
Repurposing Antimalarial Pyronaridine as a DNA Repair Inhibitor to Exploit the Full Potential of Gold-Nanoparticle-Mediated Radiation Response
 , , ,
, , ,
Abstract
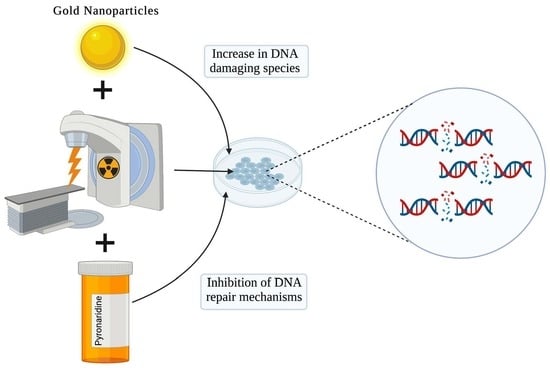
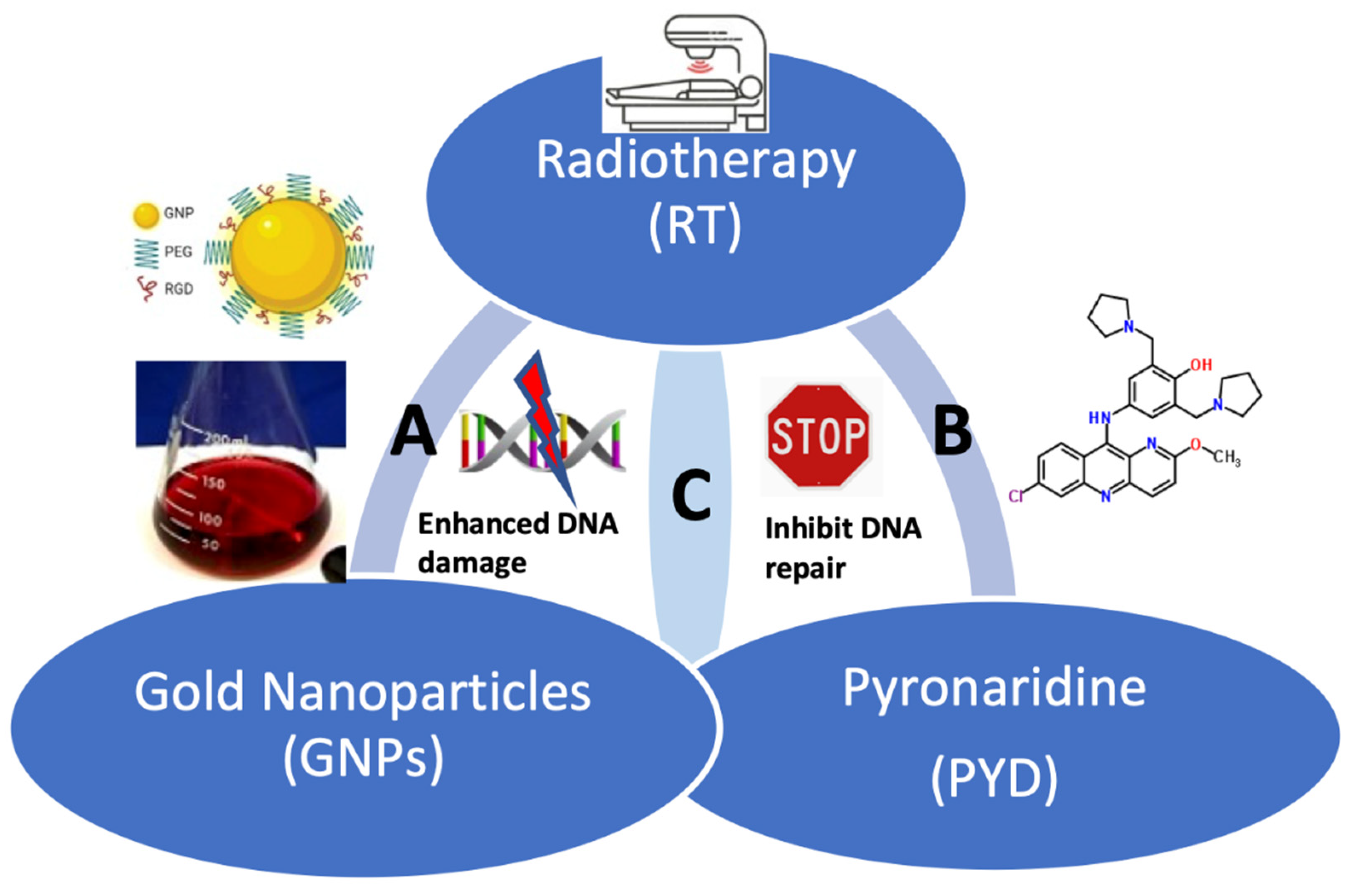
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- (1)
- Does PYD inhibit ERCC1-XPF?
- (2)
- Does PYD affect GNP uptake and transportation?
- (3)
- Can we achieve therapeutic benefit using nanomolar concentrations of PYD as opposed to micromolar concentrations?
- (4)
- Is the enhancement of cellular DNA damage of this triple combination, PYD/GNP/RT, significant compared to GNP/RT?
