1. Introduction
The World Health Organization (WHO) reported that there were an estimated 241 million cases of malaria in 2020, and estimated malaria deaths of 627,000, about 80% of which were children under the age of 5. The incidence of malaria is increasing in some regions. In 2020, about half of the world’s population was at risk of malaria. Most cases and deaths occur in sub-Saharan Africa. However, the other WHO regions also report significant numbers of cases and deaths [
1].
Plasmodium sporozoites enter the hepatocytes of susceptible human beings when an infected mosquito bites. The sporozoites injected from the salivary glands of mosquitos are moved on into the blood circulation through which they travel to the liver and invade the hepatic parenchymal cells where sporozoites develop into liver-stage schizonts.
Plasmodium (
P)
falciparum and
P. vivax are the two most common parasites causing human malaria. In human primary hepatocytes, the development of
P. falciparum takes 7 days, whereas
P. vivax completes development after 9 days [
2]. Some parasites such as
P. vivax and
P. oval can have their sporozoites differentiated into both liver-stage schizonts and hypnozoites. The schizonts then burst to release uninucleate merozoites. Hepatocyte-derived merozoites invade erythrocytes and enter a few-step cycle to develop into schizonts-containing asexual cells. Certain host erythrocytes develop into gametocytes associated with sexual cells. The hypnozoites are the dormant parasites in the liver that can cause a relapse of malaria months or even years later. The liver stage provides an ideal target for antimalarial intervention to block malarial progression before the manifestation of clinical symptoms caused by the rupture of schizonts from erythrocytes [
3,
4].
Antimalarial medications can target the
Plasmodium liver stage and/or blood stage. Artemisinin, acting on blood-stage
Plasmodium, is the first-line drug for treating severe malaria caused by
P. falciparum. It must be combined with a different class of antimalarials to avoid drug resistance. Artemisinin has not been recommended for malarial prophylaxis. Chloroquine is prescribed for the treatment of
P. vivax malaria and is no longer recommended for treating
P. falciparum malaria. Chloroquine does not have activity against liver stage
Plasmodium. It is prescribed for the intra-erythrocytic stages to prevent malaria only in a few areas with chloroquine-sensitive malaria [
5]. It is taken by travelers one to two weeks before departure and continued for four weeks after return. Mefloquine and doxycycline are also blood schizontocidal agents. Mefloquine is taken at least one week before departure and continues for 4 weeks after return. It has more side effects than chloroquine. Doxycycline is taken daily for travelers visiting chloroquine-resistant or mefloquine-resistant areas. These medications require frequent intake and good compliance of travelers with physicians’ prescriptions.
There are several antimalarials that target the
Plasmodium liver stage, including atovaquone/proguanil (Malarone), primaquine, and tafenoquine. Malarone is two combined drugs with synergistic actions that can be taken as a causal and suppressive prophylactic agent, but it is not effective in suppressing hypnozoites. Therefore, it cannot be used as a radical cure for malarial relapse caused by
P. vivax or
P. oval. It is, due to its cost, generally limited to travelers from industrialized countries. Primaquine and its derivative tafenoquine, a recently approved new drug, are the only drugs effective against hypnozoites. However, both primaquine and tafenoquine can cause fatal hemolysis in people with G6PD deficiency and should never be prescribed as prophylaxis to anyone with G6PD deficiency or unknown G6PD status [
5]. Tafenoquine has two dosage forms, one (Arakoda) used for the chemoprophylaxis of malaria in individuals traveling in endemic areas, and the other (Krintafel) as a radical cure of
P. vivax in patients ≥ 16 years. Krintafel should only be used in combination with chloroquine, not other antimalarials, against acute
P. vivax infection.
As a measure of prophylaxis, vulnerable populations take antimalarial drugs at regular intervals to prevent acute malaria attacks. Currently, all drugs used for prophylaxis require frequent administration. There have been efforts to make controlled-release formulations of antimalarials to treat experimental cerebral malaria or blood-stage
Plasmodium so that the activity of active pharmaceutical ingredients (API) can last for a longer period [
6,
7,
8]. However, there have been no reports of such slow-release formulations of liver-stage drugs for long-term causal chemoprophylaxis of malaria.
In addition to chemoprophylaxis, another important aspect of controlling malaria involves monoclonal antibodies applicable to fight against malaria. Recently, a long-acting antimalarial monoclonal antibody CIS43LS was assessed in a two-part, phase 1 clinical trial [
9]. Participants underwent controlled human malaria infection in which they were exposed to mosquitoes carrying
P. falciparum sporozoites 4 to 36 weeks after the administration of CIS43LS. The monoclonal antibody provided causal prophylaxis, which targets
Plasmodium infection at the intrahepatic stage, and the results were encouraging. The only vaccine currently available is RTS, S/AS01, which prevents infection, maturation, and multiplication within the liver but has limited efficacy [
10]. It has been used in vulnerable populations rather than travelers via the Implementation Program [
5]. Although numerous studies are ongoing to identify novel candidate antigens, there are presently no vaccines available for travelers.
One alternative that has been in progress is the use of long-acting injectable formulations of API [
11]. Long-acting injectable formulations are depot delivery systems intended for prolonged/sustained drug release over a prolonged period ranging from a few days to months. Better patient compliance may be achieved through the parenteral route of administration over the oral route for the treatment of many chronic and life-threatening diseases. Parenteral depots may also reduce the relapse rate of disease and the maintenance phase of therapy, hence improving efficacy and treatment adherence. Examples of long-acting injectables reaching commercial viability so far are the depot products mostly for antipsychotic, substance abuse, and hormonal therapy drugs [
12].
