
Role of Cyclodextrins and Drug Solid State Properties on Flufenamic Acid Dissolution Performance from Tablets



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
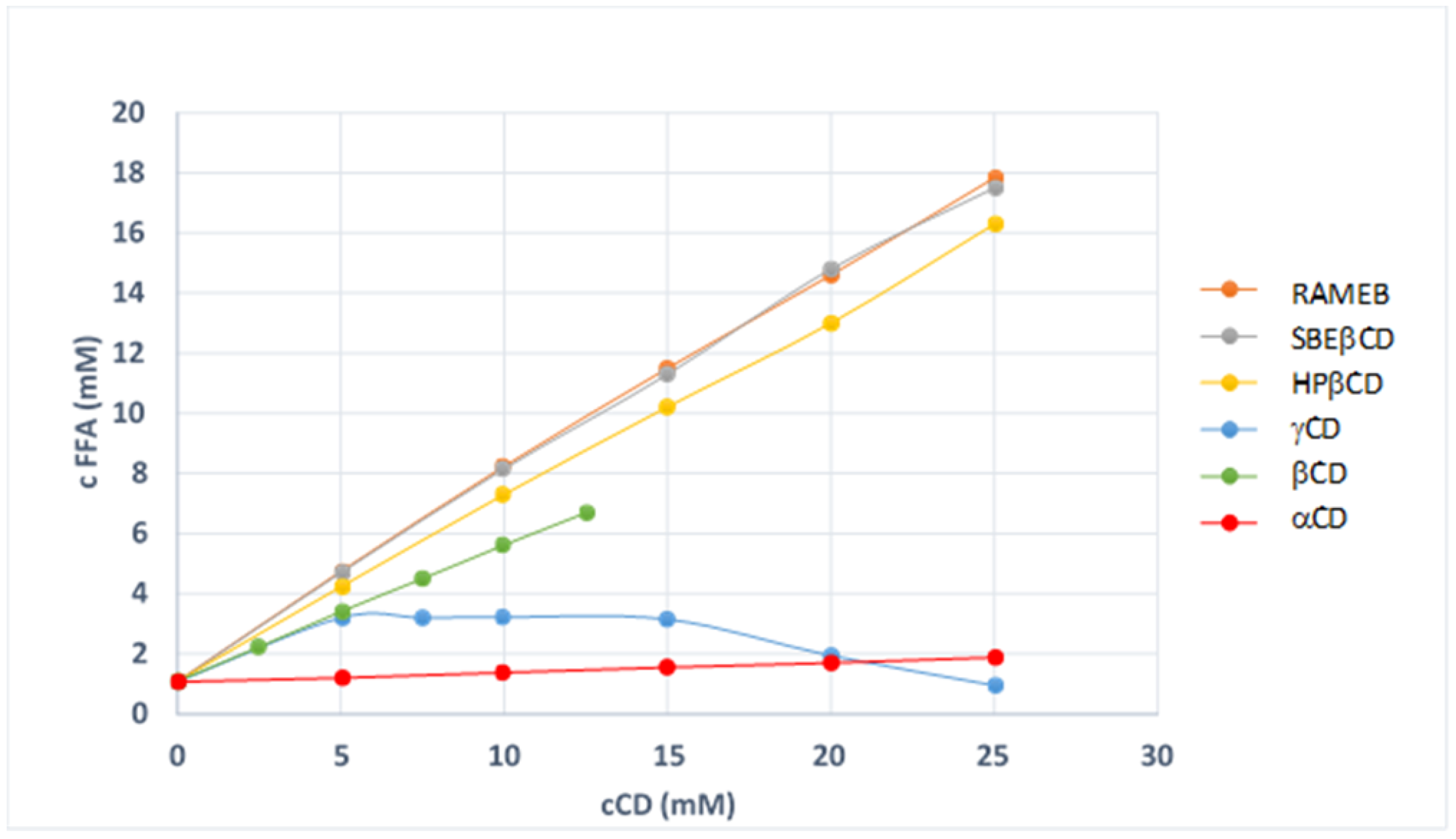
2.2. Phase Solubility Studies
2.3. Preparation Methods of Drug: CD Solid Binary Systems