2. Materials and Methods
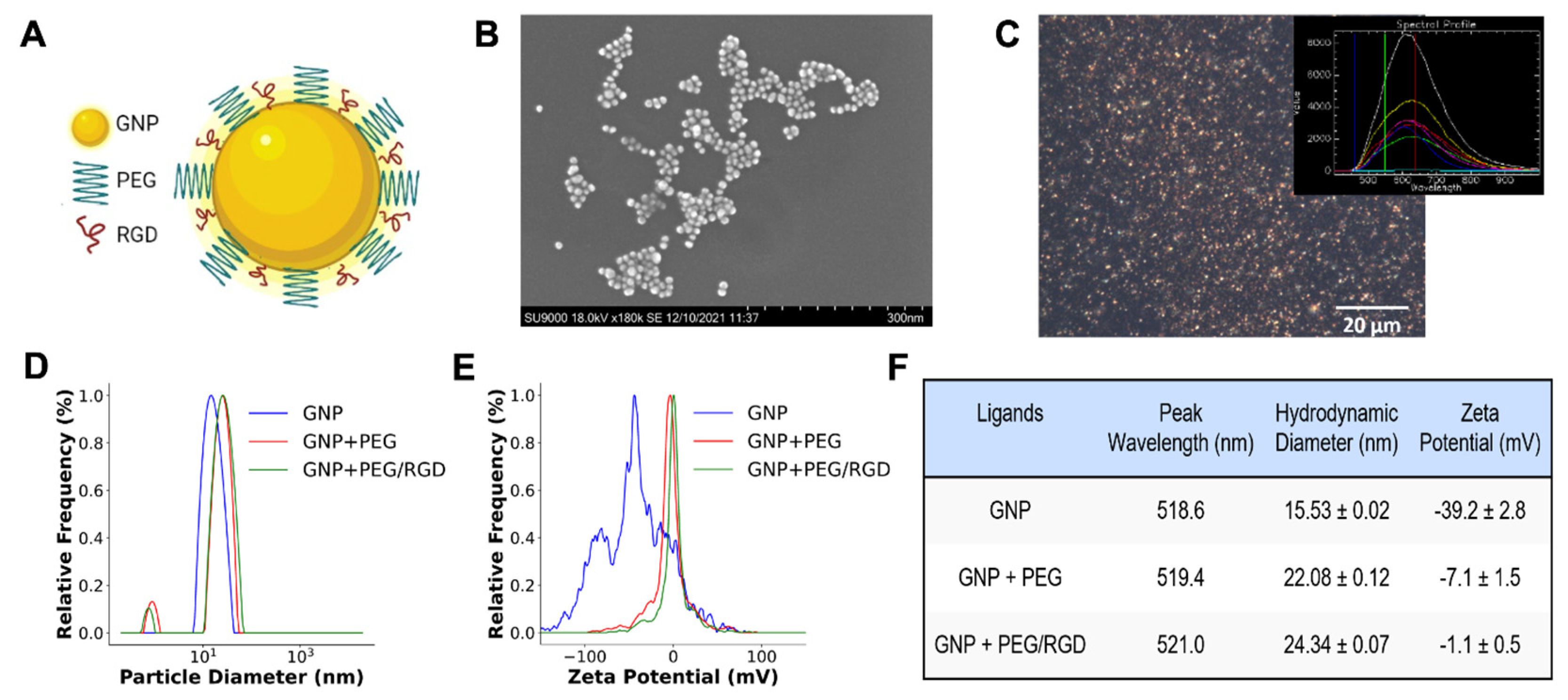
2.1. Gold Nanoparticle Synthesis, Functionalization, and Characterization
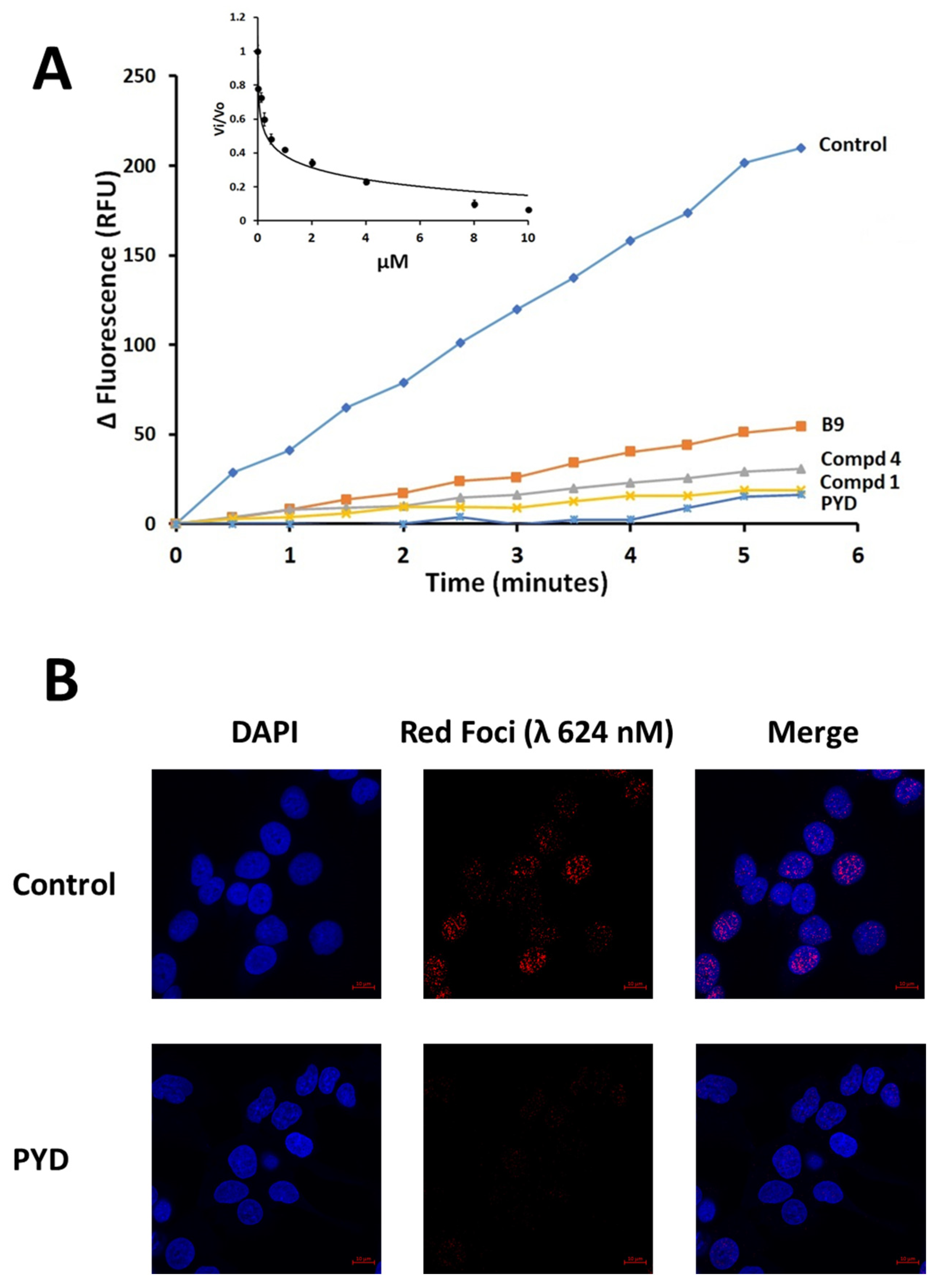
2.2. ERCC1-XPF Incision Assay
2.3. Cell and Culture Conditions
2.4. Proximity Ligation Assay
2.5. Live Cell Imaging
2.6. DNA DSB Assay/Dark-Field Imaging and HIS
2.7. Cellular Uptake of Gold Nanoparticles
2.8. Proliferation Assay
2.9. Cellular Irradiation
2.10. Statistical Analysis
3. Results and Discussion
3.1. Characterization of GNPs
3.2. PYD Is an Inhibitor of ERCC1-XPF
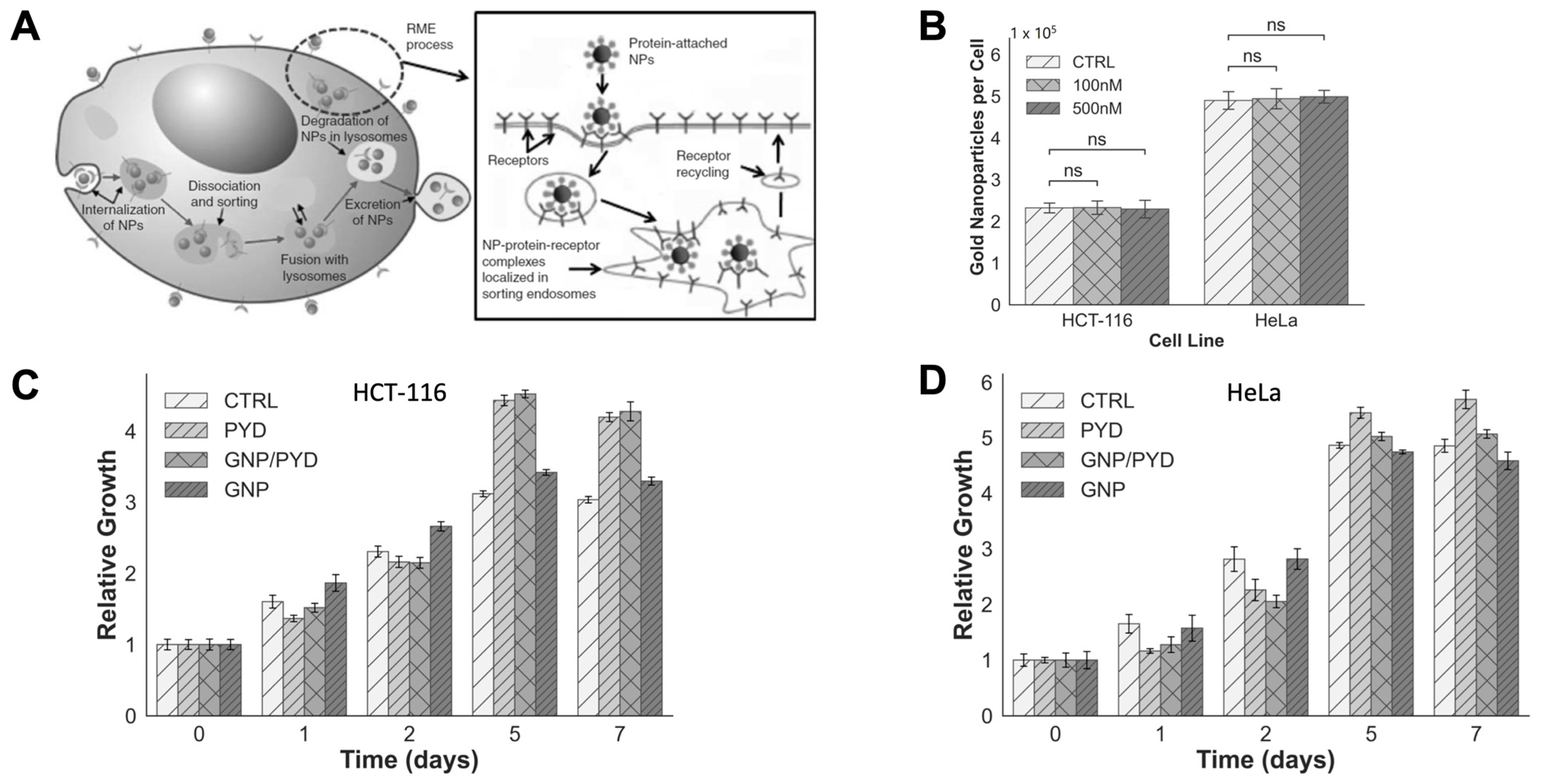
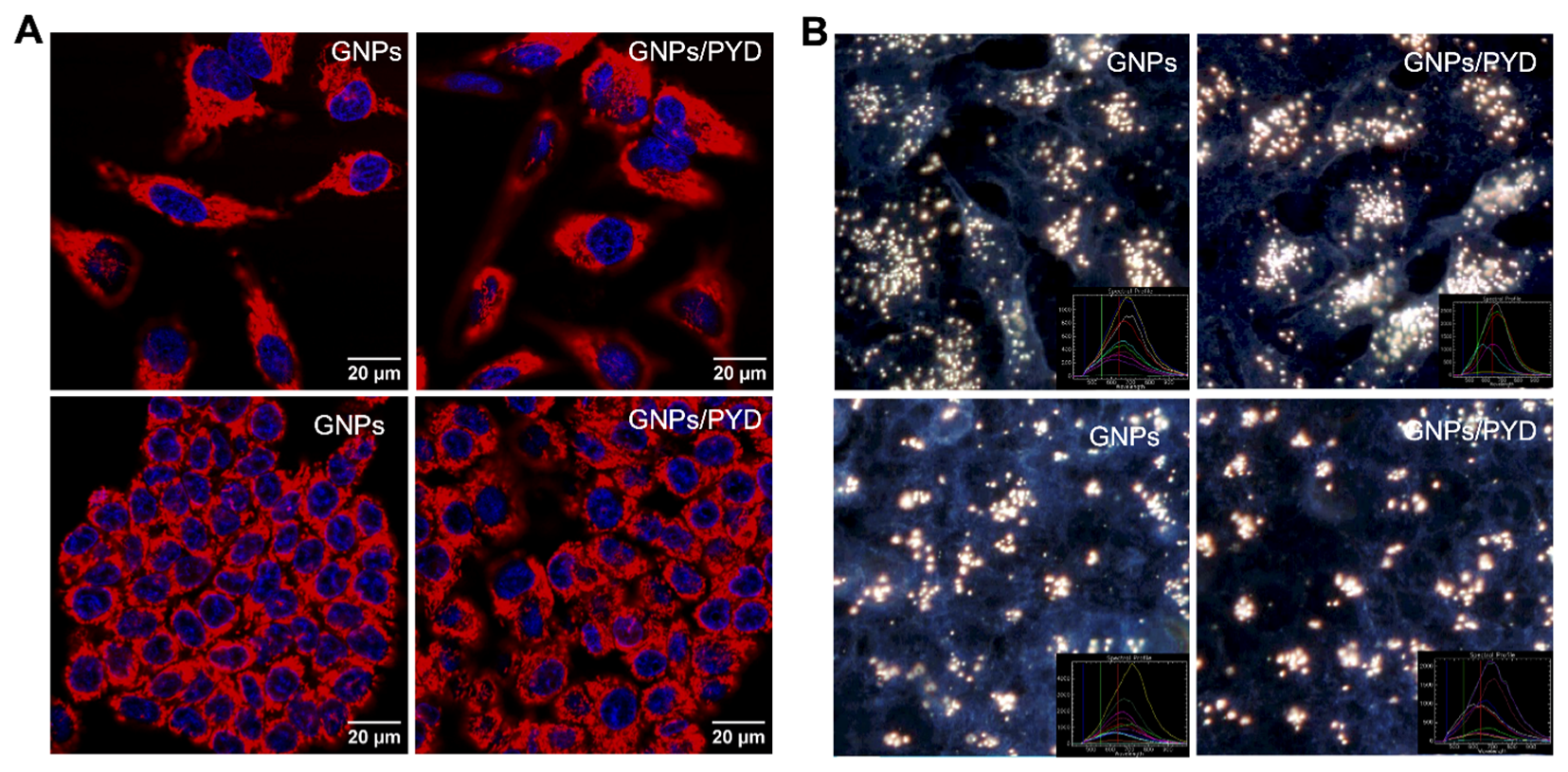
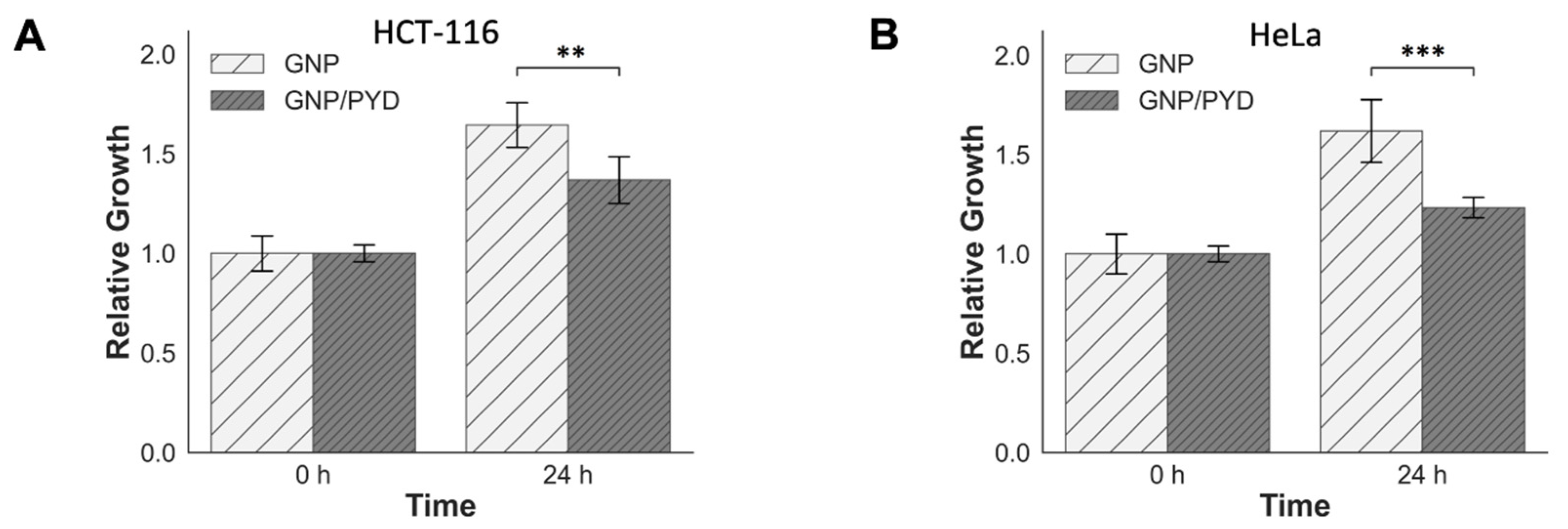
3.3. Cellular Uptake of GNPs in the Presence of PYD
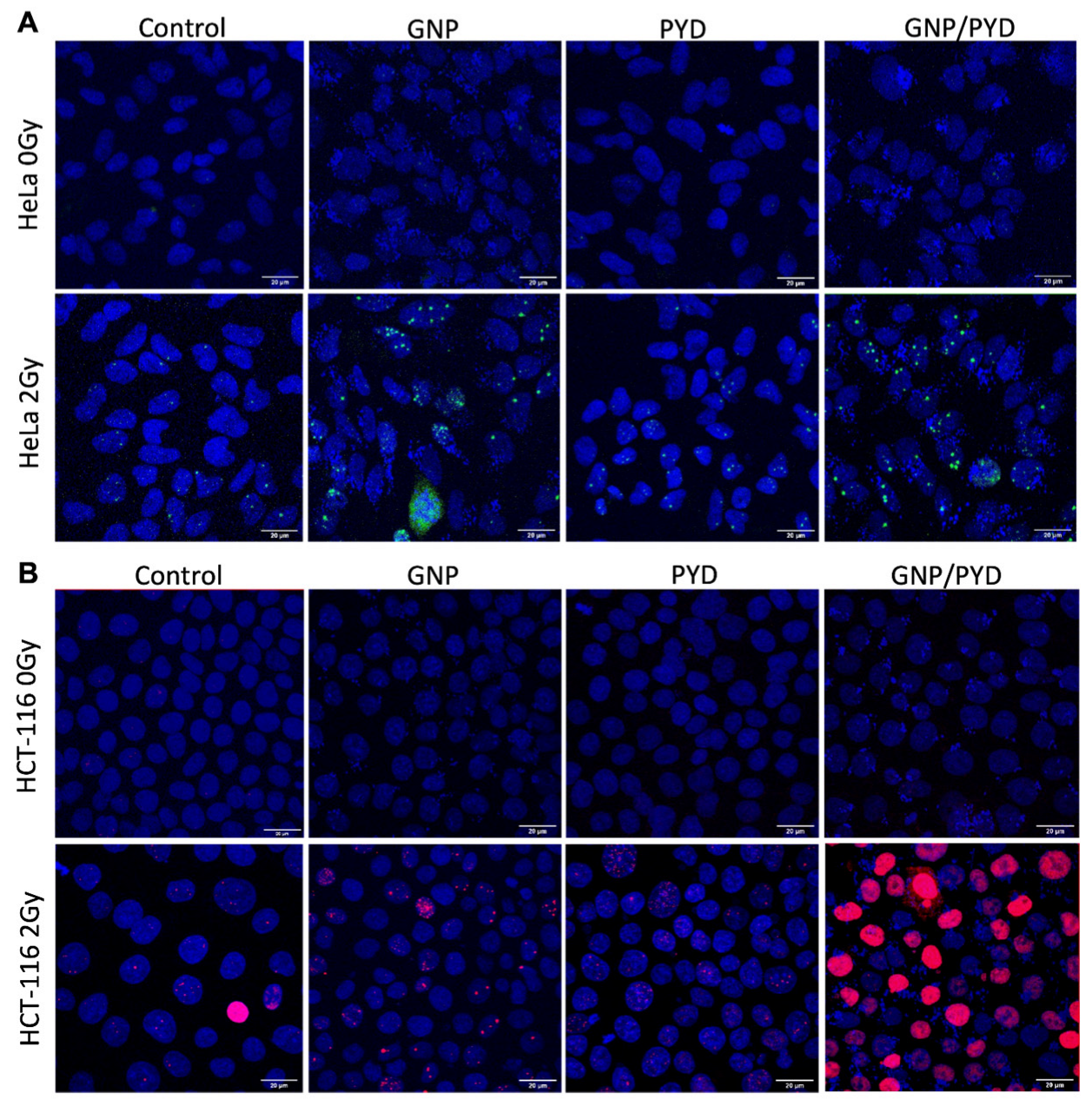
3.4. Evaluation of the Triple Combination of RT, GNPs, and Pyronaridine
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cronin, K.A.; Bs, A.J.L.; Scott, S.; Sherman, R.L.; Noone, A.-M.; Ms, N.H.; Henley, S.J.; Anderson, R.N.; Bs, A.U.F.; Ma, J.; et al. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer 2018, 124, 2785–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canadian Cancer Statistics Advisory Committee in Collaboration with the Canadian Cancer Society, Statistics Canada and the Public Health Agency of Canada. Canadian Cancer Statistics. 2021. Available online: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf (accessed on 15 October 2022).
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and Radiation Therapy: Current Advances and Future Directions. Int. J. Med Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment—Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, S.A.; Hu, C.; Sartor, O.; Gomella, L.G.; Amin, M.B.; Purdy, J.; Michalski, J.M.; Garzotto, M.G.; Pervez, N.; Balogh, A.G.; et al. Effect of Chemotherapy with Docetaxel With Androgen Suppression and Radiotherapy for Localized High-Risk Prostate Cancer: The Randomized Phase III NRG Oncology RTOG 0521 Trial. J. Clin. Oncol. 2019, 37, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Matsudaira, H.; Ueno, A.M.; Furuno, I. Iodine Contrast Medium Sensitizes Cultured Mammalian Cells to X Rays but Not to γ Rays. Radiat. Res. 1980, 84, 144. [Google Scholar] [CrossRef]
- Mello, R.S.; Callisen, H.; Winter, J.; Kagan, A.R.; Norman, A. Radiation dose enhancement in tumors with iodine. Med. Phys. 1983, 10, 75–78. [Google Scholar] [CrossRef]
- Hainfeld, J.F.; Slatkin, D.N.; Smilowitz, H.M. The use of gold nanoparticles to enhance radiotherapy in mice. Phys. Med. Biol. 2004, 49, N309. [Google Scholar] [CrossRef]
- Tudda, A.; Donzelli, E.; Nicolini, G.; Semperboni, S.; Bossi, M.; Cavaletti, G.; Castriconi, R.; Mangili, P.; del Vecchio, A.; Sarno, A.; et al. Breast radiotherapy with kilovoltage photons and gold nanoparticles as radiosensitizer: An in vitro study. Med. Phys. 2022, 49, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Chithrani, D.B.; Jelveh, S.; Jalali, F.; van Prooijen, M.; Allen, C.; Bristow, R.G.; Hill, R.P.; Jaffray, D.A. Gold Nanoparticles as Radiation Sensitizers in Cancer Therapy. Radiat. Res. 2010, 173, 719–728. [Google Scholar] [CrossRef]