A great deal of research aiming at application has been conducted by using biodegradable polymers. Poly (lactic-co-glycolic acid) (PLGA) is the most widely used polymeric biomaterial in controlled drug delivery systems including small chemical and large biopharmaceutical molecules [
13,
14]. However, the use of PLGA for the purpose of prolonged release may not meet the requirements of every agent in various cases, such as controlling encapsulated drug release rates and/or formulation instability. Drug delivery systems with high efficiency and sustained-release characteristics involve drug load and release profiles [
11].
Decoquinate (DQ) is a potent antimalarial agent targeting multistage
Plasmodium parasites [
15,
16]. As with atovaquone, its mechanism of action is to inhibit the cytochrome bc1 complex in the mitochondria. However, there was little cross-resistance between the two agents because they block different sites [
17]. As an anti-coccidiostat used for years, DQ has an excellent safety profile [
18]. DQ can effectively inhibit chloroquine-sensitive
P. falciparum as well as chloroquine- or multidrug-resistant
P. falciparum in infected human erythrocytes and protect mice from severe malaria [
19,
20]. To solve the problem of water insolubility and to improve bioavailability, DQ has been made into nanoparticles without compromising its antimalarial potency as an oral dosage form targeting liver stage
Plasmodium infection. The nanoparticle formulations of DQ are very potent at inhibiting
Plasmodium berghei in vitro at the liver stage (IC50 < 0.5 nM) and highly efficacious (1–3 mg/kg) in protecting mice from liver stage malaria [
20,
21]. The in vivo efficacy of DQ against liver stage
Plasmodium might also be associated with its pharmacokinetic feature in that there is high enrichment of DQ in the liver whether the nanoparticles are orally or intravenously administered [
22].
The lipophilic property may make DQ an appropriate candidate as a sustained-release formulation for the chemoprophylaxis of malaria. Recently, Li et al. created formulations of DQ in oil-based carriers to provide extended efficacy through continuous drug release over a prolonged period from a single injection of the depot to effectively protect mice from
Plasmodium infection [
23]. Interestingly, the microparticle formulation provided a 2.2-fold longer drug exposure and a 3–4 times longer prophylactic effect than the nanoparticle formulation. Although there have been experiments performed in animals to prove that DQ has excellent antimalarial activity, it has not become a candidate for clinical trials to evaluate its safety and efficacy in preventing or treating malaria. Long-acting injectable atovaquone nanomedicines for malaria prophylaxis have also been reported [
24], but nanoparticles may not be as sustainable as microparticles for drug release [
23].
Hot melt extrusion (HME) technology has been applied for pharmaceutical formulations to improve the dissolution rate and bioavailability of poorly water-soluble drugs and to make a sustained drug release of API over an extended period [
25]. HME has been shown to be a suitable, highly efficient method to prepare the oral dosage form of DQ targeting liver stage
Plasmodium infection [
20]. In this study, various preparations of DQ as a prophylaxis of malaria were made by different methods including HME, and evaluated in vitro and in mice. A lipids-based DQ depot was stored in the muscle and provided a long-lasting release of antimalarial activity against
Plasmodium infection.
2. Materials and Methods
Decoquinate (Pharmaceutical grade, purity ≥ 98.0%) was purchased from Genebest Pharmaceutical Co. Ltd. (Zhejiang, China). Decoquinate standard (Cat. No. 1165408, LOT F0G036) was bought from USP Rockville, MD, USA. Mono or diglycerides of medium-chain fatty acids (Capmul® MCM) were from IMCD (Abitec, Old Dixie Hwy, Miami, Florida, USA). Cholesterol was from Beijing Dingguo Changsheng Biotechnology Co., Ltd. (Beijing, China). Pharma 11 HME machine was manufactured by ThermoFisher Scientific (Thermo Electron (Karlsruhe) GmbH, Germany). PLGA 75/25 (75% polylactic acid and 25% polyglycolic acid; Mw, 72,000) and PLGA 50/50 (50% polylactic acid and 50% polyglycolic acid; Mw, 18,000) were bought from Jinan Daigang Biomaterial Co., Ltd. (Shandong, China). N-methyl-2-pyrrolidone (NMP) was from Sigma-Aldrich ((Shanghai) Trading Co., Ltd.). All other chemicals and solvents were of analytical grade. For DQ quantitation, HPLC grade acetonitrile, ethanol, and methanol were obtained from Tianjin Kemiou Chemical Reagent Company (Tianjin, China).
C57 mice were purchased from Southern Medical University, China, and employed to assess formulation efficacy, drug concentration in the blood (pharmacokinetics), and formulation toxicity. Animal studies were conducted in strict accordance with the relevant animal testing regulations (Animal Management Regulations, PR China, issued on 14 November 1988, revised version on 1 March 2007). All experimental protocols involving animals were carefully examined and approved on 4 September 2017 by the Animal Care and Ethical Committee of CAS Lamvac Biotech Co., Guangzhou, China (ZKLH/BL 0904.2017-P10, 4 September 2017).
2.1. Preparation of Sustained-Release Formulations of Decoquinate
Sustained-release formulation of DQ (SRFD) was prepared by different methods but the method eventually selected was HME. The percentage of each component was accounted for by weight. Cholesterol (40–90%) was fully mixed with DQ (10–40%). Then mono and diglycerides of medium-chain fatty acids (Capmul MCM) (5–20%) were added to the above mixture. The thoroughly mixed ingredients were loaded into the HME machine. The temperature for the HME process ranged from 70 °C to 100 °C, and the screw speed was 50 rpm. The extrudate was either made as a solid stick of a given length for subcutaneous (SQ) injection or pulverized into a powder and then suspended in normal saline (pH5.5) for intramuscular injection.
