
Impact of Cytochrome Induction or Inhibition on the Plasma and Brain Kinetics of [11C]metoclopramide, a PET Probe for P-Glycoprotein Function at the Blood-Brain Barrier
 , , , ,
, , , ,
Abstract
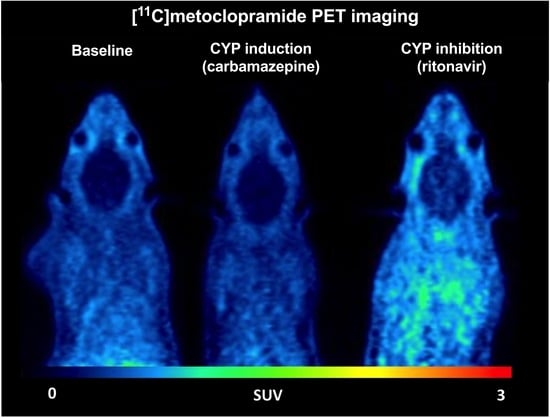
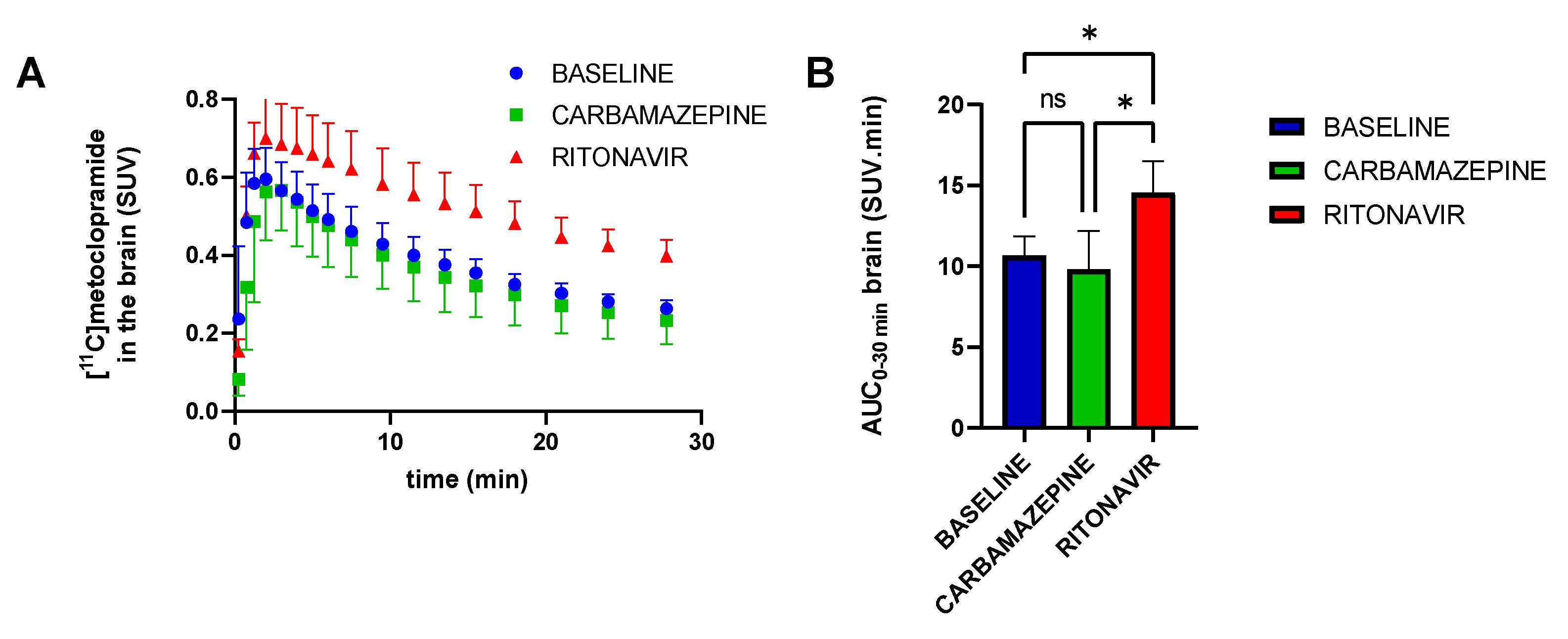
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Treatments
2.2. Radiochemistry
2.3. Animals
2.4. PET Acquisition
2.5. Arterial Input Function
2.6. PET Data Analysis
2.7. Statistical Analysis
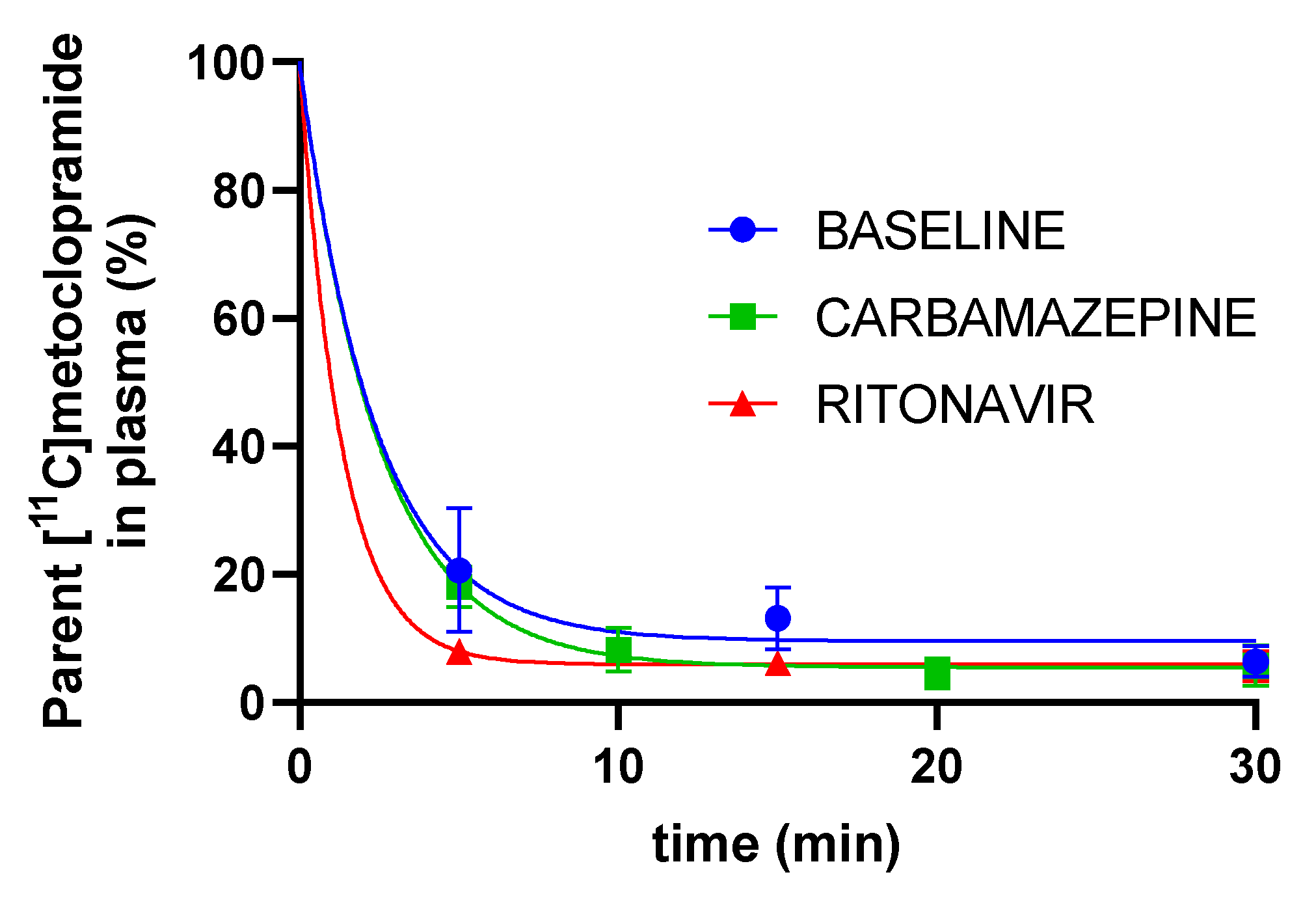
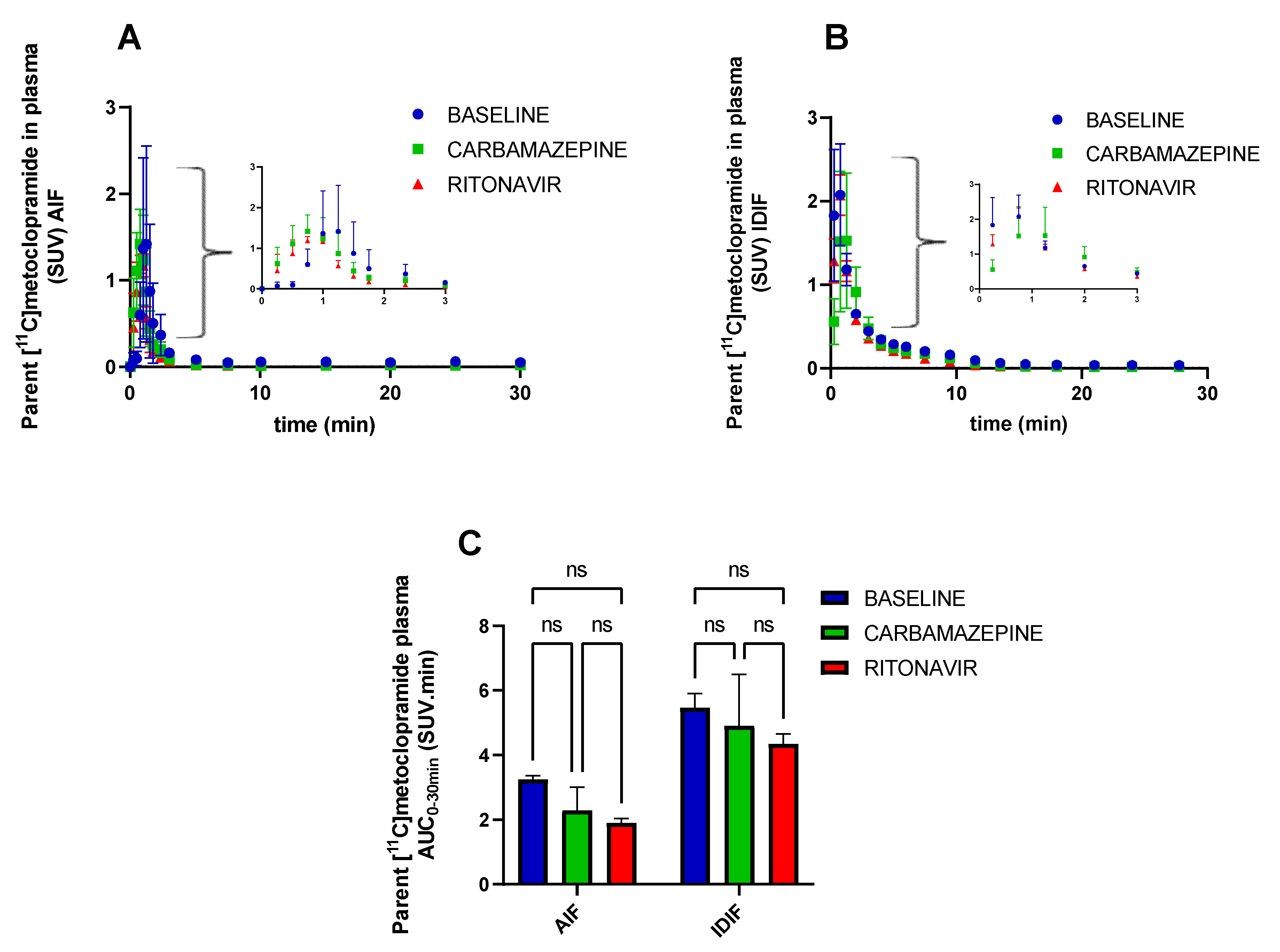
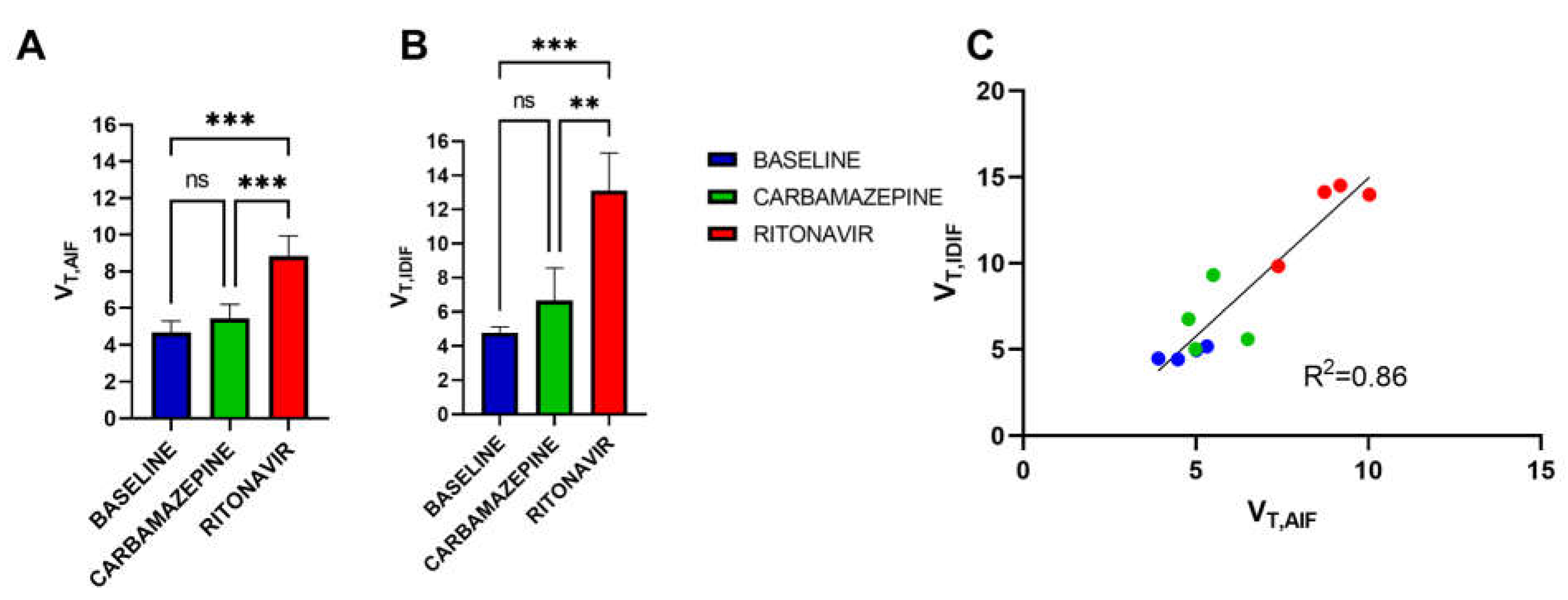
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Begley, D.J. Delivery of Therapeutic Agents to the Central Nervous System: The Problems and the Possibilities. Pharmacol. Ther. 2004, 104, 29–45. [Google Scholar] [CrossRef]
- Bauer, M.; Tournier, N.; Langer, O. Imaging P-Glycoprotein Function at the Blood-Brain Barrier as a Determinant of the Variability in Response to CNS Drugs. Clin. Pharmacol. Ther. 2019, 105, 1061–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akamine, Y.; Yasui-Furukori, N.; Ieiri, I.; Uno, T. Psychotropic Drug–Drug Interactions Involving P-Glycoprotein. CNS Drugs 2012, 26, 959–973. [Google Scholar] [CrossRef]
