gastrointestinal Behavior of Magnetically Marked Solid Dosage Forms. Adv. Drug Deliv. Rev. 2005, 57, 1210–1222. [Google Scholar] [CrossRef]
- Ibekwe, V.C.; Fadda, H.M.; McConnell, E.L.; Khela, M.K.; Evans, D.F.; Basit, A.W. Interplay between Intestinal PH, Transit Time and Feed Status on the in Vivo Performance of PH Responsive Ileo-Colonic Release Systems. Pharm. Res. 2008, 25, 1828–1835. [Google Scholar] [CrossRef]
- Amidon, G.L.; Lee, P.I.; Topp, E.M. Transport Processes in Pharmaceutical Systems; CRC Press: Boca Raton, FL, USA, 1999. [Google Scholar] [CrossRef]
- Repishti, M.; Hogan, D.L.; Pratha, V.; Davydova, L.; Donowitz, M.; Tse, C.M.; Isenberg, J.I. Human Duodenal Mucosal Brush Border Na+/H+ Exchangers NHE2 and NHE3 Alter Net Bicarbonate Movement. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, 159–163. [Google Scholar] [CrossRef] [Green Version]
- Banwell, J.G.; Gorbach, S.L.; Pierce, N.F.; Mitra, R.; Mondal, A. Acute Undifferentiated Human Diarrhea in the Tropics. II. Alterations in Intestinal Fluid and Electrolyte Movements. J. Clin. Invest. 1971, 50, 890–900. [Google Scholar] [CrossRef] [Green Version]
- McGee, L.C.; Hastings, A.B. The Carbon Dioxide Tension and Acid-Base Balance of Jejunal Secretions in Man. J. Biol. Chem. 1942, 142, 893–904. [Google Scholar] [CrossRef]
- Fadda, H.M.; Sousa, T.; Carlsson, A.S.; Abrahamsson, B.; Williams, J.G.; Kumar, D.; Basit, A.W. Drug Solubility in Luminal Fluids from Different Regions of the Small and Large Intestine of Humans. Mol. Pharm. 2010, 7, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.P.D.L.C.; Oth, M.; Deferme, S.; Lammert, F.; Tack, J.; Dressman, J.; Augustijns, P. Characterization of Fasted-State Human Intestinal Fluids Collected from Duodenum and Jejunum. J. Pharm. Pharmacol. 2010, 58, 1079–1089. [Google Scholar] [CrossRef]
- Al-Gousous, J.; Sun, K.X.; McNamara, D.P.; Hens, B.; Salehi, N.; Langguth, P.; Bermejo, M.; Amidon, G.E.; Amidon, G.L. Mass Transport Analysis of the Enhanced Buffer Capacity of the Bicarbonate-CO2 Buffer in a Phase-Heterogenous System: Physiological and Pharmaceutical Significance. Mol. Pharm. 2018, 15, 5291–5301. [Google Scholar] [CrossRef] [PubMed]
- Dinning, P.G.; Bampton, P.A.; Kennedy, M.L.; Cook, I.J. Relationship between Terminal Ileal Pressure Waves and Propagating Proximal Colonic Pressure Waves. Am. J. Physiol. Gastrointest. Liver Physiol. 1999, 277, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Koziolek, M.; Schneider, F.; Grimm, M.; Mode, C.; Seekamp, A.; Roustom, T.; Siegmund, W.; Weitschies, W. Intragastric PH and Pressure Profiles after Intake of the High-Caloric, High-Fat Meal as Used for Food Effect Studies. J. Control. Release 2015, 220, 71–78. [Google Scholar] [CrossRef]
- Rao, S.S.C.; Kavelock, R.; Beaty, J.; Ackerson, K.; Stumbo, P. Effects of Fat and Carbohydrate Meals on Colonic Motor Response. Gut 2000, 46, 205–211. [Google Scholar] [CrossRef]
- Furukawa, Y.; Cook, I.J.; Panagopoulos, V.; McEvoy, R.D.; Sharp, D.J.; Simula, M. Relationship between Sleep Patterns and Human Colonic Motor Patterns. Gastroenterology 1994, 107, 1372–1381. [Google Scholar] [CrossRef]
- Weitschies, W.; Blume, H.; Mönnikes, H. Magnetic Marker Monitoring: High Resolution Real-Time Tracking of Oral Solid Dosage Forms in the Gastrointestinal Tract. Eur. J. Pharm. Biopharm. 2010, 74, 93–101. [Google Scholar] [CrossRef]
- Murray, K.; Hoad, C.L.; Mudie, D.M.; Wright, J.; Heissam, K.; Abrehart, N.; Pritchard, S.E.; Al Atwah, S.; Gowland, P.A.; Garnett, M.C.; et al. Magnetic Resonance Imaging Quantification of Fasted State Colonic Liquid Pockets in Healthy Humans. Mol. Pharm. 2017, 14, 2629–2638. [Google Scholar] [CrossRef] [Green Version]
- Diakidou, A.; Vertzoni, M.; Goumas, K.; Söderlind, E.; Abrahamsson, B.; Dressman, J.; Reppas, C. Characterization of the Contents of Ascending Colon to Which Drugs Are Exposed after Oral Administration to Healthy Adults. Pharm. Res. 2009, 26, 2141–2151. [Google Scholar] [CrossRef] [PubMed]
- Reppas, C.; Karatza, E.; Goumas, C.; Markopoulos, C.; Vertzoni, M. Characterization of Contents of Distal Ileum and Cecum to Which Drugs/Drug Products Are Exposed during Bioavailability/Bioequivalence Studies in Healthy Adults. Pharm. Res. 2015, 32, 3338–3349. [Google Scholar] [CrossRef] [PubMed]
- Steiger, C.; Phan, N.V.; Sun, H.; Huang, H.W.; Hess, K.; Lopes, A.; Korzenik, J.; Langer, R.; Traverso, G. Controlled Delivery of Bile Acids to the Colon. Clin. Transl. Gastroenterol. 2020, 11, e00229. [Google Scholar] [CrossRef]
- Teruel, A.H.; Gonzalez-Alvarez, I.; Bermejo, M.; Merino, V.; Marcos, M.D.; Sancenon, F.; Gonzalez-Alvarez, M.; Martinez-Mañez, R. New Insights of Oral Colonic Drug Delivery Systems for Inflammatory Bowel Disease Therapy. Int. J. Mol. Sci. 2020, 21, 6502. [Google Scholar] [CrossRef]