- Zhao, Z.; Xu, H.; Li, S.; Han, Y.; Jia, J.; Han, Z.; Zhang, D.; Zhang, L.; Yu, R.; Liu, H. Hypoxic Radiosensitizer-Lipid Coated Gold Nanoparticles Enhance the Effects of Radiation Therapy on Tumor Growth. J. Biomed. Nanotechnol. 2019, 15, 1982–1993. [Google Scholar] [CrossRef]
- Rieck, K.; Bromma, K.; Sung, W.; Bannister, A.; Schuemann, J.; Chithrani, D.B. Modulation of gold nanoparticle mediated radiation dose enhancement through synchronization of breast tumor cell population. Br. J. Radiol. 2019, 92, 20190283. [Google Scholar] [CrossRef]
- Han, O.; Bromma, K.; Palmerley, N.; Bido, A.T.; Monica, M.; Alhussan, A.; Howard, P.L.; Brolo, A.G.; Beckham, W.; Alexander, A.S.; et al. Nanotechnology Driven Cancer Chemoradiation: Exploiting the Full Potential of Radiotherapy with a Unique Combination of Gold Nanoparticles and Bleomycin. Pharmaceutics 2022, 14, 233. [Google Scholar] [CrossRef]
- Cunningham, C.; de Kock, M.; Engelbrecht, M.; Miles, X.; Slabbert, J.; Vandevoorde, C. Radiosensitization Effect of Gold Nanoparticles in Proton Therapy. Front. Public Health 2021, 9, 699822. [Google Scholar] [CrossRef]
- Chithrani, B.D.; Ghazani, A.A.; Chan, W.C. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006, 6, 662–668. [Google Scholar] [CrossRef]
- Gao, H.; Shi, W.; Freund, L.B. Mechanics of receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 2005, 102, 9469–9474. [Google Scholar] [CrossRef] [Green Version]
- Abdoul-Carime, H.; Huels, M.A.; Illenberger, E.; Sanche, L. Sensitizing DNA to secondary electron damage: Resonant formation of oxidative radicals from 5-halouracils. J. Am. Chem. Soc. 2001, 123, 5354–5355. [Google Scholar] [CrossRef]
- Boudaïffa, B.; Cloutier, P.; Hunting, D.; Huels, M.A.; Sanche, L. Cross sections for low-energy (10–50 eV) electron damage to DNA. Radiat. Res. 2002, 157, 227–234. [Google Scholar] [CrossRef]
- Boudaïffa, B.; Cloutier, P.; Hunting, D.; Huels, M.A.; Sanche, L. Resonant formation of DNA strand breaks by low-energy (3 to 20 eV) electrons. Science 2000, 287, 1658–1660. [Google Scholar] [CrossRef]
- Sanche, L. Mechanisms of low energy electron damage to condensed biomolecules and DNA. Radiat. Prot. Dosim. 2002, 99, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Hunting, D.J.; Ayotte, P.; Sanche, L. Role of secondary low-energy electrons in the concomitant chemoradiation therapy of cancer. Phys. Rev. Lett. 2008, 100, 198101. [Google Scholar] [CrossRef]
- Zheng, Y.; Hunting, D.J.; Ayotte, P.; Sanche, L. Radiosensitization of DNA by gold nanoparticles irradiated with high-energy electrons. Radiat. Res. 2008, 169, 19–27. [Google Scholar] [CrossRef]
