
Pharmacogenetic Dose Modeling Based on CYP2C19 Allelic Phenotypes

Abstract
:1. Introduction
2. Materials and Methods
2.1. Literature Search
2.2. Data Selection
2.2.1. Statistical Analysis
3. Results
3.1. Selection of Studies
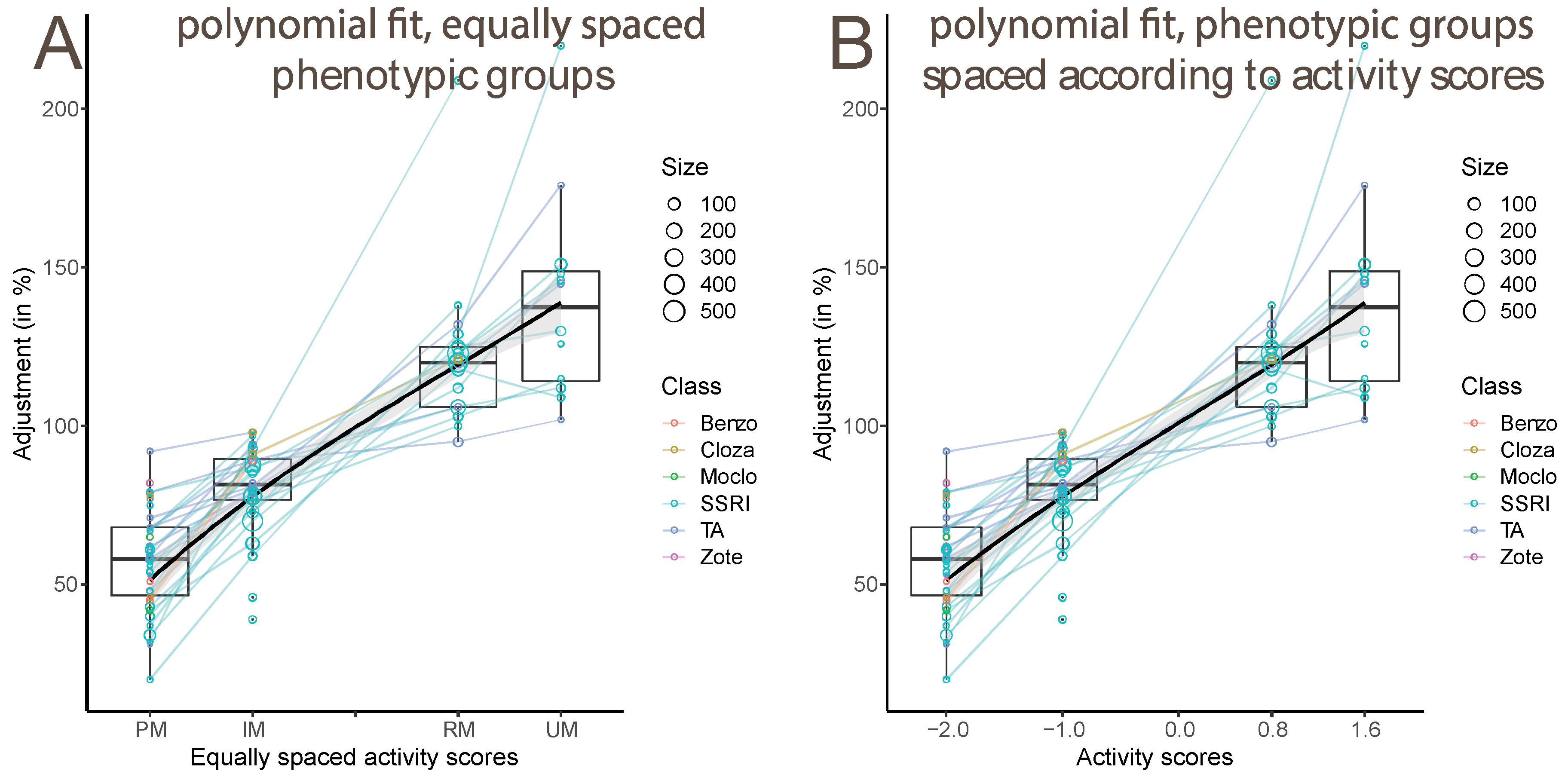
3.2. Estimate of CYP2C19 Activity Score from Dose Adjustments
3.3. Effects of Study Properties on Estimated Dose Adjustments
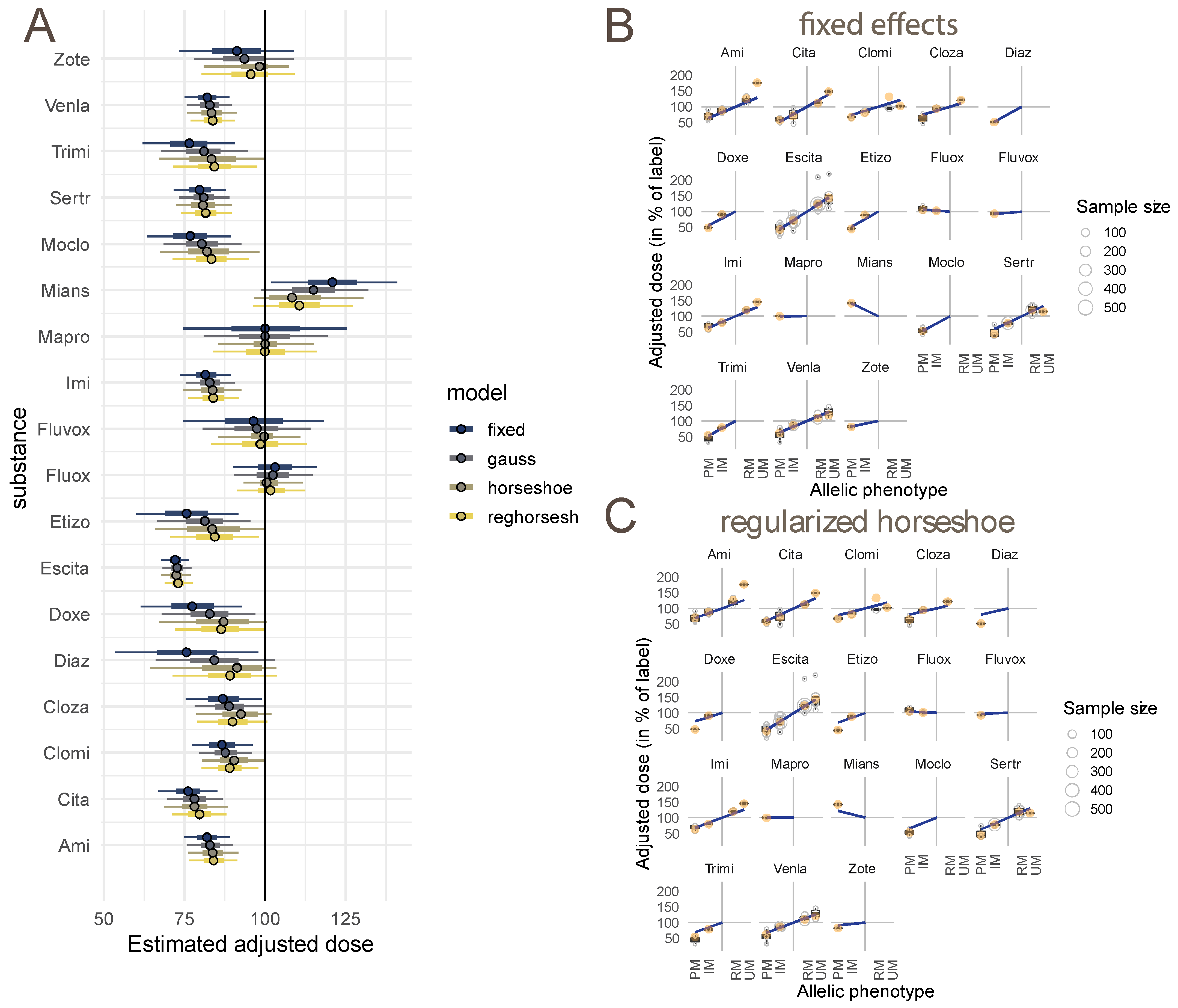
3.4. Modelled Dose Adjustments
4. Discussion
4.1. Consequences of Basing Estimates of Pharmacogenetic Effects on Estimates of Pathways: Comparison with Existing Models
4.2. Consequences of Modeling Differences between Study Methodologies
4.3. Statistical Methodology to Address Variability in the Amount of Data Available for Substances
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Regularized Horseshoe Model
/** This is the regularized horseshoe model but without the origin and added
dispersion components to weight for number of observations nobs in each
datapoint **/
data {
int N; //number of datapoints
int K; //number of shrunken predictors
int M; //total num predictors
vector<lower=1>[N] nobs; //number of observations within datapoints
//(coded as vector to avoid crash in rmarkdown)
vector[N] y;
matrix[N,M] preds; //hs shrunk first
real<lower=0> beta_scale; //prior scale non-shrunken coefs
real<lower=0> scale_global;
real<lower=1> nu_global;
real<lower=1> nu_local;
real<lower=0> slab_scale;
real<lower=0> slab_df;
//informative priors for residuals
real<lower=0> logsigmaloc;
real<lower=0> logsigmascale;
}
parameters {
//residual variance model
vector[K] z;
vector[M-K] b;
real logsigma_within; //variance within datapoints
real logsigma_betw; //variance between datapoints
//hs ridge model
real<lower=0> tau;
vector<lower=0>[K] lambda;
real<lower=0> caux;
}
transformed parameters {
//residual variance model
real sigma_within; //within
real sigma_betw; //between
vector[N] disp; //overall dispersion within + between
real mndisp;
//hs ridge
vector<lower=0>[K] lambda_tilde;
real<lower=0> c; //slab scale
//predictors
vector[M] beta; //regression coefficients
vector[N] f; //predictor
vector[N] fadjust; //predicted adjustment
//computations residual variance components model
sigma_within = exp(logsigma_within);
sigma_betw = exp(logsigma_betw);
disp = sqrt(sigma_within^2 ./ nobs + sigma_betw^2);
mndisp = sqrt(mean(sigma_within^2 ./ nobs + sigma_betw^2));
//hs ridge
c = slab_scale * sqrt(caux);
lambda_tilde = sqrt(c^2 * square(lambda) ./ (c^2 + tau^2 * square(lambda)));
for (i in 1:M) {
if (i <= K)
beta[i] = z[i] * lambda_tilde[i] * tau;
else
beta[i] = b[i-K];
}
f = preds * beta;
fadjust = preds[1:N,1:K] * beta[1:K];
}
model {
//linear model
z ~ normal(0, 1);
for (i in 1:M-K) b[i] ~ student_t(1, 0, beta_scale);
//model of the residual component variance
logsigma_within ~ normal(logsigmaloc, logsigmascale);
logsigma_betw ~ normal(logsigmaloc, logsigmascale);
// half -t priors for lambdas and tau , and inverse - gamma for c^2
tau ~ student_t(nu_global, 0, scale_global*mndisp);
lambda ~ student_t(nu_local, 0, 1);
caux ~ inv_gamma(0.5*slab_df, 0.5*slab_df);
//likelihood
y ~ normal(f, disp);
}
Appendix A.2. Non-Regularized Horseshoe
/** This is the horseshoe model (no regulrization) but without the origin and
added dispersion components to weight for number of observations nobs in
each datapoint **/
data {
int N; //number of datapoints
int K; //number of shrunken predictors
int M; //total num predictors
vector<lower=1>[N] nobs; //number of observations within datapoints
//(coded as vector to avoid crash in rmarkdown)
vector[N] y;
matrix[N,M] preds; //hs shrunk first