The PLGA 75/25 or 50/50 (10–90%) was formulated into the SRFD either via HME or by being dissolved in NMP. For the HME process, the temperature ranged from 80 °C to 135 °C, and the screw speed ranged from 50 rpm to 100 rpm. For the non-HME process, PLGA 75/25 (50%) was first dissolved in NMP, and then DQ (50%) was added to the PLGA solution. The mixture was stirred for 2 h at 200 rpm at room temperature. The mixed composition was then solidified and evaluated for the in vitro release of DQ. PLGA 75/25 (50%) alone could not be formulated with DQ via the HME process. Therefore it was combined with cholesterol and processed via HME. As a result, it was incorporated into a formulation as a minor carrier of DQ (
Table 1, F4, and 5). As shown in
Table 1, formulations 1 to 8 were processed by the HME method, whereas the emulsion method was used to prepare F9 by adding cholesterol and DQ to 5 mL triglycerides (Captex 300), heating at 80 °C degrees, and stirring to form DQ emulsion. F10 was prepared by simply dissolving DQ in organic solvent NMP. Particle sizes of SRFDs were measured by Beckman LS 13320 particle analyzer. Some evaluated SRFD preparations are shown in
Table 1.
2.2. X-ray Diffraction Analysis
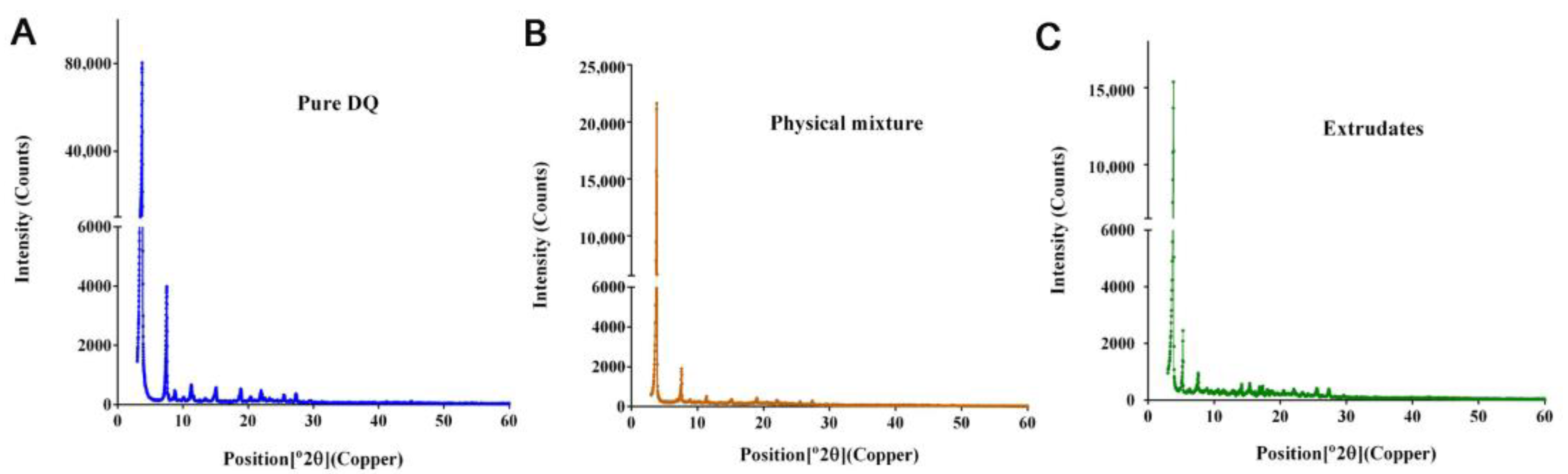
SRFD was analyzed by X-ray diffractometer (XRD, Empyrean) at the Test Center of Sun Yat-sen University in Guangzhou, China. XRD is used for observing the crystal structures of chemical compounds. The shining of an X-ray on a crystal generates a diffraction pattern characteristic of the chemical structure. A powder form of the material can be analyzed. Crystalline substance generates their characteristic sharp peaks. To obtain information on the physical property of SRFD, XRD patterns of HME product, the physical mixture of the formulation prior to the HME process, and pure DQ were observed by the standard operation rules. The targeting material used was copper with CuK α radiation, and settings were regularly used conditions.
2.3. Drug Release Test
One gram of SRFD sample was placed in a 10 mL tube filled with 6 mL phosphate-buffered saline (PBS, pH 7.4), with shaking speed at 100 rpm under 37 °C. DQ released from tested SRFDs was measured by HPLC 1260 analysis (Agilent) and compared to the total amount of the drug from each sample used in the tests. Samples were taken periodically and continuously for 52 days. The cumulative release rates of DQ against time duration were calculated by dividing the amount of the released drug measured at each time point over the total amount of the drug placed. The detected drug each time (day) is the amount of all drugs released and accumulated from preceding times.
2.4. Drug Load Determination
Depot formulations were suspended in saline or PBS. The suspensions were mixed with 3 times the volume of ethanol in Eppendorf tubes and vortexed for 2 min. After standing for half an hour, the extraction was centrifuged for 5 min at 5500× g (Eppendorf Centrifuge 5810R, Hamburg, Germany). The ethanolic supernatants were taken for DQ quantification by a high-performance liquid chromatography (HPLC) system (Agilent 1260, Santa Clara, CA, USA) (AGILENT TECHNOLOGIES (CHINA) CO. LTD, Chaoyang, Beijing) and the drug load of each SRFD was calculated. Experiments were performed in triplicate for all formulations.