- Yousfi, M.; Leger, J.; Loiseau, J.F.; Held, B.; Eichwald, O.; Defoort, B.; Dupillier, J.M. Electron beam transport in heterogeneous slab media from MeV down to eV. Radiat. Prot. Dosim. 2006, 122, 46–52. [Google Scholar] [CrossRef]
- Faridounnia, M.; Folkers, G.E.; Boelens, R. Function and Interactions of ERCC1-XPF in DNA Damage Response. Molecules 2018, 23, 3205. [Google Scholar] [CrossRef] [Green Version]
- McHugh, P.; Spanswick, V.J.; A Hartley, J. Repair of DNA interstrand crosslinks: Molecular mechanisms and clinical relevance. Lancet Oncol. 2001, 2, 483–490. [Google Scholar] [CrossRef]
- Ahmad, A.; Robinson, A.R.; Duensing, A.; van Drunen, E.; Beverloo, H.B.; Weisberg, D.B.; Hasty, P.; Hoeijmakers, J.H.J.; Niedernhofer, L.J. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol. Cell. Biol. 2008, 28, 5082–5092. [Google Scholar] [CrossRef] [Green Version]
- McNeil, E.M.; Melton, D.W. DNA repair endonuclease ERCC1-XPF as a novel therapeutic target to overcome chemo-resistance in cancer therapy. Nucleic Acids Res. 2012, 40, 9990–10004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weilbeer, C.; Jay, D.; Donnelly, J.C.; Gentile, F.; Karimi-Busheri, F.; Yang, X.; Mani, R.S.; Yu, Y.; Elmenoufy, A.H.; Barakat, K.H.; et al. Modulation of ERCC1-XPF Heterodimerization Inhibition via Structural Modification of Small Molecule Inhibitor Side-Chains. Front. Oncol. 2022, 12, 819712. [Google Scholar] [CrossRef] [PubMed]
- Elmenoufy, A.H.; Gentile, F.; Jay, D.; Karimi-Busheri, F.; Yang, X.; Soueidan, O.M.; Weilbeer, C.; Mani, R.S.; Barakat, K.H.; Tuszynski, J.A.; et al. Targeting DNA Repair in Tumor Cells via Inhibition of ERCC1–XPF. J. Med. Chem. 2019, 62, 7684–7696. [Google Scholar] [CrossRef]
- Jordheim, L.P.; Barakat, K.H.; Heinrich-Balard, L.; Matera, E.-L.; Cros-Perrial, E.; Bouledrak, K.; El Sabeh, R.; Perez-Pineiro, R.; Wishart, D.S.; Cohen, R.; et al. Small molecule inhibitors of ERCC1-XPF protein-protein interaction synergize alkylating agents in cancer cells. Mol. Pharmacol. 2013, 84, 12–24. [Google Scholar] [CrossRef]
- Villanueva, P.J.; A Gutierrez, D.; Contreras, L.; Parra, K.; Segura-Cabrera, A.; Varela-Ramirez, A.; Aguilera, R.J. The Antimalarial Drug Pyronaridine Inhibits Topoisomerase II in Breast Cancer Cells and Hinders Tumor Progression In Vivo. Clin. Cancer Drugs 2021, 8, 50–56. [Google Scholar] [CrossRef]
- Rahn, J.J.; Rowley, B.; Lowery, M.P.; Della Coletta, L.; Limanni, T.; Nairn, R.S.; Adair, G.M. Effects of varying gene targeting parameters on processing of recombination intermediates by ERCC1–XPF. DNA Repair 2011, 10, 188–198. [Google Scholar] [CrossRef] [Green Version]
- Kuraoka, I.; Kobertz, W.; Ariza, R.; Biggerstaff, M.; Essigmann, J.; Wood, R. Repair of an Interstrand DNA Cross-link Initiated by ERCC1-XPF Repair/Recombination Nuclease. J. Biol. Chem. 2000, 275, 26632–26636. [Google Scholar] [CrossRef] [Green Version]