real<lower=0> beta_scale; //prior scale non-shrunken coefs
real<lower=0> scale_global;
real<lower=0> scale_local;
real<lower=1> nu_global;
real<lower=1> nu_local;
//informative priors for residuals
real<lower=0> logsigmaloc;
real<lower=0> logsigmascale;
}
parameters {
//residual variance model
vector[K] z;
vector[M-K] b;
real logsigma_within; //variance within datapoints
real logsigma_betw; //variance between datapoints
//hs ridge model
real<lower=0> tau;
vector<lower=0>[K] lambda;
}
transformed parameters {
//residual variance model
real sigma_within; //within
real sigma_betw; //between
vector[N] disp; //overall dispersion within + between
real mndisp;
//predictors
vector[M] beta; //regression coefficients
vector[N] f; //linear predictor
vector[N] fadjust; //predicted adjustment
//computations residual variance model
sigma_within = exp(logsigma_within);
sigma_betw = exp(logsigma_betw);
disp = sqrt(sigma_within^2 ./ nobs + sigma_betw^2);
mndisp = sqrt(mean(sigma_within^2 ./ nobs + sigma_betw^2));
//hs ridge
for (i in 1:M) {
if (i <= K)
beta[i] = z[i] * lambda[i] * tau;
else
beta[i] = b[i-K];
}
f = preds * beta;
fadjust = preds[1:N,1:K] * beta[1:K];
}
model {
//linear model
z ~ normal(0, 1);
for (i in 1:M-K) b[i] ~ student_t(1, 0, beta_scale);
//model of the residual component variance
logsigma_within ~ normal(logsigmaloc, logsigmascale);
logsigma_betw ~ normal(logsigmaloc, logsigmascale);
// half -t priors for lambdas and tau , and inverse - gamma for c^2
tau ~ student_t(nu_global, 0, scale_global*mndisp);
lambda ~ student_t(nu_local, 0, scale_local);
//likelihood
y ~ normal(f, disp);
}
Appendix A.3. Fixed Effects Model
/** This is a linear (fixed effects) model but without the origin and
added dispersion components to weight for number of observations nobs
in each datapoint **/
data {
int N; //number of datapoints
int K; //number of predictors for shrinkage
int M; //total num predictors, with confounders
vector<lower=1>[N] nobs; //number of observations within datapoints
//(coded as vector to avoid crash in rmarkdown)
vector[N] y;
matrix[N,M] preds; //first predictors are shrunk
//informative priors for residuals
real<lower=0> logsigmaloc;
real<lower=0> logsigmascale;
}
parameters {
vector[M] beta; //flat prior
//residual variance model
real logsigma_within; //variance within datapoints
real logsigma_betw; //variance between datapoints
}
transformed parameters {
//residual variance model
real sigma_within; //within
real sigma_betw; //between
vector[N] disp; //overall dispersion within + between
real mndisp;
//predictors
vector[N] f; //predictor
vector[N] fadjust;
//computations residual variance model
sigma_within = exp(logsigma_within);
sigma_betw = exp(logsigma_betw);
disp = sqrt(sigma_within^2 ./ nobs + sigma_betw^2);
mndisp = sqrt(mean(sigma_within^2 ./ nobs + sigma_betw^2));
//linear predictor
f = preds * beta;
fadjust = preds[1:N,1:K] * beta[1:K];
}
model {
//model of the residual component variance
logsigma_within ~ normal(logsigmaloc, logsigmascale);
logsigma_betw ~ normal(logsigmaloc, logsigmascale);
//likelihood
y ~ normal(f, disp);
}
References
- Stingl, J.; Viviani, R. Polymorphism in CYP2D6 and CYP2C19, members of the cytochrome P450 mixed-function oxidase system, in the metabolism of psychotropic drugs. J. Intern. Med. 2015, 277, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubiaur, P.; Saiz-Rodriguez, M.; Abad-Santos, F. “Pharmacogenetics of siponimod: A systematic review” by Diaz-Villamarin et al. Information is power. Biomed. Pharmacother. 2022, 157, 114003. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.K.; Sangkuhl, K.; Swen, J.J.; Ellingrod, V.L.; Muller, D.J.; Shimoda, K.; Bishop, J.R.; Kharasch, E.D.; Skaar, T.C.; Gaedigk, A.; et al. Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin. Pharmacol. Ther. 2017, 102, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, J.K.; Bishop, J.R.; Sangkuhl, K.; Muller, D.J.; Ji, Y.; Leckband, S.G.; Leeder, J.S.; Graham, R.L.; Chiulli, D.L.; Llerena, A.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 Genotypes and Dosing of Selective Serotonin Reuptake Inhibitors. Clin. Pharmacol. Ther. 2015, 98, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchheiner, J.; Brosen, K.; Dahl, M.L.; Gram, L.F.; Kasper, S.; Roots, I.; Sjoqvist, F.; Spina, E.; Brockmoller, J. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: A first step towards subpopulation-specific dosages. Acta Psychiatr. Scand. 2001, 104, 173–192. [Google Scholar] [CrossRef] [PubMed]
- Kirchheiner, J.; Nickchen, K.; Bauer, M.; Wong, M.L.; Licinio, J.; Roots, I.; Brockmoller, J. Pharmacogenetics of antidepressants and antipsychotics: The contribution of allelic variations to the phenotype of drug response. Mol. Psychiatry 2004, 9, 442–473. [Google Scholar] [CrossRef]
- Stingl, J.C.; Brockmoller, J.; Viviani, R. Genetic variability of drug-metabolizing enzymes: The dual impact on psychiatric therapy and regulation of brain function. Mol. Psychiatry 2013, 18, 273–287. [Google Scholar] [CrossRef]
- Piironen, J.; Vehtari, A. Sparsity information and regularization in the horseshoe and other shrinkage priors. Electron. J. Stat. 2017, 11, 5018–5051. [Google Scholar] [CrossRef]
- Shimoda, K.; Someya, T.; Yokono, A.; Morita, S.; Hirokane, G.; Takahashi, S.; Okawa, M. The Impact of CYP2C19 and CYP2D6 Genotypes on Metabolism of Amitriptyline in Japanese Psychiatric Patients. J. Clin. Psychopharmacol. 2002, 22, 371–378. [Google Scholar] [CrossRef]
- Steimer, W.; Zöpf, K.; von Amelunxen, S.; Pfeiffer, H.; Bachofer, J.; Popp, J.; Messner, B.; Kissling, W.; Leucht, S. Allele-specific change of concentration and functional gene dose for the prediction of steady-state serum concentrations of amitriptyline and nortriptyline in CYP2C19 and CYP2D6 extensive and intermediate metabolizers. Clin. Chem. 2004, 50, 1623–1633. [Google Scholar] [CrossRef]
- Jiang, Z.P.; Shu, Y.; Chen, X.P.; Huang, S.L.; Zhu, R.H.; Wang, W.; He, N.; Zhou, H.H. The role of CYP2C19 in amitriptyline N-demethylation in Chinese subjects. Eur. J. Clin. Pharmacol. 2002, 58, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Jonzier-Perey, M.; Koeb, L.; Küpfer, A.; Tinguely, D.; Schöpf, J. Amitriptyline pharmacokinetics and clinical response: II. Metabolic polymorphism assessed by hydroxylation of debrisoquine and mephenytoin. Int. Clin. Psychopharmacol. 1986, 1, 102–112. [Google Scholar] [CrossRef] [PubMed]
- de Vos, A.; van der Weide, J.; Loovers, H.M. Association between CYP2C19*17 and metabolism of amitriptyline, citalopram and clomipramine in Dutch hospitalized patients. Pharm. J. 2011, 11, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, S.; Park, S.; Lee, J.H.; Kim, Y.R.; Na, H.S.; Lim, H.S.; Choi, H.Y.; Hwang, I.Y.; Lee, J.G.; Park, Z.W.; et al. A Study on CYP2C19 and CYP2D6 Polymorphic Effects on Pharmacokinetics and Pharmacodynamics of Amitriptyline in Healthy Koreans. Clin. Transl. Sci. 2017, 10, 93–101. [Google Scholar] [CrossRef]
- Matthaei, J.; Brockmöller, J.; Steimer, W.; Pischa, K.; Leucht, S.; Kullmann, M.; Jensen, O.; Ouethy, T.; Tzvetkov, M.V.; Rafehi, M. Effects of Genetic Polymorphism in CYP2D6, CYP2C19, and the Organic Cation Transporter OCT1 on Amitriptyline Pharmacokinetics in Healthy Volunteers and Depressive Disorder Patients. Front. Pharmacol. 2021, 12, 688950. [Google Scholar] [CrossRef]