2.5. Decoquinate Quantitation
DQ was quantitated by HPLC using a Diamonsil C18 column (250 × 4.6 mm, 5 µm; Beijing Dikma Technologies, Inc. China). All the in vitro and in vivo experiments including drug concentrations measured in the blood, in vivo efficacy studies, drug load determination, and drug release tests, were based on HPLC quantitation to calculate the amount of drug used or determined. For instance, in an efficacy experiment for a mouse weighing 25 g at a dose of DQ 200 mg/kg, an SRFD used was determined by HPLC to contain 5 mg DQ/12.5 mg total weight (40% drug load).
The wavelength of ultraviolet detection in HPLC was 260 nm. The isocratic mobile phase was absolute ethanol/H2O (80:20, 0.1% formic acid in both solutions). The column temperature was set to 35 °C, flow rate 1 mL/min, and injection volume 20 μL. All chemicals and solvents were of analytical grade. HPLC grade acetonitrile, ethanol, and methanol were obtained from Tianjin Kemiou Chemical Reagent Company (Tianjin, China).
2.6. Decoquinate Depot for Plasmodium Prophylaxis in Mice
C57 mice were employed to assess the chemoprophylaxis efficacy of SRFD in Plasmodium infection. Animal studies were conducted in strict accordance with the relevant animal testing regulations (National Regulations of Laboratory Animal Management, PR China, amended on 31 October 2017).
Seven-week-old C57 mice, one in each cage, were kept for at least 7 days after arrival before starting the experiments. Room temperature was 18–26 °C, the relative humidity was 34–68%, and the light and darkness alternately cycled for 12 h. Standard feeding food was provided before and during the study. Sporozoites (SPZ) of P. berghei ANKA expressing firefly luciferase were isolated from the salivary glands of laboratory-reared female Anopheles stephensi mosquitoes. The mosquitoes were maintained at 26 °C for 17 to 22 days after feeding on Plasmodium parasites from infected Kunming mice. Salivary glands separated from malaria-infected mosquitoes were manually broken to obtain SPZ. For assessment of the prophylactic efficacy of SRFD targeting hepatic Plasmodium infection, 50,000 SPZ per mouse were intravenously injected via the tail vein.