- Ghosalkar, J.; Sonawane, V.; Pisal, T.; Achrekar, S.; Pujari, R.; Chugh, A.; Shastry, P.; Joshi, K. Prostate Apoptosis Response-4 (Par-4): A Novel Target in Pyronaridine-Induced Apoptosis in Glioblastoma (GBM) Cells. Cancers 2022, 14, 3198. [Google Scholar] [CrossRef]
- Zhong, Z.-H.; Yi, Z.-Y.; Zhao, Y.-D.; Wang, J.; Jiang, Z.-B.; Xu, C.; Xie, Y.-J.; He, Q.-D.; Tong, Z.-Y.; Yao, X.-J.; et al. Pyronaridine induces apoptosis in non-small cell lung cancer cells by upregulating death receptor 5 ex-pression and inhibiting epidermal growth factor receptor. Chem. Biol. Drug Des. 2022, 99, 83–91. [Google Scholar] [CrossRef]
- Alhussan, A.; Palmerley, N.; Smazynski, J.; Karasinska, J.; Renouf, D.; Schaeffer, D.; Beckham, W.; Alexander, A.; Chithrani, D. Potential of Gold Nanoparticle in Current Radiotherapy Using a Co-Culture Model of Cancer Cells and Cancer Associated Fibroblast Cells. Cancers 2022, 14, 3586. [Google Scholar] [CrossRef]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99 Pt A, 28–51. [Google Scholar] [CrossRef] [Green Version]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.-J.; Schwaiger, M.; Weinmüller, M.; Räder, A.; Steiger, K.; Kessler, H. Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers 2017, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Li, Y.; Shen, Y.; Wang, A.; Wang, S.; Xie, T. The Functions and Applications of RGD in Tumor Therapy and Tissue Engineering. Int. J. Mol. Sci. 2013, 14, 13447–13462. [Google Scholar] [CrossRef]
- Wu, P.-H.; Onodera, Y.; Ichikawa, Y.; Rankin, E.B.; Giaccia, A.J.; Watanabe, Y.; Qian, W.; Hashimoto, T.; Shirato, H.; Nam, J.-M. Targeting integrins with RGD-conjugated gold nanoparticles in radiotherapy decreases the invasive activity of breast cancer cells. Int. J. Nanomed. 2017, 12, 5069–5085. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Li, X.; Li, H.; Zhang, W. Quantifying thiol–gold interactions towards the efficient strength control. Nat. Commun. 2014, 5, 4348. [Google Scholar] [CrossRef] [Green Version]
- Ciniero, G.; Elmenoufy, A.H.; Gentile, F.; Weinfeld, M.; Deriu, M.A.; West, F.G.; Tuszynski, J.A.; Dumontet, C.; Cros-Perrial, E.; Jordheim, L.P. Enhancing the activity of platinum-based drugs by improved inhibitors of ERCC1–XPF-mediated DNA repair. Cancer Chemother. Pharmacol. 2021, 87, 259–267. [Google Scholar] [CrossRef]
- Yang, C.; Bromma, K.; Chithrani, D. Peptide Mediated In Vivo Tumor Targeting of Nanoparticles through Optimization in Single and Multilayer In Vitro Cell Models. Cancers 2018, 10, 84. [Google Scholar] [CrossRef] [Green Version]
- Yohan, D.; Cruje, C.; Lu, X.; Chithrani, D.B. Size-Dependent Gold Nanoparticle Interaction at Nano-Micro Interface Using Both Monolayer and Multilayer (Tissue-Like) Cell Models. Nano Micro Lett. 2016, 8, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Bromma, K.; Cicon, L.; Bannister, A.; Rieck, K.; Beckham, W.; Chithrani, D. Optimization of uptake and transport of gold nanoparticles in two-dimensional and three-dimensional in-vitro cell models. In Proocedings of SPIE: Colloidal Nanoparticles for Biomedical Applications XIV; Osinski, M., Parak, W.J., Eds.; SPIE: Bellingham, WA, USA, 2019; Volume 10892, pp. 98–106. [Google Scholar]