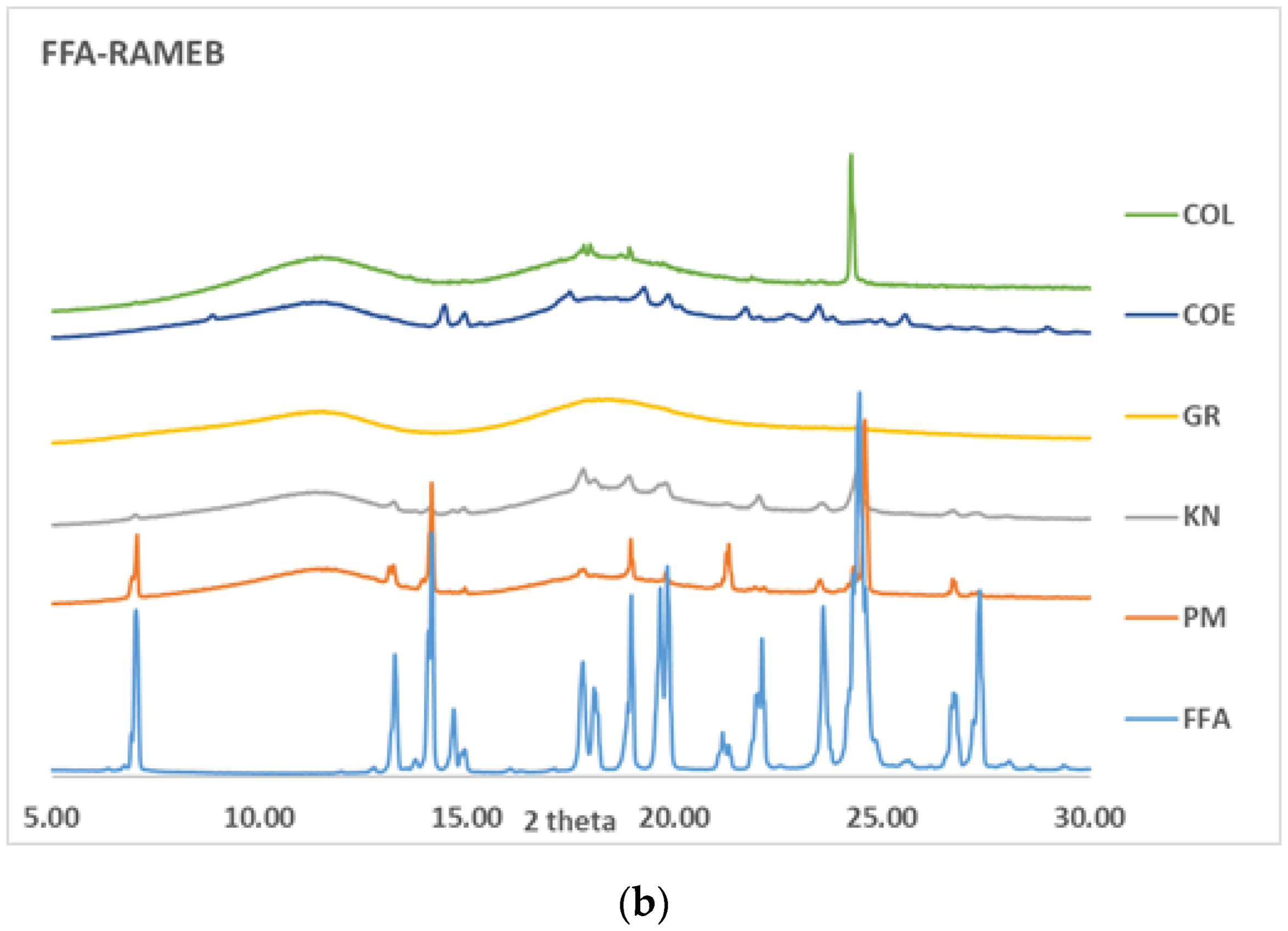
2.4. Characterisation of Drug: CD Binary Systems
2.5. Tablets Preparation
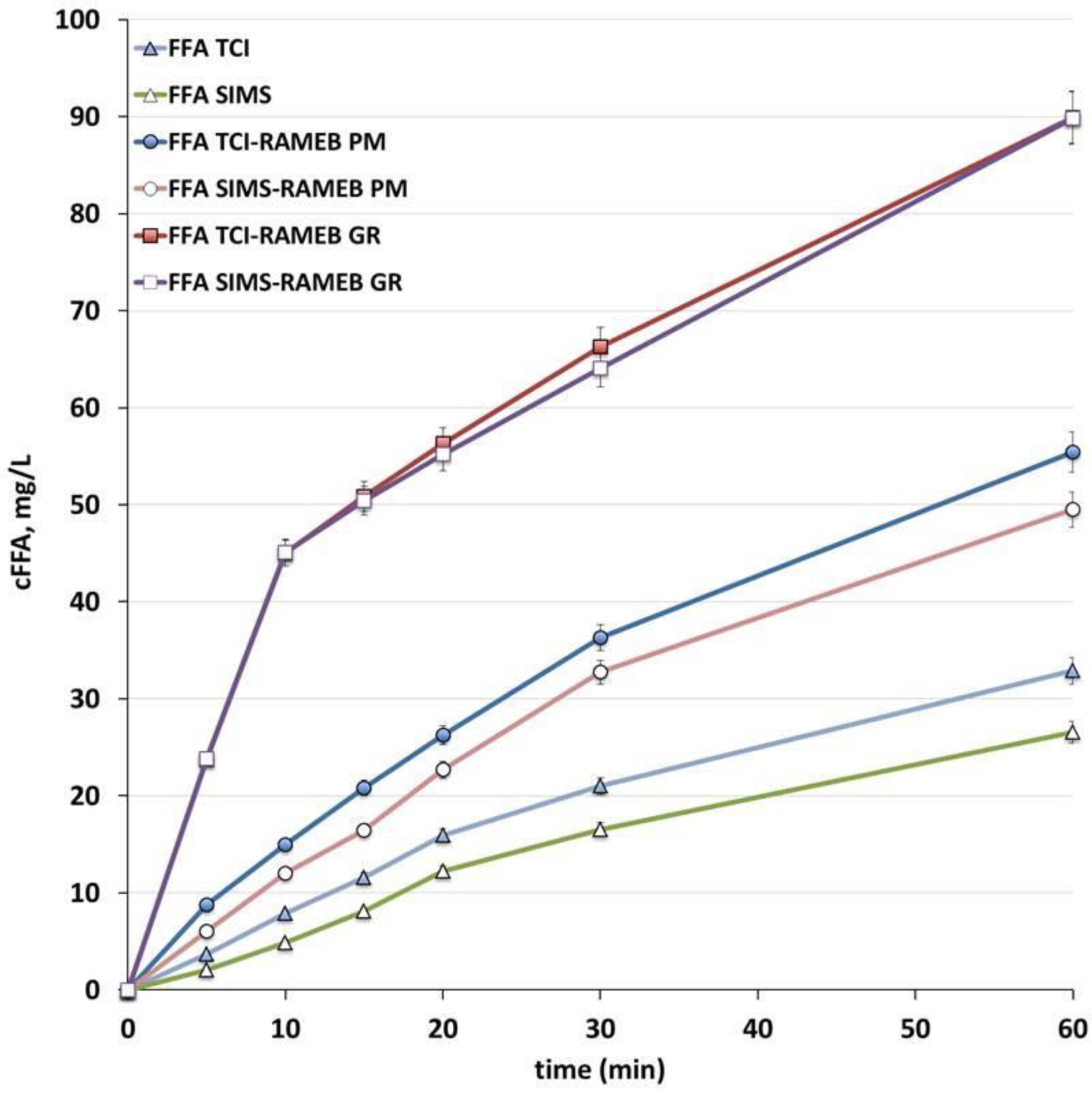
2.6. Characterisation of Tablets
3. Results
3.1. Phase-Solubility Studies
3.2. Preparation and Solid-State Characterisation of Drug-CD Binary Systems
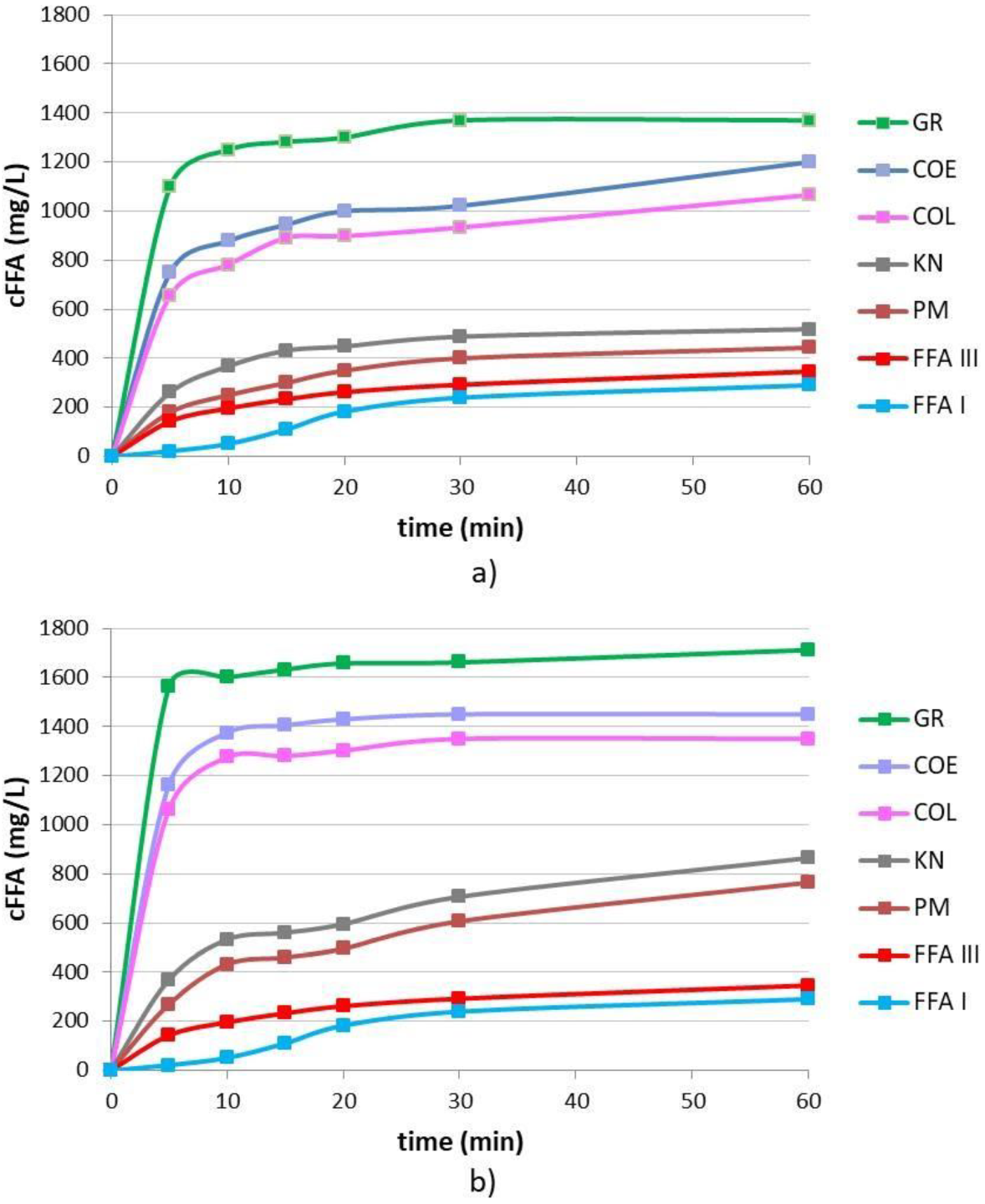
3.3. Dissolution Rate Studies of Binary Systems
3.4. Preparation and Characterisation of Tablets
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mantri, R.V.; Sanghvi, R.; Zhu, H.J. Solubility of Pharmaceutical Solids. In Developing Solid Oral Dosage Forms; Elsevier: Amsterdam, The Netherlands, 2017; pp. 3–22. ISBN 978-0-12-802447-8. [Google Scholar]
- Moreton, C. Poor Solubility—Where Do We Stand 25 Years after the “Rule of Five”? Am. Pharm Rev. 2021. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/573402-Poor-Solubility-Where-Do-We-Stand-25-Years-after-the-Rule-of-Five/ (accessed on 3 November 2021).