- Yokono, A.; Morita, S.; Someya, T.; Hirokane, G.; Okawa, M.; Shimoda, K. The effect of CYP2C19 and CYP2D6 genotypes on the metabolism of clomipramine in Japanese psychiatric patients. J. Clin. Psychopharmacol. 2001, 21, 549–555. [Google Scholar] [CrossRef]
- Nielsen, K.K.; Brøsen, K.; Hansen, M.G.; Gram, L.F. Single-dose kinetics of clomipramine: Relationship to the sparteine and S-mephenytoin oxidation polymorphisms. Clin. Pharmacol. Ther. 1994, 55, 518–527. [Google Scholar] [CrossRef]
- Kirchheiner, J.; Meineke, I.; Müller, G.; Roots, I.; Brockmöller, J. Contributions of CYP2D6, CYP2C9 and CYP2C19 to the biotransformation of E- and Z-doxepin in healthy volunteers. Pharmacogenetics 2002, 12, 571–580. [Google Scholar] [CrossRef]
- Skjelbo, E.; Brøsen, K.; Hallas, J.; Gram, L.F. The mephenytoin oxidation polymorphism is partially responsible for the N-demethylation of imipramine. Clin. Pharmacol. Ther. 1991, 49, 18–23. [Google Scholar] [CrossRef]
- Morinobu, S.; Tanaka, T.; Kawakatsu, S.; Totsuka, S.; Koyama, E.; Chiba, K.; Ishizaki, T.; Kubota, T. Effects of genetic defects in the CYP2C19 gene on the N-demethylation of imipramine, and clinical outcome of imipramine therapy. Psychiatry Clin. Neurosci. 1997, 51, 253–257. [Google Scholar] [CrossRef]
- Koyama, E.; Tanaka, T.; Chiba, K.; Kawakatsu, S.; Morinobu, S.; Totsuka, S.; Ishizaki, T. Steady-state plasma concentrations of imipramine and desipramine in relation to S-mephenytoin 4′-hydroxylation status in Japanese depressive patients. J. Clin. Psychopharmacol. 1996, 16, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Schenk, P.W.; van Vliet, M.; Mathot, R.A.; van Gelder, T.; Vulto, A.G.; van Fessem, M.A.; Verploegh-Van Rij, S.; Lindemans, J.; Bruijn, J.A.; van Schaik, R.H. The CYP2C19*17 genotype is associated with lower imipramine plasma concentrations in a large group of depressed patients. Pharm. J. 2010, 10, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Eap, C.B.; Bender, S.; Gastpar, M.; Fischer, W.; Haarmann, C.; Powell, K.; Jonzier-Perey, M.; Cochard, N.; Baumann, P. Steady state plasma levels of the enantiomers of trimipramine and of its metabolites in CYP2D6-, CYP2C19- and CYP3A4/5-phenotyped patients. Ther. Drug Monit. 2000, 22, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Kirchheiner, J.; Sasse, J.; Meineke, I.; Roots, I.; Brockmöller, J. Trimipramine pharmacokinetics after intravenous and oral administration in carriers of CYP2D6 genotypes predicting poor, extensive and ultrahigh activity. Pharmacogenetics 2003, 13, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Sindrup, S.H.; Brøsen, K.; Hansen, M.G.; Aaes-Jørgensen, T.; Overø, K.F.; Gram, L.F. Pharmacokinetics of citalopram in relation to the sparteine and the mephenytoin oxidation polymorphisms. Ther. Drug Monit. 1993, 15, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Fudio, S.; Borobia, A.M.; Piñana, E.; Ramírez, E.; Tabarés, B.; Guerra, P.; Carcas, A.; Frías, J. Evaluation of the influence of sex and CYP2C19 and CYP2D6 polymorphisms in the disposition of citalopram. Eur. J. Pharmacol. 2010, 626, 200–204. [Google Scholar] [CrossRef]
- Rudberg, I.; Hendset, M.; Uthus, L.H.; Molden, E.; Refsum, H. Heterozygous mutation in CYP2C19 significantly increases the concentration/dose ratio of racemic citalopram and escitalopram (S-citalopram). Ther. Drug Monit. 2006, 28, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Huezo-Diaz, P.; Perroud, N.; Spencer, E.P.; Smith, R.; Sim, S.; Virding, S.; Uher, R.; Gunasinghe, C.; Gray, J.; Campbell, D.; et al. CYP2C19 genotype predicts steady state escitalopram concentration in GENDEP. J. Psychopharmacol. 2012, 26, 398–407. [Google Scholar] [CrossRef]