- Simon, G.L.; Gorbach, S.L. Intestinal Flora in Health and Disease. Gastroenterology 1984, 86, 174–193. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Microbiology: Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, D.N.; Amand, A.L.S.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-Phylogenetic Characterization of Microbial Community Imbalances in Human Inflammatory Bowel Diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekirov, I.; Russell, S.L.; Caetano, M.; Antunes, L.; Finlay, B.B. Gut Microbiota in Health and Disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [Green Version]
- Abu-Ghazaleh, N.; Chua, W.J.; Gopalan, V. Intestinal Microbiota and Its Association with Colon Cancer and Red/Processed Meat Consumption. J. Gastroenterol. Hepatol. 2021, 36, 75–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadda, H.M. The Route to Palatable Fecal Microbiota Transplantation. AAPS PharmSciTech 2020, 21, 114. [Google Scholar] [CrossRef]
- Mishima, Y.; Sartor, R.B. Manipulating Resident Microbiota to Enhance Regulatory Immune Function to Treat Inflammatory Bowel Diseases. J. Gastroenterol. 2020, 55, 4–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamer, H.M.; de Preter, V.; Windey, K.; Verbeke, K. Functional Analysis of Colonic Bacterial Metabolism: Relevant to Health? Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1–G9. [Google Scholar] [CrossRef] [PubMed]
- Salyers, A.A.; West, S.E.H.; Vercellotti, J.R.; Wilkins, T.D. Fermentation of Mucins and Plant Polysaccharides by Anaerobic Bacteria from the Human Colon. Appl. Environ. Microbiol. 1977, 34, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, M.; Zimmermann-Kogadeeva, M.; Wegmann, R.; Goodman, A.L. Separating Host and Microbiome Contributions to Drug Pharmacokinetics and Toxicity. Science 2019, 363. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Zimmermann-Kogadeeva, M.; Wegmann, R.; Goodman, A.L. Mapping Human Microbiome Drug Metabolism by Gut Bacteria and Their Genes. Nature 2019, 570, 462–467. [Google Scholar] [CrossRef]
- Clarke, G.; Sandhu, K.V.; Griffin, B.T.; Dinan, T.G.; Cryan, J.F.; Hyland, N.P. Gut Reactions: Breaking down Xenobiotic–Microbiome Interactions. Pharmacol. Rev. 2019, 71, 198–224. [Google Scholar] [CrossRef]
- Tuleu, C.; Basit, A.W.; Waddington, W.A.; Ell, P.J.; Newton, J.M. Colonic Delivery of 4-Aminosalicylic Acid Using Amylose-Ethylcellulose-Coated Hydroxypropylmethylcellulose Capsules. Aliment. Pharmacol. Ther. 2002, 16, 1771–1779. [Google Scholar] [CrossRef]
- Siew, L.F.; Man, S.M.; Newton, J.M.; Basit, A.W. Amylose Formulations for Drug Delivery to the Colon: A Comparison of Two Fermentation Models to Assess Colonic Targeting Performance in Vitro. Int. J. Pharm. 2004, 273, 129–134. [Google Scholar] [CrossRef]
- Karrout, Y.; Neut, C.; Siepmann, F.; Wils, D.; Ravaux, P.; Deremaux, L.; Flament, M.P.; Dubreuil, L.; Lemdani, M.; Desreumaux, P.; et al. Enzymatically Degraded Eurylon 6 HP-PG: Ethylcellulose Film Coatings for Colon Targeting in Inflammatory Bowel Disease Patients. J. Pharm. Pharmacol. 2010, 62, 1676–1684. [Google Scholar] [CrossRef] [PubMed]
- Karrout, Y.; Dubuquoy, L.; Piveteau, C.; Siepmann, F.; Moussa, E.; Wils, D.; Beghyn, T.; Neut, C.; Flament, M.P.; Guerin-Deremaux, L.; et al. In Vivo Efficacy of Microbiota-Sensitive Coatings for Colon Targeting: A Promising Tool for IBD Therapy. J. Control. Release 2015, 197, 121–130. [Google Scholar] [CrossRef]
- Amidon, S.; Brown, J.E.; Dave, V.S. Colon-Targeted Oral Drug Delivery Systems: Design Trends and Approaches. AAPS PharmSciTech 2015, 16, 731–741. [Google Scholar] [CrossRef]
- Fallingborg, J.; Christensen, L.A.; Jacobsen, B.A.; Rasmussen, S.N. Very Low Intraluminal Colonic PH in Patients with Active Ulcerative Colitis. Dig. Dis. Sci. 1993, 38, 1989–1993. [Google Scholar] [CrossRef] [PubMed]
- Nugent, S.G.; Kumar, D.; Rampton, D.S.; Evans, D.F. Intestinal Luminal PH in Inflammatory Bowel Disease: Possible Determinants and Implications for Therapy with Aminosalicylates and Other Drugs. Gut 2001, 48, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Hebden, J.M.; Blackshaw, P.E.; Perkins, A.C.; Wilson, C.G.; Spiller, R.C. Limited Exposure of the Healthy Distal Colon to Orally-Dosed Formulation Is Further Exaggerated in Active Left-Sided Ulcerative Colitis. Aliment. Pharmacol. Ther. 2000, 14, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Haase, A.M.; Gregersen, T.; Christensen, L.A.; Agnholt, J.; Dahlerup, J.F.; Schlageter, V.; Krogh, K. Regional Gastrointestinal Transit Times in Severe Ulcerative Colitis. Neurogastroenterol. Motil. 2016, 28, 217–224. [Google Scholar] [CrossRef]