- Cho, W.-S.; Cho, M.; Jeong, J.; Choi, M.; Han, B.S.; Shin, H.-S.; Hong, J.; Chung, B.H.; Jeong, J.; Cho, M.-H. Size-dependent tissue kinetics of PEG-coated gold nanoparticles. Toxicol. Appl. Pharmacol. 2010, 245, 116–123. [Google Scholar] [CrossRef]
- Bromma, K.; Alhussan, A.; Perez, M.; Howard, P.; Beckham, W.; Chithrani, D. Three-Dimensional Tumor Spheroids as a Tool for Reliable Investigation of Combined Gold Nanoparticle and Docetaxel Treatment. Cancers 2021, 13, 1465. [Google Scholar] [CrossRef]
- Bromma, K.; Cicon, L.; Beckham, W.; Chithrani, D.B. Gold nanoparticle mediated radiation response among key cell components of the tumour microenvironment for the advancement of cancer nanotechnology. Sci. Rep. 2020, 10, 12096. [Google Scholar] [CrossRef]
- Zuber, A.; Purdey, M.; Schartner, E.; Forbes, C.; van der Hoek, B.; Giles, D.; Abell, A.; Monro, T.; Ebendorff-Heidepriem, H. Detection of gold nanoparticles with different sizes using absorption and fluorescence based method. Sens. Actuators B Chem. 2016, 227, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Haiss, W.; Thanh, N.T.K.; Aveyard, J.; Fernig, D.G. Determination of Size and Concentration of Gold Nanoparticles from UV−Vis Spectra. Anal. Chem. 2007, 79, 4215–4221. [Google Scholar] [CrossRef]
- Bowles, M.; Lally, J.; Fadden, A.J.; Mouilleron, S.; Hammonds, T.; McDonald, N.Q. Fluorescence-based incision assay for human XPF-ERCC1 activity identifies important elements of DNA junction recognition. Nucleic Acids Res. 2012, 40, e101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeil, E.M.; Astell, K.R.; Ritchie, A.-M.; Shave, S.; Houston, D.R.; Bakrania, P.; Jones, H.M.; Khurana, P.; Wallace, C.; Chapman, T.; et al. Inhibition of the ERCC1–XPF structure-specific endonuclease to overcome cancer chemoresistance. DNA Repair 2015, 31, 19–28. [Google Scholar] [CrossRef]
- Elmenoufy, A.H.; Gentile, F.; Jay, D.; Karimi-Busheri, F.; Yang, X.; Soueidan, O.M.; Mani, R.S.; Ciniero, G.; Tuszynski, J.A.; Weinfeld, M.; et al. Design, synthesis and in vitro cell-free/cell-based biological evaluations of novel ERCC1-XPF inhibitors targeting DNA repair pathway. Eur. J. Med. Chem. 2020, 204, 112658. [Google Scholar] [CrossRef]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.A.; Alkawareek, M.Y.; Dreaden, E.C.; Brown, D.; Alkilany, A.M.; Farokhzad, O.C.; Mahmoudi, M. Cellular uptake of nanoparticles: Journey inside the cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef]