- Noehr-Jensen, L.; Zwisler, S.T.; Larsen, F.; Sindrup, S.H.; Damkier, P.; Nielsen, F.; Brosen, K. Impact of CYP2C19 phenotypes on escitalopram metabolism and an evaluation of pupillometry as a serotonergic biomarker. Eur. J. Clin. Pharmacol. 2009, 65, 887–894. [Google Scholar] [CrossRef] [Green Version]
- Ohlsson Rosenborg, S.; Mwinyi, J.; Andersson, M.; Baldwin, R.M.; Pedersen, R.S.; Sim, S.C.; Bertilsson, L.; Ingelman-Sundberg, M.; Eliasson, E. Kinetics of omeprazole and escitalopram in relation to the CYP2C19*17 allele in healthy subjects. Eur. J. Clin. Pharmacol. 2008, 64, 1175–1179. [Google Scholar] [CrossRef]
- Rudberg, I.; Mohebi, B.; Hermann, M.; Refsum, H.; Molden, E. Impact of the ultrarapid CYP2C19*17 allele on serum concentration of escitalopram in psychiatric patients. Clin. Pharmacol. Ther. 2008, 83, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Faraj, P.; Hermansen, A.; Molden, E.; Hole, K. Identification of Escitalopram Metabolic Ratios as Potential Biomarkers for Predicting CYP2C19 Poor Metabolizers. Ther. Drug Monit. 2022, 44, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Marshe, V.S.; Magarbeh, L.; Frey, B.N.; Milev, R.V.; Soares, C.N.; Parikh, S.V.; Placenza, F.; Strother, S.C.; Hassel, S.; et al. Effects of CYP2C19 and CYP2D6 gene variants on escitalopram and aripiprazole treatment outcome and serum levels: Results from the CAN-BIND 1 study. Transl. Psychiatry 2022, 12, 366. [Google Scholar] [CrossRef] [PubMed]
- Bråten, L.S.; Haslemo, T.; Jukic, M.M.; Ivanov, M.; Ingelman-Sundberg, M.; Molden, E.; Kringen, M.K. A Novel CYP2C-Haplotype Associated with Ultrarapid Metabolism of Escitalopram. Clin. Pharmacol. Ther. 2021, 110, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Tsuchimine, S.; Ochi, S.; Tajiri, M.; Suzuki, Y.; Sugawara, N.; Inoue, Y.; Yasui-Furukori, N. Effects of Cytochrome P450 (CYP) 2C19 Genotypes on Steady-State Plasma Concentrations of Escitalopram and its Desmethyl Metabolite in Japanese Patients with Depression. Ther. Drug Monit. 2018, 40, 356–361. [Google Scholar] [CrossRef]
- Jukić, M.M.; Haslemo, T.; Molden, E.; Ingelman-Sundberg, M. Impact of CYP2C19 Genotype on Escitalopram Exposure and Therapeutic Failure: A Retrospective Study Based on 2087 Patients. Am. J. Psychiatry 2018, 175, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Uckun, Z.; Baskak, B.; Ozel-Kizil, E.T.; Ozdemir, H.; Devrimci Ozguven, H.; Suzen, H.S. The impact of CYP2C19 polymorphisms on citalopram metabolism in patients with major depressive disorder. J. Clin. Pharm. Ther. 2015, 40, 672–679. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Cheng, Z.N.; Huang, S.L.; Chen, X.P.; Ou-Yang, D.S.; Jiang, C.H.; Zhou, H.H. Effect of the CYP2C19 oxidation polymorphism on fluoxetine metabolism in Chinese healthy subjects. Br. J. Clin. Pharmacol. 2001, 52, 96–99. [Google Scholar] [CrossRef] [Green Version]
- Scordo, M.G.; Spina, E.; Dahl, M.L.; Gatti, G.; Perucca, E. Influence of CYP2C9, 2C19 and 2D6 genetic polymorphisms on the steady-state plasma concentrations of the enantiomers of fluoxetine and norfluoxetine. Basic Clin. Pharmacol. Toxicol. 2005, 97, 296–301. [Google Scholar] [CrossRef]
- Jan, M.W.; ZumBrunnen, T.L.; Kazmi, Y.R.; VanDenBerg, C.M.; Desai, H.D.; Weidler, D.J.; Flockhart, D.A. Pharmacokinetics of fluvoxamine in relation to CYP2C19 phenotype and genotype. Drug Metab. Drug Interact. 2002, 19, 1–11. [Google Scholar] [CrossRef]