- Ibekwe, V.C.; Liu, F.; Fadda, H.M.; Khela, M.K.; Evans, D.F.; Parsons, G.E.; Basit, A.W. An Investigation into the In Vivo Performance Variability of PH Responsive Polymers for Ileo- Colonic Drug Delivery Using Gamma Scintigraphy in Humans. J. Pharm. Sci. 2006, 95, 2760–2766. [Google Scholar] [CrossRef]
- McConnell, E.L.; Short, M.D.; Basit, A.W. An in Vivo Comparison of Intestinal PH and Bacteria as Physiological Trigger Mechanisms for Colonic Targeting in Man. J. Control. Release 2008, 130, 154–160. [Google Scholar] [CrossRef]
- Baker, D.E. MMX (TM) Mesalamine. Rev. Gastroenterol. Disord. 2006, 6, 146–152. [Google Scholar]
- Liu, F.; Moreno, P.; Basit, A.W. A Novel Double-Coating Approach for Improved PH-Triggered Delivery to the Ileo-Colonic Region of the Gastrointestinal Tract. Eur. J. Pharm. Biopharm. 2010, 74, 311–315. [Google Scholar] [CrossRef]
- Varum, F.; Freire, A.C.; Bravo, R.; Basit, A.W. OPTICORETM, an Innovative and Accurate Colonic Targeting Technology. Int. J. Pharm. 2020, 583, 119372. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, R.C.A.; Stellaard, F.; Olsder, G.G.; Woerdenbag, H.J.; Frijlink, H.W.; Kosterink, J.G.W. Oral Ileocolonic Drug Delivery by the Colopulse-System: A Bioavailability Study in Healthy Volunteers. J. Control. Release 2010, 146, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Maurer, J.M.; Hofman, S.; Schellekens, R.C.A.; Tonnis, W.F.; Dubois, A.O.T.; Woerdenbag, H.J.; Hinrichs, W.L.J.; Kosterink, J.G.W.; Frijlink, H.W. Development and Potential Application of an Oral ColoPulse Infliximab Tablet with Colon Specific Release: A Feasibility Study. Int. J. Pharm. 2016, 505, 175–186. [Google Scholar] [CrossRef]
- Maroni, A.; Del Curto, M.D.; Zema, L.; Foppoli, A.; Gazzaniga, A. Film Coatings for Oral Colon Delivery. Int. J. Pharm. 2013, 457, 372–394. [Google Scholar] [CrossRef]
- Sinha, V.R.; Kumria, R. Polysaccharides in Colon-Specific Drug Delivery. Int. J. Pharm. 2001, 224, 19–38. [Google Scholar] [CrossRef]
- Maderuelo, C.; Lanao, J.M.; Zarzuelo, A. Enteric Coating of Oral Solid Dosage Forms as a Tool to Improve Drug Bioavailability. Eur. J. Pharm. Sci. 2019, 138, 105019. [Google Scholar] [CrossRef]
- Dulin, W. Oral Targeted Drug Delivery Systems: Enteric Coating, In Oral Controlled Release Formulation Design and Drug Delivery: Theory to Practice; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp. 205–223. [Google Scholar] [CrossRef]
- Awad, A.; Madla, C.M.; McCoubrey, L.E.; Ferraro, F.; Gavins, F.K.H.; Buanz, A.; Gaisford, S.; Orlu, M.; Siepmann, F.; Siepmann, J.; et al. Clinical Translation of Advanced Colonic Drug Delivery Technologies. Adv. Drug Deliv. Rev. 2022, 181, 114076. [Google Scholar] [CrossRef]
- Keohane, K.; Rosa, M.; Coulter, I.S.; Griffin, B.T. Enhanced Colonic Delivery of Ciclosporin A Self-Emulsifying Drug Delivery System Encapsulated in Coated Minispheres. Drug Dev. Ind. Pharm. 2016, 42, 245–253. [Google Scholar] [CrossRef]
- Hodges, L.A.; Connolly, S.M.; Band, J.; O’Mahony, B.; Ugurlu, T.; Turkoglu, M.; Wilson, C.G.; Stevens, H.N.E. Scintigraphic Evaluation of Colon Targeting Pectin-HPMC Tablets in Healthy Volunteers. Int. J. Pharm. 2009, 370, 144–150. [Google Scholar] [CrossRef]
- Freire, C.; Podczeck, F.; Ferreira, D.; Veiga, F.; Sousa, J.; Pena, A. Assessment of the In-Vivo Drug Release from Pellets Film-Coated with a Dispersion of High Amylose Starch and Ethylcellulose for Potential Colon Delivery. J. Pharm. Pharmacol. 2010, 62, 55–61. [Google Scholar] [CrossRef]
- Freire, C.; Podczeck, F.; Veiga, F.; Sousa, J. Starch-Based Coatings for Colon-Specific Delivery. Part II: Physicochemical Properties and in Vitro Drug Release from High Amylose Maize Starch Films. Eur. J. Pharm. Biopharm. 2009, 72, 587–594. [Google Scholar] [CrossRef]
- Varum, F.; Freire, A.C.; Fadda, H.M.; Bravo, R.; Basit, A.W. A Dual PH and Microbiota-Triggered Coating (PhloralTM) for Fail-Safe Colonic Drug Release. Int. J. Pharm. 2020, 583, 119379. [Google Scholar] [CrossRef] [PubMed]
- Ibekwe, V.C.; Khela, M.K.; Evans, D.F.; Basit, A.W. A New Concept in Colonic Drug Targeting: A Combined PH-Responsive and Bacterially-Triggered Drug Delivery Technology. Aliment. Pharmacol. Ther. 2008, 28, 911–916. [Google Scholar] [CrossRef]