- Chithrani, B.D.; Chan, W.C. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007, 7, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Jeremic, B.; Aguerri, A.R.; Filipovic, N. Radiosensitization by gold nanoparticles. Clin. Transl. Oncol. 2013, 15, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Oh, N.; Park, J.H. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int. J. Nanomed. 2014, 9 (Suppl. 1), 51–63. [Google Scholar]
- Jin, H.; Heller, D.A.; Sharma, R.; Strano, M.S. Size-dependent cellular uptake and expulsion of single-walled carbon nanotubes: Single particle tracking and a generic uptake model for nanoparticles. ACS Nano 2009, 3, 149–158. [Google Scholar] [CrossRef]
- Bannister, A.; Dissanayake, D.; Kowalewski, A.; Cicon, L.; Bromma, K.; Chithrani, D.B. Modulation of the Microtubule Network for Optimization of Nanoparticle Dynamics for the Advancement of Cancer Nanomedicine. Bioengineering 2020, 7, 56. [Google Scholar] [CrossRef]
- Cruje, C.; Chithrani, B.D. Integration of Peptides for Enhanced Uptake of PEGylayed Gold Nanoparticles. J. Nanosci. Nanotechnol. 2015, 15, 2125–2131. [Google Scholar] [CrossRef]
- Cruje, C.; Chithrani, D.B. Polyethylene Glycol Functionalized Nanoparticles for Improved Cancer Treatment. Rev. Nanosci. Nanotechnol. 2014, 3, 20–30. [Google Scholar] [CrossRef]
- Cruje, C.; Yang, C.; Uertz, J.; van Prooijen, M.; Chithrani, B.D. Optimization of PEG coated nanoscale gold particles for enhanced radiation therapy. RSC Adv. 2015, 5, 101525–101532. [Google Scholar] [CrossRef]
- Bromma, K.; Dos Santos, N.; Barta, I.; Alexander, A.; Beckham, W.; Krishnan, S.; Chithrani, D.B. Enhancing nanoparticle accumulation in two dimensional, three dimensional, and xenograft mouse cancer cell models in the presence of docetaxel. Sci. Rep. 2022, 12, 13508. [Google Scholar] [CrossRef] [PubMed]
- Cheresh, D.A.; Desgrosellier, J.S. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar]
- Connor, E.E.; Mwamuka, J.; Gole, A.; Murphy, C.J.; Wyatt, M.D. Gold Nanoparticles Are Taken Up by Human Cells but Do Not Cause Acute Cytotoxicity. Small 2005, 1, 325–327. [Google Scholar] [CrossRef]
- Zhang, X.-D.; Guo, M.-L.; Wu, H.-Y.; Sun, Y.-M.; Ding, Y.-Q.; Feng, X. Irradiation stability and cytotoxicity of gold nanoparticles for radiotherapy. Int. J. Nanomed. 2009, 4, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, Z.; Xu, S.; Wang, F.; Shen, Y.; Huang, S.; Guo, S. pH, redox and photothermal tri-responsive DNA/polyethylenimine conjugated gold nanorods as nanocarriers for specific intracellular co-release of doxorubicin and chemosensitizer pyronaridine to combat multidrug resistant cancer. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1785–1795. [Google Scholar] [CrossRef]
- Schuemann, J.; Berbeco, R.; Chithrani, D.; Cho, S.H.; Kumar, R.; McMahon, S.; Sridhar, S.; Krishnan, S. Roadmap to Clinical Use of Gold Nanoparticles for Radiation Sensitization. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 189–205. [Google Scholar] [CrossRef] [Green Version]