HME extrudates were dosed either in solid sticks given to mice by SC to each side of hind legs by using trocar needles or in liquid form administered to mice by IM after being pulverized into a powder and suspended in saline. The liquid form for administration by intramuscular injection was made by suspending the HME extrudates in normal saline with pH 5.5. The solid stick with a diameter of about 2 mm was shaped by HME output and the length of the stick was determined by weight based on the drug content equivalent to the amount of drug administered. The liquid suspension was given to both hind legs to cut the total volume in half to alleviate discomfort.
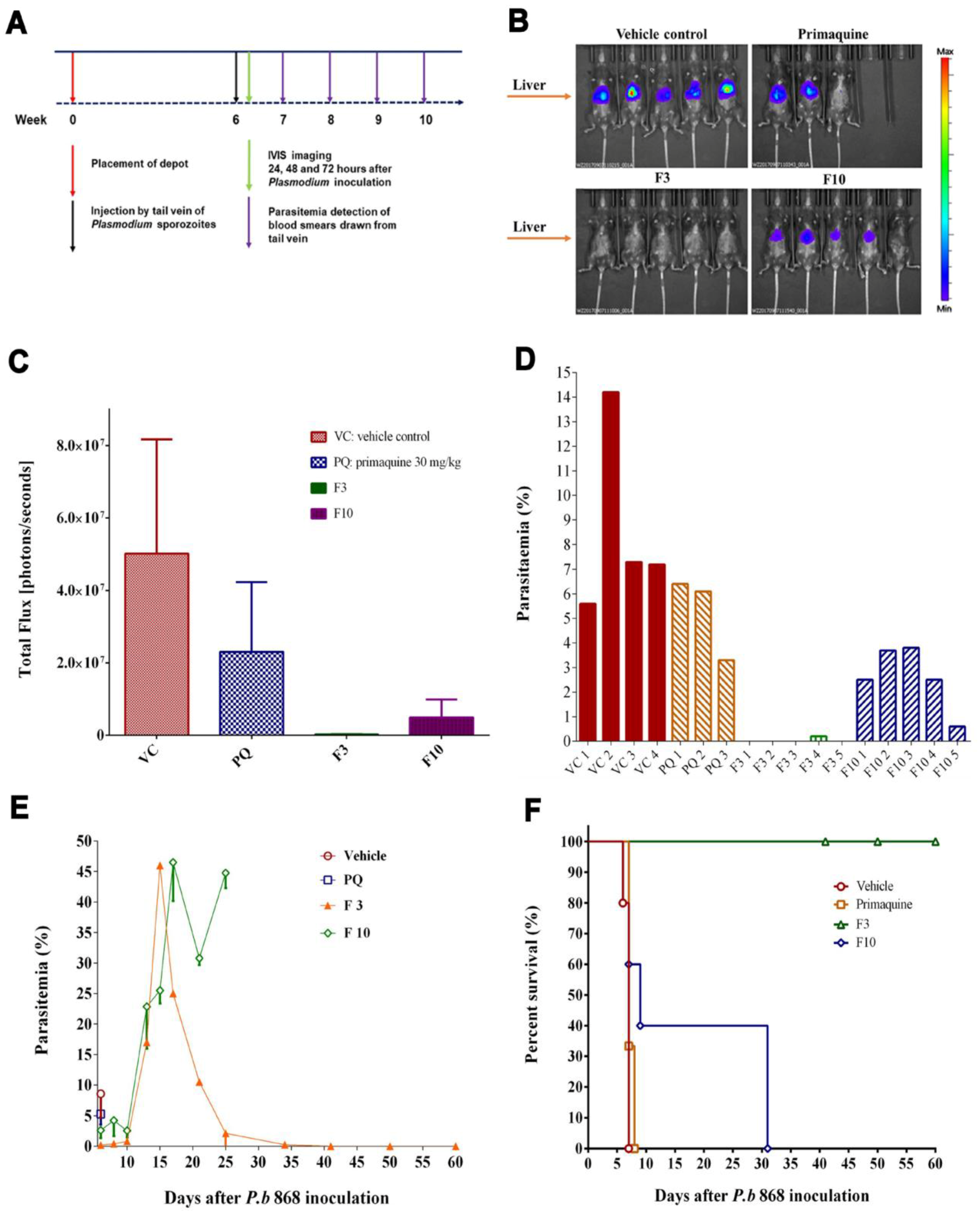
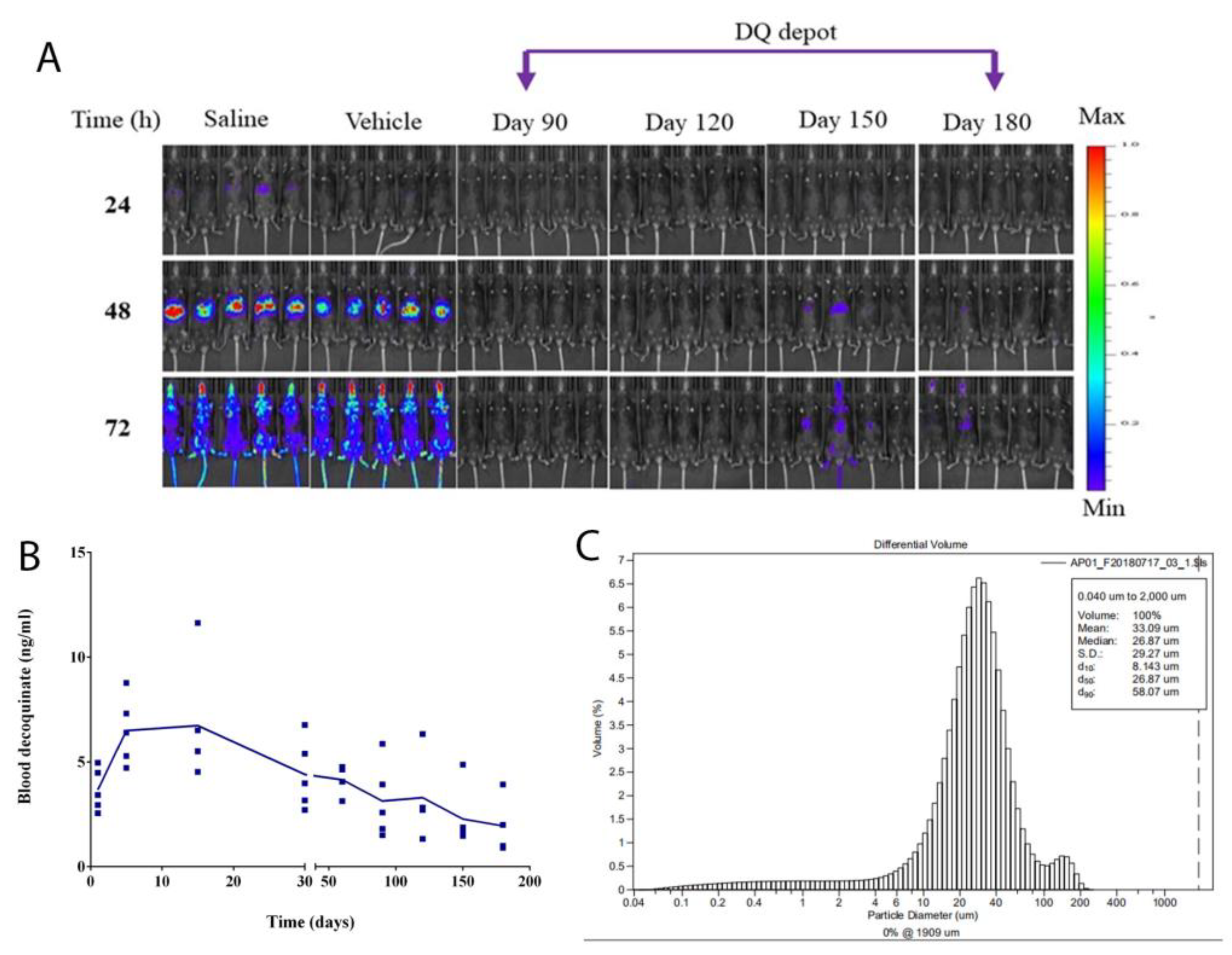
Liver stage infection of P. berghei was monitored by using the in vivo imaging system (IVIS). The images were obtained at 24 h, 48 h, and 72 h post SPZ inoculation. P. berghei infecting mice reside in the liver for intrahepatic development in the first 48 h and then spread from the liver to the whole body after 48 h. This time course is reflected in images characteristic of bioluminescent signals in different regions of the body. The signals indicative of the location of parasites expressing firefly luciferase were generated by giving the mice 150 mg/kg luciferin (Gold Biotechnology, St. Louis, MO) intraperitoneally. Meanwhile, fluorescent flux counts were also acquired to represent quantitative data. The counts measured were bioluminescent photons emitted from whole bodies or regions of intensity (ROI). The ROI settings of the Living Image® 4.0 software reflect the luminescent intensity. A 3-D bioluminescent imaging tomography was performed with the software to obtain sequential images with filters ranging from 580 to 660 nm.