- Dodoo, C.C.; Wang, J.; Basit, A.W.; Stapleton, P.; Gaisford, S. Targeted Delivery of Probiotics to Enhance Gastrointestinal Stability and Intestinal Colonisation. Int. J. Pharm. 2017, 530, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, J.R.; Fischer, M.; Sagi, S.V.; Bohm, M.E.; Fadda, H.M.; Ranmal, S.R.; Budree, S.; Basit, A.W.; Glettig, D.L.; de la Serna, E.L.; et al. Fecal Microbiota Transplantation Capsules with Targeted Colonic Versus Gastric Delivery in Recurrent Clostridium Difficile Infection: A Comparative Cohort Analysis of High and Lose Dose. Dig. Dis. Sci. 2019, 64, 1672–1678. [Google Scholar] [CrossRef] [PubMed]
- Preisig, D.; Varum, F.; Bravo, R.; Hartig, C.; Spleiss, J.; Abbes, S.; Caobelli, F.; Wild, D.; Puchkov, M.; Huwyler, J.; et al. Colonic Delivery of Metronidazole-Loaded Capsules for Local Treatment of Bacterial Infections: A Clinical Pharmacoscintigraphy Study. Eur. J. Pharm. Biopharm. 2021, 165, 22–30. [Google Scholar] [CrossRef]
- Varum, F.; Bravo, R.; Basit, A. OPTICORETM: A First-in-Class Colonic Targeting Technology. ONdrugDelivery 2020, 2020, 40–44. [Google Scholar]
- Council of Experts; Comitees, E.U.S. Pharmacopeia National Formulary. USP 42 NF 37, 37th ed.; The United States Pharmacopeial convention: Rockville, MD, USA, 2019. [Google Scholar]
- Klein, S.; Rudolph, M.W.; Skalsky, B.; Petereit, H.U.; Dressman, J.B. Use of the BioDis to Generate a Physiologically Relevant IVIVC. J. Control. Release 2008, 130, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, L.; Ferguson, S.M.; Hudson, T.J.; Watanabe, S.; Katsuma, M.; Fix, J.A. In Vitro Evaluation of Dissolution Behavior for a Colon-Specific Drug Delivery System (CODESTM) in Multi-PH Media Using United States Pharmacopeia Apparatus II and III. AAPS PharmSciTech 2002, 3, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.; Stein, J.; Dressman, J. Site-Specific Delivery of Anti-Inflammatory Drugs in the Gastrointestinal Tract: An in-Vitro Release Model. J. Pharm. Pharmacol. 2005, 57, 709–719. [Google Scholar] [CrossRef]
- Merchant, H.A.; Frost, J.A.; Basit, A.W. Apparatus and Method for Testing Medicaments. U.S. Patent US010234467B2, 19 March 2019. [Google Scholar]
- Goyanes, A.; Hatton, G.B.; Merchant, H.A.; Basit, A.W. Gastrointestinal Release Behaviour of Modified-Release Drug Products: Dynamic Dissolution Testing of Mesalazine Formulations. Int. J. Pharm. 2015, 484, 103–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Gousous, J.; Amidon, G.L.; Langguth, P. Toward Biopredictive Dissolution for Enteric Coated Dosage Forms. Mol. Pharm. 2016, 13, 1927–1936. [Google Scholar] [CrossRef]
- Karkossa, F.; Klein, S. A Biopredictive In Vitro Comparison of Oral Locally Acting Mesalazine Formulations by a Novel Dissolution Model for Assessing Intraluminal Drug Release in Individual Subjects. J. Pharm. Sci. 2018, 107, 1680–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bott, C.; Rudolph, M.W.; Schneider, A.R.J.; Schirrmacher, S.; Skalsky, B.; Petereit, H.U.; Langguth, P.; Dressman, J.B.; Stein, J. In Vivo Evaluation of a Novel PH- and Time-Based Multiunit Colonic Drug Delivery System. Aliment. Pharmacol. Ther. 2004, 20, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Fadda, H.M.; Basit, A.W. Dissolution of PH Responsive Formulations in Media Resembling Intestinal Fluids: Bicarbonate versus Phosphate Buffers. J. Drug Deliv. Sci. Technol. 2005, 15, 273–279. [Google Scholar] [CrossRef]
- Liu, F.; Merchant, H.A.; Kulkarni, R.P.; Alkademi, M.; Basit, A.W. Evolution of a Physiological PH 6.8 Bicarbonate Buffer System: Application to the Dissolution Testing of Enteric Coated Products. Eur. J. Pharm. Biopharm. 2011, 78, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galia, E.; Nicolaides, E.; Hörter, D.; Löbenberg, R.; Reppas, C.; Dressman, J.B. Evaluation of Various Dissolution Media for Predicting In Vivo Performance of Class I and II Drugs. Pharm. Res. 1998, 15, 698–705. [Google Scholar] [CrossRef]
- Kalantzi, L.; Persson, E.; Polentarutti, B.; Abrahamsson, B.; Goumas, K.; Dressman, J.B.; Reppas, C. Canine Intestinal Contents vs. Simulated Media for the Assessment of Solubility of Two Weak Bases in the Human Small Intestinal Contents. Pharm. Res. 2006, 23, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, C.; Andreas, C.J.; Vertzoni, M.; Dressman, J.; Reppas, C. In-Vitro Simulation of Luminal Conditions for Evaluation of Performance of Oral Drug Products: Choosing the Appropriate Test Media. Eur. J. Pharm. Biopharm. 2015, 93, 173–182. [Google Scholar] [CrossRef]
- Karkossa, F.; Klein, S. Assessing the Influence of Media Composition and Ionic Strength on Drug Release from Commercial Immediate-Release and Enteric-Coated Aspirin Tablets. J. Pharm. Pharmacol. 2017, 69, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Blechar, J.A.; Al-Gousous, J.; Wilhelmy, C.; Postina, A.M.; Getto, M.; Langguth, P. Toward Mechanistic Design of Surrogate Buffers for Dissolution Testing of Ph-Dependent Drug Delivery Systems. Pharmaceutics 2020, 12, 1197. [Google Scholar] [CrossRef] [PubMed]