- Banks, W.A. From Blood–Brain Barrier to Blood–Brain Interface: New Opportunities for CNS Drug Delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, Maintenance and Disruption of the Blood-Brain Barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan, P.; John, C.; Zoghbi, S.S.; Halldin, C.; Gottesman, M.M.; Innis, R.B.; Hall, M.D. Imaging the Function of P-Glycoprotein with Radiotracers: Pharmacokinetics and in Vivo Applications. Clin. Pharmacol. Ther. 2009, 86, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Toornvliet, R.; van Berckel, B.N.M.; Luurtsema, G.; Lubberink, M.; Geldof, A.A.; Bosch, T.M.; Oerlemans, R.; Lammertsma, A.A.; Franssen, E.J.F. Effect of Age on Functional P-Glycoprotein in the Blood-Brain Barrier Measured by Use of (R)-[(11)C]Verapamil and Positron Emission Tomography. Clin. Pharmacol. Ther. 2006, 79, 540–548. [Google Scholar] [CrossRef]
- Bauer, M.; Karch, R.; Neumann, F.; Abrahim, A.; Wagner, C.C.; Kletter, K.; Müller, M.; Zeitlinger, M.; Langer, O. Age Dependency of Cerebral P-Gp Function Measured with (R)-[11C]Verapamil and PET. Eur. J. Clin. Pharmacol. 2009, 65, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.; Wulkersdorfer, B.; Karch, R.; Philippe, C.; Jäger, W.; Stanek, J.; Wadsak, W.; Hacker, M.; Zeitlinger, M.; Langer, O. Effect of P-Glycoprotein Inhibition at the Blood-Brain Barrier on Brain Distribution of (R)-[11 C]Verapamil in Elderly vs. Young Subjects. Br. J. Clin. Pharmacol. 2017, 83, 1991–1999. [Google Scholar] [CrossRef] [Green Version]