- Carter, J.D.; Cheng, N.N.; Qu, Y.; Suarez, G.D.; Guo, T. Nanoscale energy deposition by X-ray absorbing nanostructures. J. Phys. Chem. B 2007, 111, 11622–11625. [Google Scholar] [CrossRef]
- Zheng, Y.; Sanche, L. Gold nanoparticles enhance DNA damage induced by anti-cancer drugs and radiation. Radiat. Res. 2009, 172, 114–119. [Google Scholar] [CrossRef]
- Zhang, Z.; Berg, A.; Levanon, H.; Fessenden, A.R.W.; Meisel, D. On the interactions of free radicals with gold nanoparticles. J. Am. Chem. Soc. 2003, 125, 7959–7963. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Broma, K.; Sung, W.; Schuemann, J.; Chithrani, D. Determining the Radiation Enhancement Effects of Gold Nanoparticles in Cells in a Com-bined Treatment with Cisplatin and Radiation at Therapeutic Megavoltage Energies. Cancers 2018, 10, 150. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Coulter, J.A.; Hounsell, A.R.; Butterworth, K.T.; McMahon, S.J.; Hyland, W.B.; Muir, M.F.; Dickson, G.R.; Prise, K.M.; Currell, F.J.; et al. Cell-Specific Radiosensitization by Gold Nanoparticles at Mega-voltage Radiation Energies. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 531–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löbrich, M.; Shibata, A.; Beucher, A.; Fisher, A.; Ensminger, M.; Goodrazi, A.A.; Barton, O.; Jeggo, P.A. γH2AX foci analysis for monitoring DNA double-strand break repair: Strengths, limitations and optimiza-tion. Cell Cycle 2010, 9, 662–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdiglesias, V.; Giunta, S.; Fenech, M.; Neri, M.; Bonassi, S. γH2AX as a marker of DNA double strand breaks and genomic instability in human population studies. Mutat. Res. Rev. Mutat. Res. 2013, 753, 24–40. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jackson, N.; Alhussan, A.; Bromma, K.; Jay, D.; Donnelly, J.C.; West, F.G.; Lavasanifar, A.; Weinfeld, M.; Beckham, W.; Chithrani, D.B. Repurposing Antimalarial Pyronaridine as a DNA Repair Inhibitor to Exploit the Full Potential of Gold-Nanoparticle-Mediated Radiation Response. Pharmaceutics 2022, 14, 2795. https://doi.org/10.3390/pharmaceutics14122795
Jackson N, Alhussan A, Bromma K, Jay D, Donnelly JC, West FG, Lavasanifar A, Weinfeld M, Beckham W, Chithrani DB. Repurposing Antimalarial Pyronaridine as a DNA Repair Inhibitor to Exploit the Full Potential of Gold-Nanoparticle-Mediated Radiation Response. Pharmaceutics. 2022; 14(12):2795. https://doi.org/10.3390/pharmaceutics14122795
Chicago/Turabian StyleJackson, Nolan, Abdulaziz Alhussan, Kyle Bromma, David Jay, James C. Donnelly, Frederick G. West, Afsaneh Lavasanifar, Michael Weinfeld, Wayne Beckham, and Devika B. Chithrani. 2022. "Repurposing Antimalarial Pyronaridine as a DNA Repair Inhibitor to Exploit the Full Potential of Gold-Nanoparticle-Mediated Radiation Response" Pharmaceutics 14, no. 12: 2795. https://doi.org/10.3390/pharmaceutics14122795