- Fadda, H.M.; Merchant, H.A.; Arafat, B.T.; Basit, A.W. Physiological Bicarbonate Buffers: Stabilisation and Use as Dissolution Media for Modified Release Systems. Int. J. Pharm. 2009, 382, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Vertzoni, M.; Diakidou, A.; Chatzilias, M.; Söderlind, E.; Abrahamsson, B.; Dressman, J.B.; Reppas, C. Biorelevant Media to Simulate Fluids in the Ascending Colon of Humans and Their Usefulness in Predicting Intracolonic Drug Solubility. Pharm. Res. 2010, 27, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- Garbacz, G.; Wedemeyer, R.S.; Nagel, S.; Giessmann, T.; Mönnikes, H.; Wilson, C.G.; Siegmund, W.; Weitschies, W. Irregular Absorption Profiles Observed from Diclofenac Extended Release Tablets Can Be Predicted Using a Dissolution Test Apparatus That Mimics in Vivo Physical Stresses. Eur. J. Pharm. Biopharm. 2008, 70, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Garbacz, G.; Rappen, G.M.; Koziolek, M.; Weitschies, W. Dissolution of Mesalazine Modified Release Tablets under Standard and Bio-Relevant Test Conditions. J. Pharm. Pharmacol. 2015, 67, 199–208. [Google Scholar] [CrossRef]
- Al-Gousous, J.; Salehi, N.; Amidon, G.E.; Zi, R.M.; Langguth, P.; Amidon, G.L. Mass Transport Analysis of Bicarbonate Buffer: Effect of the CO2 − H2CO3 Hydration − Dehydration Kinetics in the Fluid Boundary Layer and the Apparent Effective PKa Controlling Dissolution of Acids and Bases. Mol. Pharm. 2019, 16, 2626–2635. [Google Scholar] [CrossRef]
- Al-Gousous, J.; Ruan, H.; Blechar, J.A.; Sun, K.X.; Salehi, N.; Langguth, P.; Job, N.M.; Lipka, E.; Loebenberg, R.; Bermejo, M.; et al. Mechanistic Analysis and Experimental Verification of Bicarbonate-Controlled Enteric Coat Dissolution: Potential in Vivo Implications. Eur. J. Pharm. Biopharm. 2019, 139, 47–58. [Google Scholar] [CrossRef]
- Ibekwe, V.C.; Fadda, H.M.; Parsons, G.E.; Basit, A.W. A Comparative in Vitro Assessment of the Drug Release Performance of PH-Responsive Polymers for Ileo-Colonic Delivery. Int. J. Pharm. 2006, 308, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Krieg, B.J.; Taghavi, S.M.; Amidon, G.L.; Amidon, G.E. In Vivo Predictive Dissolution: Transport Analysis of the CO2, Bicarbonate in Vivo Buffer System. J. Pharm. Sci. 2014, 103, 3473–3490. [Google Scholar] [CrossRef] [Green Version]
- Wray, H.; Joseph, R.; Palmen, M.; Pierce, D. A Pharmacokinetic and Scintigraphic Comparison of MMXTM Mesalamine and Delayed-Release Mesalamine. Am. J. Gastroenterol. 2008, 103, S433–S434. [Google Scholar] [CrossRef]
- Sakamoto, A.; Izutsu, K.I.; Yoshida, H.; Abe, Y.; Inoue, D.; Sugano, K. Simple Bicarbonate Buffer System for Dissolution Testing: Floating Lid Method and Its Application to Colonic Drug Delivery System. J. Drug Deliv. Sci. Technol. 2021, 63, 102447. [Google Scholar] [CrossRef]
- Scott, N.; Patel, K.; Sithole, T.; Xenofontos, K.; Mohylyuk, V.; Liu, F. Regulating the PH of Bicarbonate Solutions without Purging Gases: Application to Dissolution Testing of Enteric Coated Tablets, Pellets and Microparticles. Int. J. Pharm. 2020, 585, 119562. [Google Scholar] [CrossRef] [PubMed]
- Garbacz, G.; Kołodziej, B.; Koziolek, M.; Weitschies, W.; Klein, S. An Automated System for Monitoring and Regulating the PH of Bicarbonate Buffers. AAPS PharmSciTech 2013, 14, 517–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, H.; Yoshida, H.; Izutsu, K.I.; Goda, Y. Use of Bicarbonate Buffer Systems for Dissolution Characterization of Enteric-Coated Proton Pump Inhibitor Tablets. J. Pharm. Pharmacol. 2016, 68, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Karkossa, F.; Krueger, A.; Urbaniak, J.; Klein, S. Simulating Different Dosing Scenarios for a Child-Appropriate Valproate ER Formulation in a New Pediatric Two-Stage Dissolution Model. AAPS PharmSciTech 2017, 18, 309–316. [Google Scholar] [CrossRef]
- Garbacz, G.; Kołodziej, B.; Koziolek, M.; Weitschies, W.; Klein, S. A Dynamic System for the Simulation of Fasting Luminal PH-Gradients Using Hydrogen Carbonate Buffers for Dissolution Testing of Ionisable Compounds. Eur. J. Pharm. Sci. 2014, 51, 224–231. [Google Scholar] [CrossRef]
- Effinger, A.; O’Driscoll, C.M.; McAllister, M.; Fotaki, N. Impact of Gastrointestinal Disease States on Oral Drug Absorption—Implications for Formulation Design—A PEARRL Review. J. Pharm. Pharmacol. 2019, 71, 674–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freire, A.C.; Basit, A.W.; Choudhary, R.; Piong, C.W.; Merchant, H.A. Does Sex Matter? The Influence of Gender on Gastrointestinal Physiology and Drug Delivery. Int. J. Pharm. 2011, 415, 15–28. [Google Scholar] [CrossRef]