- Deo, A.K.; Borson, S.; Link, J.M.; Domino, K.; Eary, J.F.; Ke, B.; Richards, T.L.; Mankoff, D.A.; Minoshima, S.; O’Sullivan, F.; et al. Activity of P-Glycoprotein, a β-Amyloid Transporter at the Blood-Brain Barrier, Is Compromised in Patients with Mild Alzheimer Disease. J. Nucl. Med. 2014, 55, 1106–1111. [Google Scholar] [CrossRef]
- Feldmann, M.; Asselin, M.-C.; Liu, J.; Wang, S.; McMahon, A.; Anton-Rodriguez, J.; Walker, M.; Symms, M.; Brown, G.; Hinz, R.; et al. P-Glycoprotein Expression and Function in Patients with Temporal Lobe Epilepsy: A Case-Control Study. Lancet Neurol. 2013, 12, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Pottier, G.; Marie, S.; Goutal, S.; Auvity, S.; Peyronneau, M.-A.; Stute, S.; Boisgard, R.; Dollé, F.; Buvat, I.; Caillé, F.; et al. Imaging the Impact of the P-Glycoprotein (ABCB1) Function on the Brain Kinetics of Metoclopramide. J. Nucl. Med. 2016, 57, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tournier, N.; Bauer, M.; Pichler, V.; Nics, L.; Klebermass, E.-M.; Bamminger, K.; Matzneller, P.; Weber, M.; Karch, R.; Caillé, F.; et al. Impact of P-Glycoprotein Function on the Brain Kinetics of the Weak Substrate 11C-Metoclopramide Assessed with PET Imaging in Humans. J. Nucl. Med. 2019, 60, 985–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breuil, L.; Marie, S.; Goutal, S.; Auvity, S.; Truillet, C.; Saba, W.; Langer, O.; Caillé, F.; Tournier, N. Comparative