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridgeirsdottir, G.A.; Harris, R.; Fischer, P.M.; Roberts, C.J. Support Tools in Formulation Development for Poorly Soluble Drugs. J. Pharm. Sci. 2016, 105, 2260–2269. [Google Scholar] [CrossRef] [PubMed]
- Guinamard, R.; Simard, C.; Del Negro, C. Flufenamic acid as an ion channel modulator. Pharmacol. Ther. 2013, 138, 272–284. [Google Scholar] [CrossRef] [Green Version]
- Schattling, B.; Steinbach, K.; Thies, E.; Kruse, M.; Menigoz, A.; Ufer, F.; Flockerzi, V.; Brück, W.; Pongs, O.; Vennekens, R.; et al. TRPM4 cation channel mediates axonal and neuronal degeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2012, 18, 1805–1811. [Google Scholar] [CrossRef]
- Angelucci, L.; Petrangeli, B.; Celletti, P.; Favilli, S. Bioavailability of Flufenamic Acid in Hard and Soft Gelatin Capsules. J. Pharm. Sci. 1976, 65, 455–456. [Google Scholar] [CrossRef]
- Kaniwa, N.; Ogata, H.; Aoyagi, N.; Shibazaki, T.; Ejima, A.; Watanabe, Y.; Motohashi, K.; Sasahara, K.; Nakajima, E.; Morioka, T.; et al. The bioavailability of flufenamic acid and its dissolution rate from capsules. Int. J. Clin. Pharmacol. Ther. Toxicol. 1983, 21, 56–63. [Google Scholar]
- Akbuğa, J.; Gülhan, S.; Bayraktar-Alpmen, G. Studies on flufenamic acid capsules and tablets. Pharmazie 1983, 38, 478–480. [Google Scholar]
- Sood, J.; Sapra, B.; Bhandari, S.; Jindal, M.; Tiwary, A.K. Understanding pharmaceutical polymorphic transformations I: Influence of process variables and storage conditions. Ther. Deliv. 2014, 5, 1123–1142. [Google Scholar] [CrossRef]
- Zhang, K.; Fellah, N.; López-Mejías, V.; Ward, M.D. Polymorphic Phase Transformation Pathways under Nanoconfinement: Flufenamic Acid. Cryst. Growth Des. 2020, 20, 7098–7103. [Google Scholar] [CrossRef]
- Censi, R.; Di Martino, P. Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef] [Green Version]
- Maestrelli, F.; Rossi, P.; Paoli, P.; De Luca, E.; Mura, P. The role of solid state properties on the dissolution performance of flufenamic acid. J. Pharm. Biomed. Anal. 2020, 180, 113058. [Google Scholar] [CrossRef] [PubMed]
- Brewster, M.E.; Loftsson, T. Cyclodextrins as pharmaceutical solubilizers. Adv. Drug Deliv. Rev. 2007, 59, 645–666. [Google Scholar] [CrossRef] [PubMed]
- Kurkov, S.V.; Loftsson, T. Cyclodextrins. Int. J. Pharm. 2013, 453, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Floare, C.G.; Pirnau, A.; Bogdan, M. 1H NMR spectroscopic characterization of inclusion complexes of tolfenamic and flufenamic acids with β-cyclodextrin. J. Mol. Struct. 2013, 1044, 72–78. [Google Scholar] [CrossRef]
- Alshehri, S.; Shakeel, F.; Ibrahim, M.; Elzayat, E.; Altamimi, M.; Shazly, G.; Mohsin, K.; Alkholief, M.; Alsulays, B.; Alshetaili, A.; et al. Influence of the microwave technology on solid dispersions of mefenamic acid and flufenamic acid. PLoS ONE 2017, 12, e0182011. [Google Scholar] [CrossRef] [Green Version]