Blood-stage Plasmodium infection was monitored by microscopic examination of either a thin film or a thick film. Blood smears were stained with 3% Giemsa for 20 min. Parasite density was estimated by counting parasites against RBCs on the thin film:
Parasite density per μL = Number of parasites counted × RBC count per μL ÷ Number of RBCs counted. Infected red blood cells per 10,000 red blood cells were counted in thin films. In the cases where parasitemia was undetectable on the thin film, the thick film was prepared to count parasites against WBCs:
Parasite density per μL = Number of parasites counted × WBC count per μL ÷ Number of WBCs counted. Infected red blood cells per 100 WBCs were obtained. Survival rates were calculated from the date of Plasmodium SPZ inoculation to the endpoint of the efficacy of each SRFD, to determine the time lengths of SRFD protection in various dosages.
2.7. Measurement of Decoquinate Concentrations in the Blood
Propranolol 5% in purity, purchased from Sigma, St. Louis, MO. USA, was used as an internal standard. DQ standards were made by dissolving it in ethanol at 50 μg/mL as stock solution, which was then diluted to a series of 10 gradient concentrations from 0.5 ng/mL to 500 ng/mL. Each standard (20 μL) was mixed with blank blood 100 μL in a 1.5 mL tube by vortex for 3 min. Protein precipitation solution 400 μL (ethanol /acetonitrile (1:1) containing propranolol 1000 ng/mL) was added and then mixed again for 5 min. After standing at room temperature for 8 h, the tubes were vortexed for 5 min and centrifuged at 16,000× g for 60 min at 4 °C. The supernatant 100 μL was transferred to each well of the 96-well plate and DQ was measured by liquid chromatography–mass spectrometry (LC-MS) system (Applied Biosystems-SCIEX model for API 3000 mass spectrometer).
To measure DQ in the blood, the whole blood sample (100 μL) was mixed with 20 μL ethanol for 3 min. Precipitation solution (400 μL) was added and mixed for another 5 min. The rest of the procedure was the same as that for standard curve preparation. Data were analyzed by one-way analysis of variance (ANOVA) and expressed as mean ± SD.
The same species of mice (C57) as those used in efficacy experiments was dosed with F2 at DQ 200 mg/kg by IM. After the drug administration, blood samples were collected from mice by cardiac puncture on different days (day 1, day 5, day 15, day 30, day 60, day 90, day 120, day 150, and day 180) with a group of 5 mice for each time point. Due to the limited sample size, DQ was measured by LC-MS, only one point per animal sample. Original data points were plotted to represent the drug level in the blood on that day.
4. Discussion
Intragastric administration of nanoparticle formulations of DQ to mice provided an excellent efficacy of causal prophylaxis targeting the intrahepatic stage by killing parasites before they enter the blood [