- Merchant, H.A.; Liu, F.; Orlu Gul, M.; Basit, A.W. Age-Mediated Changes in the Gastrointestinal Tract. Int. J. Pharm. 2016, 512, 382–395. [Google Scholar] [CrossRef]
- Goyanes, A.; Hatton, G.B.; Basit, A.W. A Dynamic in Vitro Model to Evaluate the Intestinal Release Behaviour of Modified-Release Corticosteroid Products. J. Drug Deliv. Sci. Technol. 2015, 25, 36–42. [Google Scholar] [CrossRef]
- Goyanes, A.; Chang, H.; Sedough, D.; Hatton, G.B.; Wang, J.; Buanz, A.; Gaisford, S.; Basit, A.W. Fabrication of Controlled-Release Budesonide Tablets via Desktop (FDM) 3D Printing. Int. J. Pharm. 2015, 496, 414–420. [Google Scholar] [CrossRef]
- Fina, F.; Goyanes, A.; Gaisford, S.; Basit, A.W. Selective Laser Sintering (SLS) 3D Printing of Medicines. Int. J. Pharm. 2017, 529, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyanes, A.; Fina, F.; Martorana, A.; Sedough, D.; Gaisford, S.; Basit, A.W. Development of Modified Release 3D Printed Tablets (Printlets) with Pharmaceutical Excipients Using Additive Manufacturing. Int. J. Pharm. 2017, 527, 21–30. [Google Scholar] [CrossRef]
- Merchant, H.A.; Goyanes, A.; Parashar, N.; Basit, A.W. Predicting the Gastrointestinal Behaviour of Modified-Release Products: Utility of a Novel Dynamic Dissolution Test Apparatus Involving the Use of Bicarbonate Buffers. Int. J. Pharm. 2014, 475, 585–591. [Google Scholar] [CrossRef]
- Sheng, J.J.; McNamara, D.P.; Amidon, G.L. Toward an In Vivo Dissolution Methodology: A Comparison of Phosphate and Bicarbonate Buffers. Mol. Pharm. 2009, 6, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Andreas, C.J.; Chen, Y.C.; Markopoulos, C.; Reppas, C.; Dressman, J. In Vitro Biorelevant Models for Evaluating Modified Release Mesalamine Products to Forecast the Effect of Formulation and Meal Intake on Drug Release. Eur. J. Pharm. Biopharm. 2015, 97, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.W.; Klein, S.; Beckert, T.E.; Petereit, H.U.; Dressman, J.B. A New 5-Aminosalicylic Acid Multi-Unit Dosage Form for the Therapy of Ulcerative Colitis. Eur. J. Pharm. Biopharm. 2001, 51, 183–190. [Google Scholar] [CrossRef]
- French, D.L.; Mauger, J.W. Evaluation of the Physicochemical Properties and Dissolution Characteristics of Mesalamine: Relevance to Controlled Intestinal Drug Delivery. Pharm. Res. Off. J. Am. Assoc. Pharm. Sci. 1993, 10, 1285–1290. [Google Scholar] [CrossRef]
- Jantratid, E.; De Maio, V.; Ronda, E.; Mattavelli, V.; Vertzoni, M.; Dressman, J.B. Application of Biorelevant Dissolution Tests to the Prediction of in Vivo Performance of Diclofenac Sodium from an Oral Modified-Release Pellet Dosage Form. Eur. J. Pharm. Sci. 2009, 37, 434–441. [Google Scholar] [CrossRef]
- Hofmann, M.; García, M.A.; Al-Gousous, J.; Ruiz-Picazo, A.; Thieringer, F.; Nguyen, M.A.; Månsson, W.; Galle, P.R.; Langguth, P. In Vitro Prediction of in Vivo Absorption of Ibuprofen from Suspensions through Rational Choice of Dissolution Conditions. Eur. J. Pharm. Biopharm. 2020, 149, 229–237. [Google Scholar] [CrossRef]
- Wilding, I.R. A Scintigraphic Study to Evaluate What Happens to Pentasa and Asacol in the Human Gut. Pract. Gastroenterol. 1999, 23 (Suppl. 11), 1–8. [Google Scholar]
- Yu, A.; Baker, J.R.; Fioritto, A.F.; Wang, Y.; Luo, R.; Li, S.; Wen, B.; Bly, M.; Tsume, Y.; Koenigsknecht, M.J.; et al. Measurement of in Vivo Gastrointestinal Release and Dissolution of Three Locally Acting Mesalamine Formulations in Regions of the Human Gastrointestinal Tract. Mol. Pharm. 2017, 14, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Milojevic, S.; Newton, J.M.; Cummings, J.H.; Gibson, G.R.; Botham, R.L.; Ring, S.G.; Stockham, M.; Allwood, M.C. Amylose as a Coating for Drug Delivery to the Colon: Preparation and in Vitro Evaluation Using Glucose Pellets. J. Control. Release 1996, 38, 85–94. [Google Scholar] [CrossRef]
- Karrout, Y.; Neur, C.; Wils, D.; Siepmann, F.; Deremaux, L.; Flament, M.P.; Dubreuil, L.; Desreumaux, P.; Siepmann, J. Peas Starch-Based Film Coatings for Site-Specific Drug Delivery to the Colon. J. Appl. Polym. Sci. 2011, 119, 1176–1184. [Google Scholar] [CrossRef]
- Macfarlane, G.T.; Englyst, H.N. Starch Utilization by the Human Large Intestinal Microflora. J. Appl. Bacteriol. 1986, 60, 195–201. [Google Scholar] [CrossRef]
- Englyst, H.N.; Hay, S.; Macfarlane, G.T. Polysaccharide Breakdown by Mixed Populations of Human Faecal Bacteria. FEMS Microbiol. Lett. 1987, 45, 163–171. [Google Scholar] [CrossRef]
- Wahlgren, M.; Axenstrand, M.; Håkansson, Å.; Marefati, A.; Pedersen, B.L. In Vitro Methods to Study Colon Release: State of the Art and an Outlook on New Strategies for Better in-Vitro Biorelevant Release Media. Pharmaceutics 2019, 11, 95. [Google Scholar] [CrossRef] [Green Version]