- Mura, P.; Corti, G.; Cirri, M.; Maestrelli, F.; Mennini, N.; Bragagni, M. Development of Mucoadhesive Films for Buccal Administration of Flufenamic Acid: Effect of Cyclodextrin Complexation. J. Pharm. Sci. 2010, 99, 3019–3029. [Google Scholar] [CrossRef]
- Domańska, U.; Pelczarska, A.; Pobudkowska, A. Effect of 2-Hydroxypropyl-β-cyclodextrin on Solubility of Sparingly Soluble Drug Derivatives of Anthranilic Acid. Int. J. Mol. Sci. 2011, 12, 2383–2394. [Google Scholar] [CrossRef] [Green Version]
- De Marco, I.; Reverchon, E. Supercritical antisolvent micronization of cyclodextrins. Powder Technol. 2008, 183, 239–246. [Google Scholar] [CrossRef]
- Wongmekiat, A.; Tozuka, Y.; Oguchi, T.; Yamamoto, K. Formation of Fine Drug Particles by Cogrinding with Cyclodextrins. I. The Use of β-Cyclodextrin Anhydrate and Hydrate. Pharm. Res. 2002, 19, 1867–1872. [Google Scholar] [CrossRef]
- Turunen, E.; Mannila, J.; Laitinen, R.; Riikonen, J.; Lehto, V.-P.; Järvinen, T.; Ketolainen, J.; Järvinen, K.; Jarho, P. Fast-dissolving sublingual solid dispersion and cyclodextrin complex increase the absorption of perphenazine in rabbits. J. Pharm. Pharmacol. 2010, 63, 19–25. [Google Scholar] [CrossRef]
- Loftsson, T.; Hreinsdóttir, D.; Másson, M. The complexation efficiency. J. Incl. Phenom. Macrocycl. Chem. 2007, 57, 545–552. [Google Scholar] [CrossRef]
- Anderson, N.H.; Bauer, M.; Boussac, N.; Khan-Malek, R.; Munden, P.; Sardaro, M. An evaluation of fit factors and dissolution efficiency for the comparison of in vitro dissolution profiles. J. Pharm. Biomed. Anal. 1998, 17, 811–822. [Google Scholar] [CrossRef]
- Higuchi, T.; Connors, K.A. Phase Solubility Techniques. Adv. Anal. Chem. Instrum. 1965, 4, 117–212. [Google Scholar]
- Iqubal, M.K. Recent Advances in Direct Compression Technique for Pharmaceutical Tablet Formulation. Int. J. Pharm. Res. Dev. 2014, 6, 49–57. [Google Scholar]
- Dokala, G.K.; Pallavi, C. Direct Compression—An Overview. Int. J. Res. Pharm. Biom. Sci. 2013, 4, 155–158. [Google Scholar]
- Sharma, N.; Pahuja, S.; Sharma, N. Immediate release tablets: A review. Int. J. Pharm. Sci. Res. 2019, 11, 3607–3618. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD | K1:1 (M−1) | CE (M−1) | FFA Solubility Increase * |
|---|---|---|---|
| αCD | 32 ± 0.8 | 34 ± 0.9 | 1.62 |
| βCD | 760 ± 18 | 815 ± 20 | 6.22 |
| γCD | n.c. | n.c. | ---- |
| HPβCD | 1410 ± 35 | 1511 ± 37 | 15.43 |
| RAMEB | 1860 ± 45 | 1999 ± 49 | 17.64 |
| SBEβCD | 1812 ± 43 | 1946 ± 47 | 16.29 |
| Sample | Tm (°C) | ΔH (J/g) | % Rc |
|---|---|---|---|
| FFA | 135.4 ± 0.3 | 124.6 ± 6.0 | 100.0 ± 4.8 |
| PM FFA βCD | 135.2 ± 0.5 | 115.2 ± 5.7 | 92.5 ± 4.6 |
| KN FFA βCD | 135.1 ± 0.4 | 82.2 ± 3.9 | 65.9 ± 3.2 |
| GR FFA βCD | 134.8 ± 0.5 | 35.3 ± 1.7 | 24.4 ± 1.2 |
| COE FFA βCD | 125.4 ± 0.3 | 74.6 ± 3.6 | 59.8 ± 2.9 |