- Wang, J.H.; Liu, Z.Q.; Wang, W.; Chen, X.P.; Shu, Y.; He, N.; Zhou, H.H. Pharmacokinetics of sertraline in relation to genetic polymorphism of CYP2C19. Clin. Pharmacol. Ther. 2001, 70, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Rudberg, I.; Hermann, M.; Refsum, H.; Molden, E. Serum concentrations of sertraline and N-desmethyl sertraline in relation to CYP2C19 genotype in psychiatric patients. Eur. J. Clin. Pharmacol. 2008, 64, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Bråten, L.S.; Haslemo, T.; Jukic, M.M.; Ingelman-Sundberg, M.; Molden, E.; Kringen, M.K. Impact of CYP2C19 genotype on sertraline exposure in 1200 Scandinavian patients. Neuropsychopharmacology 2020, 45, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Saiz-Rodríguez, M.; Belmonte, C.; Román, M.; Ochoa, D.; Koller, D.; Talegón, M.; Ovejero-Benito, M.C.; López-Rodríguez, R.; Cabaleiro, T.; Abad-Santos, F. Effect of Polymorphisms on the Pharmacokinetics, Pharmacodynamics and Safety of Sertraline in Healthy Volunteers. Basic Clin. Pharmacol. Toxicol. 2018, 122, 501–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabris, G.; Baumann, P.; Janzier-perey, M.; Bosshart, P.; Woggon, B.; Küpfer, A. N-methylation of maprotiline in debrisoquine/mephenytoin-phenotyped depressive patients. Biochem. Pharmacol. 1985, 34, 409–410. [Google Scholar] [CrossRef]
- Dahl, M.L.; Tybring, G.; Elwin, C.E.; Alm, C.; Andreasson, K.; Gyllenpalm, M.; Bertilsson, L. Stereoselective disposition of mianserin is related to debrisoquin hydroxylation polymorphism. Clin. Pharmacol. Ther. 1994, 56, 176–183. [Google Scholar] [CrossRef]
- Gram, L.F.; Guentert, T.W.; Grange, S.; Vistisen, K.; Brøsen, K. Moclobemide, a substrate of CYP2C19 and an inhibitor of CYP2C19, CYP2D6, and CYP1A2: A panel study. Clin. Pharmacol. Ther. 1995, 57, 670–677. [Google Scholar] [CrossRef]
- McAlpine, D.E.; Biernacka, J.M.; Mrazek, D.A.; O’Kane, D.J.; Stevens, S.R.; Langman, L.J.; Courson, V.L.; Bhagia, J.; Moyer, T.P. Effect of cytochrome P450 enzyme polymorphisms on pharmacokinetics of venlafaxine. Ther. Drug Monit. 2011, 33, 14–20. [Google Scholar] [CrossRef]
- Scherf-Clavel, M.; Weber, H.; Wurst, C.; Stonawski, S.; Hommers, L.; Unterecker, S.; Wolf, C.; Domschke, K.; Rost, N.; Brückl, T.; et al. Effects of Pharmacokinetic Gene Variation on Therapeutic Drug Levels and Antidepressant Treatment Response. Pharmacopsychiatry 2022, 55, 246–254. [Google Scholar] [CrossRef]
- Kringen, M.K.; Bråten, L.S.; Haslemo, T.; Molden, E. The Influence of Combined CYP2D6 and CYP2C19 Genotypes on Venlafaxine and O-Desmethylvenlafaxine Concentrations in a Large Patient Cohort. J. Clin. Psychopharmacol. 2020, 40, 137–144. [Google Scholar] [CrossRef]
- Dahl, M.L.; Llerena, A.; Bondesson, U.; Lindström, L.; Bertilsson, L. Disposition of clozapine in man: Lack of association with debrisoquine and S-mephenytoin hydroxylation polymorphisms. Br. J. Clin. Pharmacol. 1994, 37, 71–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaquenoud Sirot, E.; Knezevic, B.; Morena, G.P.; Harenberg, S.; Oneda, B.; Crettol, S.; Ansermot, N.; Baumann, P.; Eap, C.B. ABCB1 and cytochrome P450 polymorphisms: Clinical pharmacogenetics of clozapine. J. Clin. Psychopharmacol. 2009, 29, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Ammar, H.; Chadli, Z.; Mhalla, A.; Khouadja, S.; Hannachi, I.; Alshaikheid, M.; Slama, A.; Ben Fredj, N.; Ben Fadhel, N.; Ben Romdhane, H.; et al. Clinical and genetic influencing factors on clozapine pharmacokinetics in Tunisian schizophrenic patients. Pharm. J. 2021, 21, 551–558. [Google Scholar] [CrossRef]