- Hughes, S.A.; Shewry, P.R.; Gibson, G.R.; McCleary, B.V.; Rastall, R.A. In Vitro Fermentation of Oat and Barley Derived β-Glucans by Human Faecal Microbiota. FEMS Microbiol. Ecol. 2008, 64, 482–493. [Google Scholar] [CrossRef] [Green Version]
- Sousa, T.; Yadav, V.; Zann, V.; Borde, A.; Abrahamsson, B.; Basit, A.W. On the Colonic Bacterial Metabolism of Azo-Bonded Prodrugs of 5-Aminosalicylic Acid. J. Pharm. Sci. 2014, 103, 3171–3175. [Google Scholar] [CrossRef]
- Hatton, G.B.; Madla, C.M.; Rabbie, S.C.; Basit, A.W. All Disease Begins in the Gut: Influence of Gastrointestinal Disorders and Surgery on Oral Drug Performance. Int. J. Pharm. 2018, 548, 408–422. [Google Scholar] [CrossRef]
- Yadav, V.; Gaisford, S.; Merchant, H.A.; Basit, A.W. Colonic Bacterial Metabolism of Corticosteroids. Int. J. Pharm. 2013, 457, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Karatza, E.; Vertzoni, M.; Muenster, U.; Reppas, C. The Impact of Handling and Storage of Human Fecal Material on Bacterial Activity. J. Pharm. Sci. 2016, 105, 3458–3461. [Google Scholar] [CrossRef] [PubMed]
- Molly, K.; Woestyne, M.V.; De Smet, I.; Verstraete, W. Validation of the Simulator of the Human Intestinal Microbial Ecosystem (SHIME) Reactor Using Microorganism-Associated Activities. Microb. Ecol. Health Dis. 1994, 7, 191–200. [Google Scholar] [CrossRef]
- Van den Abbeele, P.; Roos, S.; Eeckhaut, V.; Mackenzie, D.A.; Derde, M.; Verstraete, W.; Marzorati, M.; Possemiers, S.; Vanhoecke, B.; Van Immerseel, F.; et al. Incorporating a Mucosal Environment in a Dynamic Gut Model Results in a More Representative Colonization by Lactobacilli. Microb. Biotechnol. 2012, 5, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Ghyselinck, J.; Verstrepen, L.; Moens, F.; Van den Abbeele, P.; Said, J.; Smith, B.; Bjarnason, I.; Basit, A.W.; Gaisford, S. A 4-Strain Probiotic Supplement Influences Gut Microbiota Composition and Gut Wall Function in Patients with Ulcerative Colitis. Int. J. Pharm. 2020, 587, 119648. [Google Scholar] [CrossRef]
- García-Villalba, R.; Vissenaekens, H.; Pitart, J.; Romo-Vaquero, M.; Espín, J.C.; Grootaert, C.; Selma, M.V.; Raes, K.; Smagghe, G.; Possemiers, S.; et al. Gastrointestinal Simulation Model TWIN-SHIME Shows Differences between Human Urolithin-Metabotypes in Gut Microbiota Composition, Pomegranate Polyphenol Metabolism, and Transport along the Intestinal Tract. J. Agric. Food Chem. 2017, 65, 5480–5493. [Google Scholar] [CrossRef] [Green Version]
- Beeck, R.; Glöckl, G.; Krause, J.; Schick, P.; Weitschies, W. Mimicking the Dynamic Colonic Microbiota in Vitro to Gain a Better Understanding on the in Vivo Metabolism of Xenobiotics: Degradation of Sulfasalazine. Int. J. Pharm. 2021, 603, 120704. [Google Scholar] [CrossRef]
- Fässler, C.; Arrigoni, E.; Venema, K.; Hafner, V.; Brouns, F.; Amadò, R. Digestibility of Resistant Starch Containing Preparations Using Two in Vitro Models. Eur. J. Nutr. 2006, 45, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Van Nuenen, M.H.M.C.; Meyer, P.D.; Venema, K. The Effect of Various Inulins and Clostridium Difficile on the Metabolic Activity of the Human Colonic Microbiota in Vitro. Microb. Ecol. Health Dis. 2003, 15, 137–144. [Google Scholar] [CrossRef]
- Minekus, M.; Smeets-Peeters, M.; Havenaar, R.; Bernalier, A.; Fonty, G.; Marol-Bonnin, S.; Alric, M.; Marteau, P.; Huis In’t Veld, J.H.J. A Computer-Controlled System to Simulate Conditions of the Large Intestine with Peristaltic Mixing, Water Absorption and Absorption of Fermentation Products. Appl. Microbiol. Biotechnol. 1999, 53, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Bordonaro, M.; Venema, K.; Putri, A.K.; Lazarova, D. Approaches That Ascertain the Role of Dietary Compounds in Colonic Cancer Cells. World J. Gastrointest. Oncol. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sferrazza, G.; Siviero, P.D.; Nicotera, G.; Turella, P.; Serafino, A.; Blandizzi, C.; Pierimarchi, P. Regulatory Framework on Bioequivalence Criteria for Locally Acting Gastrointestinal Drugs: The Case for Oral Modified Release Mesalamine Formulations. Expert Rev. Clin. Pharmacol. 2017, 10, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- FDA. Code of Federal Regulations Title 21: Food and Drugs. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=320.24 (accessed on 24 October 2021).
- FDA. Product-Specific Guidances for Generic Drug Development. Available online: https://www.fda.gov/drugs/guidances-drugs/product-specific-guidances-generic-drug-development (accessed on 24 October 2021).