Vulnerability of PET Radioligands to Partial Inhibition of P-Glycoprotein at the Blood-Brain Barrier: A Criterion of Choice? J. Cereb. Blood Flow Metab. 2022, 42, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Bamminger, K.; Pichler, V.; Weber, M.; Binder, S.; Maier-Salamon, A.; Tahir, A.; Jäger, W.; Haslacher, H.; Tournier, N.; et al. Impaired Clearance from the Brain Increases the Brain Exposure to Metoclopramide in Elderly Subjects. Clin. Pharmacol. Ther. 2020, 109, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Zoufal, V.; Mairinger, S.; Brackhan, M.; Krohn, M.; Filip, T.; Sauberer, M.; Stanek, J.; Wanek, T.; Tournier, N.; Bauer, M.; et al. Imaging P-Glycoprotein Induction at the Blood-Brain Barrier of a β-Amyloidosis Mouse Model with 11C-Metoclopramide PET. J. Nucl. Med. 2020, 61, 1050–1057. [Google Scholar] [CrossRef]
- Breuil, L.; Goutal, S.; Marie, S.; Del Vecchio, A.; Audisio, D.; Soyer, A.; Goislard, M.; Saba, W.; Tournier, N.; Caillé, F. Comparison of the Blood-Brain Barrier Transport and Vulnerability to P-Glycoprotein-Mediated Drug-Drug Interaction of Domperidone versus Metoclopramide Assessed Using In Vitro Assay and PET Imaging. Pharmaceutics 2022, 14, 1658. [Google Scholar] [CrossRef]