- Kondo, T.; Tanaka, O.; Otani, K.; Mihara, K.; Tokinaga, N.; Kaneko, S.; Chiba, K.; Ishizaki, T. Possible inhibitory effect of diazepam on the metabolism of zotepine, an antipsychotic drug. Psychopharmacology 1996, 127, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, T.; Yasui-Furukori, N.; Suzuki, A.; Inoue, Y.; Tateishi, T.; Otani, K. Pharmacokinetics and pharmacodynamics of etizolam are influenced by polymorphic CYP2C19 activity. Eur. J. Clin. Pharmacol. 2005, 61, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Bertilsson, L.; Henthorn, T.K.; Sanz, E.; Tybring, G.; Säwe, J.; Villén, T. Importance of genetic factors in the regulation of diazepam metabolism: Relationship to S-mephenytoin, but not debrisoquin, hydroxylation phenotype. Clin. Pharmacol. Ther. 1989, 45, 348–355. [Google Scholar] [CrossRef]
- Carvalho, C.M.; Polson, N.G.; Scott, J.G. The horseshoe estimator for sparse signals. Biometrika 2010, 97, 465–480. [Google Scholar] [CrossRef] [Green Version]
- George, E.I.; McCulloch, R.E. Variable selection via Gibbs sampling. J. Am. Stat. Assoc. 1993, 123, 881–889. [Google Scholar] [CrossRef]
- Stan Development Team. Stan Modeling Language User Guide and Reference Manual; Version 2.26.1; Stan Development Team: Washington, DC, USA, 2017. [Google Scholar]
- Sim, S.C.; Risinger, C.; Dahl, M.L.; Aklillu, E.; Christensen, M.; Bertilsson, L.; Ingelman-Sundberg, M. A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin. Pharmacol. Ther. 2006, 79, 103–113. [Google Scholar] [CrossRef]
- Milosavljevic, F.; Bukvic, N.; Pavlovic, Z.; Miljevic, C.; Pesic, V.; Molden, E.; Ingelman-Sundberg, M.; Leucht, S.; Jukic, M.M. Association of CYP2C19 and CYP2D6 Poor and Intermediate Metabolizer Status with Antidepressant and Antipsychotic Exposure: A Systematic Review and Meta-analysis. JAMA Psychiatry 2021, 78, 270–280. [Google Scholar] [CrossRef]
- Magliocco, G.; Desmeules, J.; Matthey, A.; Quiros-Guerrero, L.M.; Bararpour, N.; Joye, T.; Marcourt, L.; Queiroz, E.F.; Wolfender, J.L.; Gloor, Y.; et al. Metabolomics reveals biomarkers in human urine and plasma to predict cytochrome P450 2D6 (CYP2D6) activity. Br. J. Pharmacol. 2021, 178, 4708–4725. [Google Scholar] [CrossRef]
- Stingl Kirchheiner, J.C.; Brockmoller, J. Why, when, and how should pharmacogenetics be applied in clinical studies? Current and future approaches to study designs. Clin. Pharmacol. Ther. 2011, 89, 198–209. [Google Scholar] [CrossRef]
- Li-Wan-Po, A.; Girard, T.; Farndon, P.; Cooley, C.; Lithgow, J. Pharmacogenetics of CYP2C19: Functional and clinical implications of a new variant CYP2C19*17. Br. J. Clin. Pharmacol. 2010, 69, 222–230. [Google Scholar] [CrossRef]
- Robinson, G.K. That BLUP is a Good Thing: The Estimation of Random Effects. Stat. Sci. 1991, 6, 15–32. [Google Scholar]



{kind=link}
{kind=link}
| Drug | Parameter | Study Subjects/Dosing | Dose | Identification of Metabolizer Groups | Reference |
|---|---|---|---|---|---|
| Amitriptyline | Css | Patients, MD | 25–225 mg | CYP2C19*2, CYP2C19*3 | [9] |
| Amitriptyline | Css | Patients, MD | 150 mg | CYP2C19*2 | [10] |
| Amitriptyline | AUC | Healthy, SD | 50 mg | CYP2C19*2 | [11] |
| Amitriptyline | Css | Patients, MD | 150 mg | Phenotyping | [12] |
| Amitriptyline | Css | Patients, MD | 100–150 mg | CYP2C19*2, CYP2C19*17 | [13] |
| Amitriptyline | AUC, MR | Patients, MD | 25 mg | CYP2C19*2, CYP2C19*3 | [14] |
| Amitriptyline | AUC | Healthy, SD | 25 mg | CYP2C19*2, CYP2C19*17 | [15] |
| Clomipramine | Css | Patients, MD | 10–250 mg | CYP2C19*2, CYP2C19*3 | [16] |
| Clomipramine | 1/CL | Healthy, SD | 100 mg | Phenotyping | [17] |
| Clomipramine | Css | Patients, MD | 25–300 mg | CYP2C19*2, CYP2C19*17 | [13] |
| Doxepine | 1/CL | Healthy, SD | 75 mg | CYP2C19*2 | [18] |
| Imipramine | 1/CL | Healthy, SD | 100 mg | Phenotyping | [19] |
| Imipramine | Css | Patients, MD | 70 mg | Phenotyping | [20] |
| Imipramine | Css | Patients, MD | 50 mg | Phenotyping | [21] |