20]. Only when DQ is made into nanoparticles and sufficiently absorbed [
20,
21] can DQ reach and act on the sites where parasites are. Alternatively, DQ may be made in sustained-release formulations suitable for parenteral administration, long-term storage, and slow release of API, which can provide causal prophylaxis of
Plasmodium infection [
23]. This approach may help those who often forget to take the prophylactic medications as prescribed by physicians. The creation of SRFD could uniquely provide long-term prophylaxis for malaria.
PLGA has been widely used for sustained drug delivery [
24]. Formulations made of PLGA as a carrier provide effective treatment of various medical conditions such as psychiatric diseases [
12]. However, PLGA and DQ may not be compatible in forming a formulation of releasing API sufficient for the chemoprophylaxis of malarial infection. DQ is practically water-insoluble and extremely lipophilic. It is not an easy task of finding an appropriate carrier for DQ in sustained-release formulation. Originally, cholesterol was used as a control excipient for the SRFD. Surprisingly, it became a major carrier of DQ in SRFD, which provided excellent protection for mice inoculated with
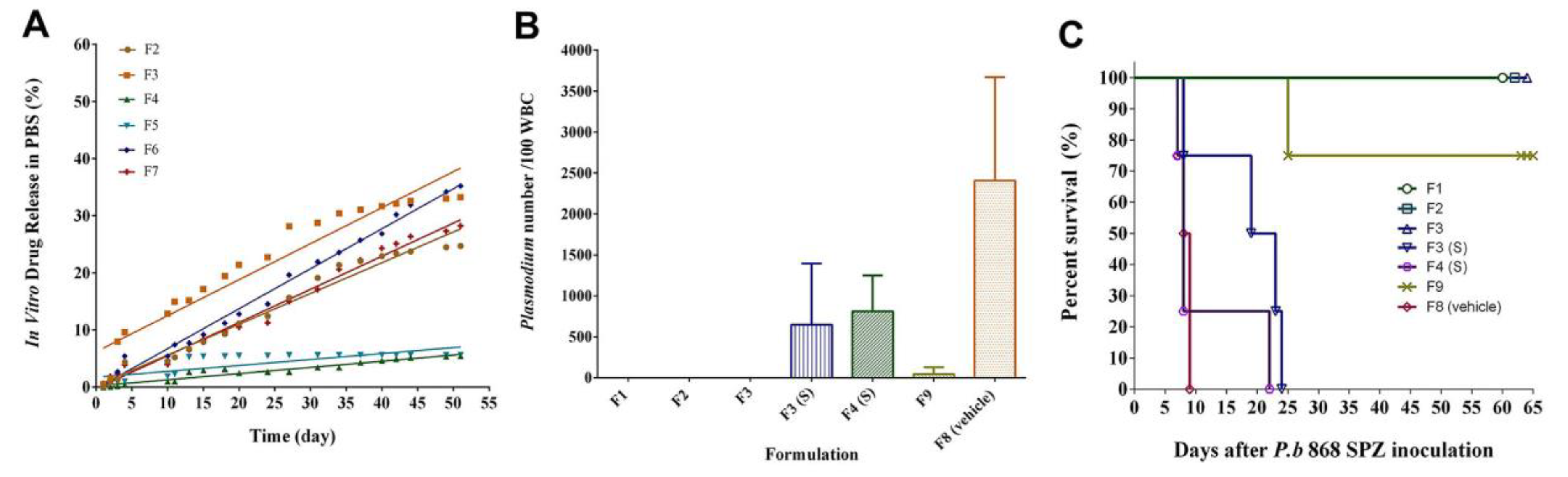
Plasmodium berghei SPZ. Furthermore, the SRFD prepared by HME such as F1, F2, and F3 released DQ (
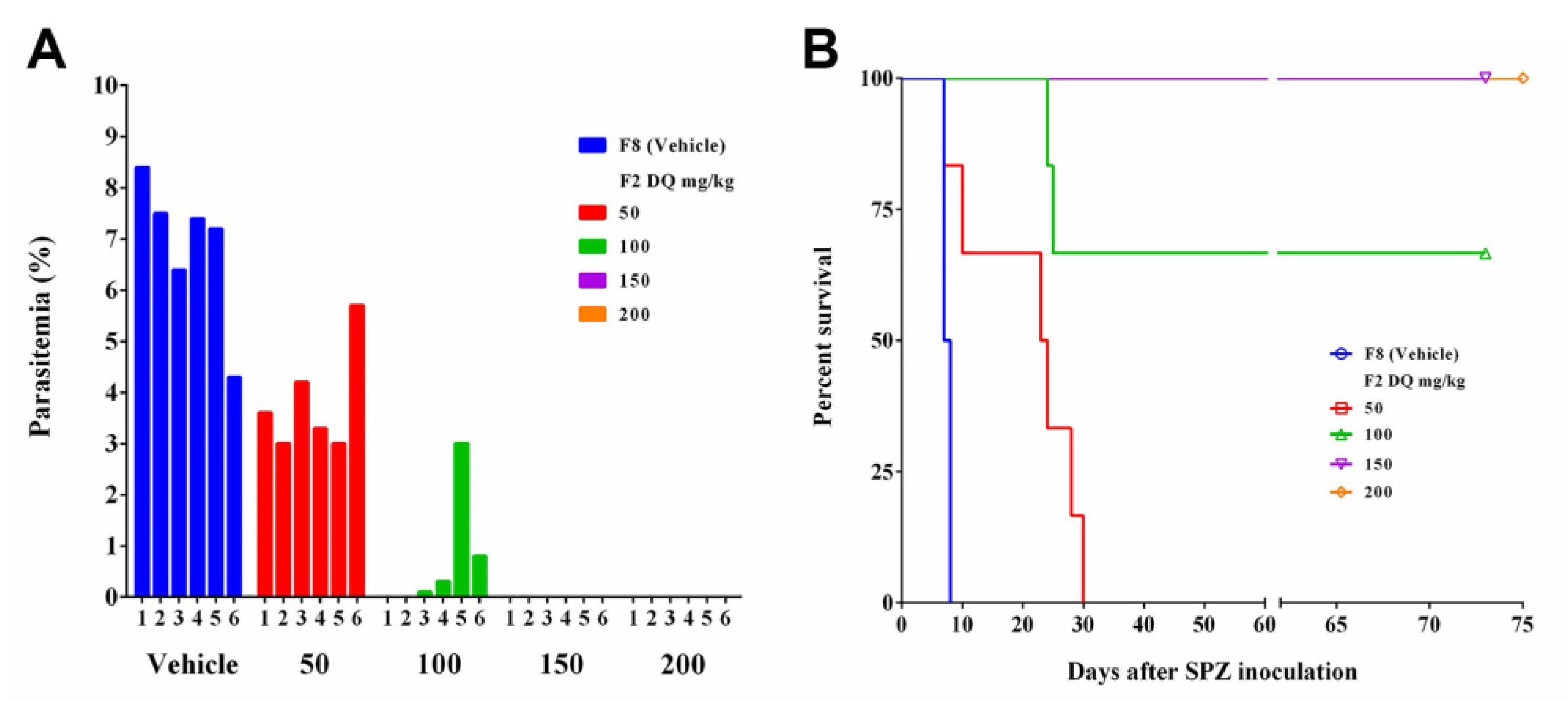
Figure 1A) sufficient for prophylactic efficacy (
Figure 1B,C), better than F9, which was prepared by the emulsion method. Formulations containing PLGA, whether prepared by HME (F4) or by dissolving the components in organic solvent NMP (F10), did not release sufficient DQ to fight against
Plasmodium SPZ invasion (
Figure 1 and
Figure 2). Other polymers (PLA, PGA, PCL) were also assessed and proved to be unsuccessful in making an SRFD for the long-term prophylaxis of malaria (data not shown).
Mild conditions such as the range of temperatures set well below the melting points of each component of the SRFD were chosen in the HME process so that active as well as inactive ingredients could be kept intact without structural changes in the molecules. Under such HME settings, the drug particles of the preparations could be obtained in the micrometer range (
Figure 3A) to meet the requirement of the sustained release of DQ. There have been reports that large drug particles as the intramuscular reservoir, not fine particles such as nanometer particles, favor long-lasting drug exposure [
23,
26,
27,
28]. Large particles derived from different drug compositions can lead to the delayed release of API to last for many days in vitro (
Figure 1A) and for as long as 180 days in vivo (
Figure 4B). In contrast, nanoparticles of DQ formulation prepared by HME had drug dissolution in a short period of time from 1 to 9 h [
20].