| COL FFA βCD | 135.2 ± 0.4 | 27.8 ± 1.3 | 20.7 ± 1.0 |
| PM FFA HPβCD | 135.0 ± 0.4 | 78.2 ± 3.8 | 62.8 ± 3.1 |
| KN FFA HPβCD | 135.7 ± 0.6 | 53.3 ± 2.7 | 42.7 ± 2.1 |
| GR FFA HPβCD | 135.8 ± 0.5 | 25.8 ± 1.3 | 14.7 ± 0.7 |
| COE FFA HPβCD | 125.6 ± 0.4 | 38.6 ± 1.9 | 30.9 ± 1.5 |
| COL FFA HPβCD | 135.7 ± 0.4 | 18.6 ± 0.9 | 13.7 ± 0.6 |
| PM FFA SBEβCD | 135.6 ± 0.6 | 92.8 ± 4.5 | 74.5 ± 3.6 |
| KN FFA SBEβCD | 135.5 ± 0.4 | 40.8 ± 2.0 | 32.8 ± 1.6 |
| GR FFA SBEβCD | 135.2 ± 0.3 | 12.2 ± 0.6 | 9.8 ± 0.5 |
| COE FFA SBEβCD | 126.3 ± 0.7 | 39.5 ± 2.0 | 31.6 ± 1.6 |
| COL FFA SBEβCD | 135.1 ± 0.5 | 13.7 ± 0.7 | 10.4 ± 0.5 |
| PM FFA RAMEB | 134.2 ± 0.8 | 29.3 ± 1.5 | 23.5 ± 1.2 |
| KN FFA RAMEB | n.d. | n.d. | n.d. |
| GR FFA RAMEB | n.d. | n.d. | n.d. |
| COE FFA RAMEB | n.d. | n.d. | n.d. |
| COL FFA RAMEB | n.d. | n.d. | n.d. |
| Batch | Drug Content (%) | Mean Weight (mg) | Thickness (cm) | Hardness (Kg/cm2) | Disagg. Time (s) | PD 10 | PD 30 | DE 10 | DE 60 |
|---|---|---|---|---|---|---|---|---|---|
| IRT | |||||||||
| FFA | 100.2 ± 0.9 | 200 ± 0.7 | 0.10 ± 0.01 | 1.5 ± 0.5 | 100 | 15.9 ± 0.6 | 29.6 ± 1.1 | 7.9 ± 0.3 | 26.6 ± 1.0 |
| PM | 99.9 ± 0.8 | 433.8 ± 0.9 | 0.25 ± 0.02 | 5.0 ± 0.5 | 160 | 39.3 ± 1.5 | 68.5 ± 2.6 | 22.9 ± 0.8 | 58.6 ± 2.2 |
| GR | 98.9 ± 1.2 | 433.2 ± 0.5 | 0.25 ± 0.02 | 4.0 ± 0.3 | 180 | 62.9 ± 2.3 | 92.8 ± 3.5 | 38.9 ± 1.5 | 82.2 ± 3.1 |
| CT | |||||||||
| FFA | 99.8 ± 0.8 | 250.8 ± 0.9 | 0.20 ± 0.01 | 15.0 ± 0.5 | 300 | 7.9 ± 0.3 | 21.0 ± 0.7 | 3.8 ± 0.1 | 19.1 ± 0.7 |
| PM | 98.7 ± 1.3 | 487.2 ± 0.5 | 0.30 ± 0.02 | 17.0 ± 0.3 | 420 | 15.0 ± 0.5 | 36.3 ± 1.3 | 8.1 ± 0.3 | 32.9 ± 1.2 |
| GR | 99.2 ± 0.9 | 488 ± 0.4 | 0.30 ± 0.02 | 16.5 ± 0.5 | 450 | 45.0 ± 1.7 | 66.3 ± 2.5 | 23.2 ± 0.8 | 61.6 ± 2.3 |
| CT | True Density (g/cm3) | Coefficient of Variation CV (%) |
|---|---|---|
| FFA SIMS | 1.426 | 0.178 |
| FFA TCI | 1.379 | 0.046 |
| FFA SIMS-RAMEB PM | 1.431 | 0.050 |
| FFA TCI-RAMEB PM | 1.360 | 0.040 |
| FFA SIMS-RAMEB GR | 1.438 | 0.029 |
| FFA TCI-RAMEB GR | 1.439 | 0.039 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maestrelli, F.; Cirri, M.; De Luca, E.; Biagi, D.; Mura, P. Role of Cyclodextrins and Drug Solid State Properties on Flufenamic Acid Dissolution Performance from Tablets. Pharmaceutics 2022, 14, 284. https://doi.org/10.3390/pharmaceutics14020284
Maestrelli F, Cirri M, De Luca E, Biagi D, Mura P. Role of Cyclodextrins and Drug Solid State Properties on Flufenamic Acid Dissolution Performance from Tablets. Pharmaceutics. 2022; 14(2):284. https://doi.org/10.3390/pharmaceutics14020284
Chicago/Turabian StyleMaestrelli, Francesca, Marzia Cirri, Enrico De Luca, Diletta Biagi, and Paola Mura. 2022. "Role of Cyclodextrins and Drug Solid State Properties on Flufenamic Acid Dissolution Performance from Tablets" Pharmaceutics 14, no. 2: 284. https://doi.org/10.3390/pharmaceutics14020284