- U.S. Department of Health and Human Services; Food and Drug Adminstration. Guidance for Industry Bioequivalence Recommendations for Specific Products. Evaluation 2010. [Google Scholar]
- EMA (European Medicines Agency); (EMA) Committee for Medicinal Products for Human Use (CHMP). Guideline on Equivalence Studies for the Demonstration of Therapeutic Equivalence for Locally Applied, Locally Acting Products in the Gastrointestinal Tract; European Medicines Agency: Amsterdam, The Netherlands, 2018; Volume 44, pp. 1–13. [Google Scholar]
- EMEA. Note for Guidance on the Clinical Requirements for Locally Applied, Locally Acting Products Containing Known Constituents. Eur. Med. Agency 1995, 1–4. [Google Scholar]
- Richardson, J.C.; Bowtell, R.W.; Mäder, K.; Melia, C.D. Pharmaceutical Applications of Magnetic Resonance Imaging (MRI). Adv. Drug Deliv. Rev. 2005, 57, 1191–1209. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | API | Dosage Form | Coating Polymer | Polymer Brand Name | Release Mechanism |
|---|---|---|---|---|---|
| Asacol® | Mesalazine | Delayed-release tablets | Methacrylic acid copolymer type B | Eudragit® S | pH-dependent release (pH > 7.0) |
| Mezavant®/ Lialda® | Mesalazine | Delayed-release tablets | Methacrylic acid copolymer type A & B | Eudragit® L Eudragit® S | pH-dependent release (pH > 6.0) pH-dependent release (pH > 7.0) |
| Uceris® | Budesonide | Extended-release tablets | Methacrylic acid copolymer type A & B | Eudragit® L Eudragit® S | pH-dependent release (pH > 5.0) pH-dependent release (pH > 7.0) |
| Salofalk® | Mesalazine | Delayed-release granules | Methacrylic acid copolymer type A | Eudragit® L | pH-dependent release (pH > 6.0) |
| Pentasa® | Mesalazine | Controlled-release capsules | Ethylcellulose Hydroxypropyl Methylcellulose | Surelease® | Time-controlled sustained release |
| Asacol®/ Yaldigo®/ Asacolon® (OPTICORETM) | Mesalazine | Delayed-release tablets | Methacrylic acid copolymer type B & Resistant Starch | Eudragit® S | pH-dependent release (pH > 7.0) & Enzymatic triggered release |
| Buffer Name | Resembling Segment | Buffer Species and Molarities | Other Relevant Contents 3 | pH | IS (mM) | Buffer Capacity (mmol/L/ΔpH) | Media Type a |
|---|---|---|---|---|---|---|---|
| Hanks [90] | Proximal small intestine | Bicarbonate 4.2 mM b | Mg2+, Ca2+ | 7.4 | 155 | 1 | Bio-relevant |
| Phosphate 0.3 mM b | |||||||
| mHanks [91] | Proximal small intestine | Bicarbonate 4.2 mM b | Mg2+, Ca2+ | 6.8 c | 155 | 3.1 | Bio-relevant |
| Phosphate 0.8 mM | |||||||
| FaSSIF (V-1) [92] | Proximal small intestine | Phosphate 28.7 mM d | TC, LC | 6.5 | 12 [93] | Bio-relevant | |
| (V-2) | Maleate 19.1 mM e | 10 [94] | |||||
| CarbSIF [95] | Proximal small intestine | Bicarbonate 15 mM | 6.0–6.8 c | 140 | Bio-relevant | ||
| - [87] | Proximal small intestine | Phosphate 15 mM | 6.0 | 139 | Surrogate | ||
| - [96] | Proximal small intestine | Succinate 15 mM b | 6.8 | 139 | Surrogate | ||
| - [96] | Proximal small intestine | Citrate 15 mM b | 6.8 | 139 | Surrogate | ||
| Krebs [97] | Distal small Intestine | Bicarbonate 25 mM | Mg2+, Ca2+ | 161 | 3.7 | Bio-relevant | |
| mKrebs [97] | Distal small Intestine | Bicarbonate 25 mM | Mg2+, Ca2+ | 7.4 c | 161 | 5.5 | Bio-relevant |
| SIFileum [94] | Distal small Intestine | Maleate 52.8 mM | TC, LC | 7.5 | 10 | Bio-relevant | |
| - [87] | Distal small Intestine | Phosphate 19.5–23.5 mM | 6.8–6.95 | 139 | Surrogate | ||
| - [96] | Distal small Intestine | Bicarbonate 30 mM b | 7.4 | 139 | Surrogate | ||
| - [96] | Distal small Intestine | Succinate 15 mM b | 7.4 | 139 | Surrogate | ||
| -[96] | Distal small intestine | Citrate 15 mM b | 7.4 | 139 | Surrogate | ||
| - [83] | Colon | Citrate | 5.0 | Compendial-based | |||
| Phosphate | |||||||
| SCoF [84] | Colon | Acetic acid 170 mM | 5.8 | 160 | 29.1 | Compendial-based | |
| FaSSCoF [98] | Ascending colon | Tris 45.4 mM | BSA, PA, PC | 7.8 | 16 | Bio-relevant | |
| Maleate 75.8 mM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García, M.A.; Varum, F.; Al-Gousous, J.; Hofmann, M.; Page, S.; Langguth, P. In Vitro Methodologies for Evaluating Colon-Targeted Pharmaceutical Products and Industry Perspectives for Their Applications. Pharmaceutics 2022, 14, 291. https://doi.org/10.3390/pharmaceutics14020291
García MA, Varum F, Al-Gousous J, Hofmann M, Page S, Langguth P. In Vitro Methodologies for Evaluating Colon-Targeted Pharmaceutical Products and Industry Perspectives for Their Applications. Pharmaceutics. 2022; 14(2):291. https://doi.org/10.3390/pharmaceutics14020291
Chicago/Turabian StyleGarcía, Mauricio A., Felipe Varum, Jozef Al-Gousous, Michael Hofmann, Susanne Page, and Peter Langguth. 2022. "In Vitro Methodologies for Evaluating Colon-Targeted Pharmaceutical Products and Industry Perspectives for Their Applications" Pharmaceutics 14, no. 2: 291. https://doi.org/10.3390/pharmaceutics14020291