The SRFD was optimized through the screening of various formulations as shown in
Figure 1 and
Figure 2. The HME method is highly efficient for large-scale preparation and may have improved product quality by enhancing the mixing of all components and controlling the temperature as well as the whole processing [
29]. XRD analyses showed crystalline peaks of a small fraction of DQ from HME extrudates, suggesting that DQ had not been completely miscible with other formulation components. This might negatively impact the release of DQ and compromise long-lasting prophylaxis. However, compared to the XDR pattern of pure DQ (
Figure 3A), there was still more than 80% of loaded DQ (
Figure 3C) complexed with the formulation components, which ensured the sustained-release of DQ over a long-extended period and protected the infected mice. Further assessment, as shown in
Figure 4 and
Figure 5, demonstrated that the SRFD at a dose of DQ 150–200 mg/kg could release DQ from the depot formulation (F2) stored in the muscle over 120 days, which had been enough for mice protection from
Plasmodium SPZ infection. The scenario is different from previous studies in that nanoparticle formulations of DQ were prepared for the oral route, which significantly improved the bioavailability and efficacy of DQ [
20,
21]. The effective dose of DQ by the oral route daily was much lower than that of the SRFD: only 3 to 5 mg/kg for causal prophylaxis of malaria in mice. For long-term prophylaxis of malaria, a relatively large dose of DQ was used to sustain the drug release for as long as possible.
The study conducted by Li et. al. used peanut oil as a vehicle to prepare a microparticle-sized depot formulation of DQ and high-pressure homogenization to reduce the particle size to less than 15 µM [
20]. SRFDs prepared by HME in this study were not further processed by additional physical manipulation such as ultrasonication and high-pressure homogenization. The HME extrudates could be homogeneously suspended and easily handled in an aqueous solution. Partial protection (67%) (
Figure 5B) for mice inoculated with
P. berghei SPZ one month after SRFD placement was 100 mg/kg, which is equivalent to the effective dose of 120 mg/kg from the study of assessing oily suspended DQ for the long-term (8 weeks) chemoprophylaxis of malaria [
23].
In pharmaceutical applications, cholesterol is commonly used in liposomal preparation [
30,
31]. Cholesterol was mixed well with DQ in constructing SRFDs via HME. It might also help release DQ from the muscle depot. The SRFD with cholesterol as an inactive ingredient prevented
Plasmodium infection much better than expected. The use of cholesterol in the sustainable formulation of drug release has not been reported previously and it is the first time that it has been used for forming antimalarial agents, specifically DQ, in the depot formulation.
Cholesterol is a natural molecule that constitutes a structural component of about 30% of all animal cell membranes [
32]. The molecule is essential to maintaining both the integrity and fluidity of the membrane structure. In the clinic, as compared to the cholesterol content of the HDL particles, abnormal cholesterol levels such as higher blood LDL, especially higher LDL particle concentrations and smaller LDL particle sizes, contribute to myocardial infarction (heart attack), stroke, and peripheral vascular disease. There has been very little correlation between the amount of cholesterol used in the drug formulation and an increased risk of cardiovascular disease. Given the fact that the pool size of cholesterol in the human body is so large and its metabolic routes are so diverse, the amount of cholesterol used should be safe. On the other hand, since cholesterol has the structure of the tetracyclic ring in a trans conformation, it makes all but the side chain of cholesterol rigid and planar. In this structural role, cholesterol also reduces the permeability of the plasma membrane to neutral solutes, hydrogen ions, and sodium ions. The nature of cholesterol might make it suitable and valuable for its role within the drug formulation to control the release of antimalarial agents as well as other therapeutic drugs. Compared to polymers commercially available, cholesterol is cheap and readily available.
The implant made by melt exclusion traditionally needs to be placed surgically or by using a large-diameter needle [
33]. As for SRFD F3, it was injected in the forms of an aqueous suspension and a solid stick. The aqueous suspension could be handled more easily and provided a better outcome than the solid form. DQ was injected intramuscularly and resided in situ for slow and constant release. Cholesterol and medium-chain fatty acids could favor the release of DQ slowly but efficiently for the inhibition of
Plasmodium parasites. These lipids could be absorbed by surrounding tissues faster than DQ, and then metabolized or excreted. Therefore, there was no need to remove any injected materials. The DQ of the SRFD residing in the muscle tissue could be diffused to local surrounding tissues or could enter the bloodstream and then spread to the sites where the killing event of
Plasmodium parasites occurred. The target sites were the peripheral tissues, especially the liver and the skin. The dose of DQ ranging from 100 to 150 mg/kg in the SRFD was fully effective in preventing
Plasmodium infection. To translate this dose of DQ for human beings, API 10 to 15 mg/kg or 1050 mg may be needed for injection.
Primaquine and tafenoquine are used as primary and terminal prophylaxes for malaria [
34]. Potential dose-dependent acute hemolytic anemia in individuals with glucose-6-phosphate dehydrogenase deficiency (G6PDd), and often the unavailability of testing for G6PDd, limits their use [
35]. The combination of atovaquone–proguanil has been used as a causal and suppressive prophylactic agent [
36] to delay the emergency. These medications prescribed as a prophylactic agent of malaria require frequent administration. DQ has multistage antimalarial activity and might also be used as a causal prophylactic and suppressive agent but with little cross-resistance with atovaquone [
37]. A DQ depot may provide an excellent alternative to current prophylactic remedies to overcome the incompliance of patients. DQ has been proven to be safe as a veterinary drug for many years [
38]. No eminent adverse effects were found when the nanoparticle formulations were given to mice either by intragastric administration or by intravenous injection [
21,
22]. Adverse effects or toxic signs were not observed in mice when SRFD suspension was placed by IM. Here, the SRFD was produced by HME, a process that is readily scalable and highly efficient. Nevertheless, preclinical studies should not begin until the full toxicity results of decoquinate in the blood upon being dosed intramuscularly are obtained and understood and the maximum concentration without causing health complications in long-term administration has been determined. The model of large animals such as monkeys and dogs may be necessary to further assess the long-acting efficacy of DQ and the adverse effects of drug exposure in the blood or key tissues such as the liver and the skin.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}