| Imipramine | Css | Patients, MD | dose adjusted | CYP2C19*2 | [22] |
| Trimipramine | Css | Patients, MD | 350 mg | Phenotyping | [23] |
| Trimipramine | 1/CL | Healthy, SD | 75 mg | CYP2C19*2 | [24] |
| Citalopram | AUC | Healthy, MD | 40 mg | Phenotyping | [25] |
| Citalopram | Css | Patients, MD | 10–60 mg | CYP2C19*2, CYP2C19*17 | [13] |
| Citalopram | CL | Healthy, SD | 20 mg | CYP2C19*2 | [26] |
| Citalopram | Css | Patients, MD | 35 ± 20/34 ± 17 | CYP2C19*2 | [27] |
| Escitalopram | Css | Patients, MD | 4.8–7.4 mg | CYP2C19*2, CYP2C19*17 | [28] |
| Escitalopram | CL | Healthy, MD | 10 mg | Phenotyping | [29] |
| Escitalopram | AUC | Healthy, SD | 5 mg | CYP2C19*2, CYP2C19*17 | [30] |
| Escitalopram | Css | Patients, MD | 16 ± 5/21 ± 13 | CYP2C19*2 | [31] |
| Escitalopram | Css | Patients, MD | 20 ± 9/22 ± 10 | CYP2C19*2 | [27] |
| Escitalopram | Css, MR | Patients, MD | 5–40 mg | CYP2C19*2, CYP2C19*17 | [32] |
| Escitalopram | Css | Patients, MD | 10–20 | CYP2C19*2, CYP2C19*17 | [33] |
| Escitalopram | Css | Patients, MD | 10 mg | CYP2C19*2, CYP2C19*17 | [34] |
| Escitalopram | Css | Patients, MD | 5–20 mg | CYP2C19*2, *3, CYP2C19*17 | [35] |
| Escitalopram | Css | Patients, MD | 12.6–18.1 mg | CYP2C19*2, CYP2C19*17 | [36] |
| Escitalopram | MR | Patients, MD | 10–80 mg | CYP2C19*2, CYP2C19*17 | [37] |
| Fluoxetine | AUC, CL | Healthy, SD | 40 mg | CYP2C19*2, CYP2C19*17 | [38] |
| Fluoxetine | Css | Patients, MD | 10–60 mg | CYP2C19*2, CYP2C19*17 | [39] |
| Fluvoxamine | AUC | Healthy, SD | 100 mg | Phenotyping | [40] |
| Sertraline | AUC, CL | Healthy, SD | 100 mg | Phenotyping | [41] |
| Sertraline | CL | Patients, MD | dose–adjusted | CYP2C19*2, CYP2C19*17 | [42] |
| Sertraline | Css | Patients, MD | dose–adjusted | CYP2C19*2, CYP2C19*17 | [43] |
| Sertraline | AUC | Healthy, SD | CYP2C19*2, CYP2C19*17 | [44] | |
| Maprotiline | Css | Patients, MD | 150 mg | Phenotyping | [45] |
| Mianserin | AUC | Healthy, SD | 30 mg | Phenotyping | [46] |
| Moclobemide | 1/Cl | Healthy, MD | 300 mg; 600 mg | Phenotyping | [47] |
| Venlafaxine | Css | Patients, MD | <225 mg; >225 mg | CYP2C19*2, CYP2C19*17 | [48] |
| Venlafaxine | Css | Patients, MD | dose–corrected TDM | CYP2C19*2, CYP2C19*17 | [49] |
| Venlafaxine | Css | Patients, MD | 150 mg | CYP2C19*2, CYP2C19*17 | [50] |
| Clozapine | AUC | Healthy, SD | 10 mg | Phenotyping | [51] |
| Clozapine | Css | Patients, MD | 250 (25–800) mg | CYP2C19*2, CYP2C19*17 | [52] |
| Clozapine | Css | Patients, MD | 433 mg | CYP2C19*2, CYP2C19*17 | [53] |
| Zotepine | 1/CL | Healthy, SD | 25 mg | Phenotyping | [54] |
| Etizolam | AUC | Healthy, SD | 1 mg | CYP2C19*2, *3 | [55] |
| Diazepam | CL | Healthy, SD | 10 mg | Phenotyping | [56] |
| Estimate | EM | IM | PM | RM | UM | CYP2C19*17 + |
|---|---|---|---|---|---|---|
| Median | 0 | −1 | −1.96 | 0.70 | 1.75 | 0.79 |
| 5% lower | 0 | −1 | 2.91 | 0.26 | 1.13 | 0.48 |
| 95% upper | 0 | −1 | −1.42 | 1.28 | 2.76 | 1.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stingl, J.C.; Radermacher, J.; Wozniak, J.; Viviani, R. Pharmacogenetic Dose Modeling Based on CYP2C19 Allelic Phenotypes. Pharmaceutics 2022, 14, 2833. https://doi.org/10.3390/pharmaceutics14122833
Stingl JC, Radermacher J, Wozniak J, Viviani R. Pharmacogenetic Dose Modeling Based on CYP2C19 Allelic Phenotypes. Pharmaceutics. 2022; 14(12):2833. https://doi.org/10.3390/pharmaceutics14122833
Chicago/Turabian StyleStingl, Julia Carolin, Jason Radermacher, Justyna Wozniak, and Roberto Viviani. 2022. "Pharmacogenetic Dose Modeling Based on CYP2C19 Allelic Phenotypes" Pharmaceutics 14, no. 12: 2833. https://doi.org/10.3390/pharmaceutics14122833