- Gil-Martins, E.; Barbosa, D.J.; Silva, V.; Remião, F.; Silva, R. Dysfunction of ABC Transporters at the Blood-Brain Barrier: Role in Neurological Disorders. Pharmacol. Ther. 2020, 213, 107554. [Google Scholar] [CrossRef]
- Pike, V.W. PET Radiotracers: Crossing the Blood-Brain Barrier and Surviving Metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Hefner, G.; Wolff, J.; Hahn, M.; Hiemke, C.; Toto, S.; Roll, S.C.; Messer, T.; Klimke, A. Prevalence and Sort of Pharmacokinetic Drug-Drug Interactions in Hospitalized Psychiatric Patients. J. Neural Transm. 2020, 127, 1185–1198. [Google Scholar] [CrossRef]
- Dalic, L.; Cook, M.J. Managing Drug-Resistant Epilepsy: Challenges and Solutions. Neuropsychiatr. Dis. Treat. 2016, 12, 2605–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, L.T.; Yang, S.; Tran, Q.T.; Kim, S.K.; Yun, H.; Chae, J. Effects of Carbamazepine and Phenytoin on Pharmacokinetics and Pharmacodynamics of Rivaroxaban. Pharmaceutics 2020, 12, 1040. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, T.; Asoh, M.; Nakura, H.; Watanabe, M.; Tanaka, M.; Kumai, T.; Kobayashi, S. Carbamazepine Induces Multiple Cytochrome P450 Subfamilies in Rats. Chem. Biol. Interact. 1999, 117, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Rioux, N.; Bellavance, E.; Bourg, S.; Garneau, M.; Ribadeneira, M.D.; Duan, J. Assessment of CYP3A-Mediated Drug–Drug Interaction Potential for Victim Drugs Using an in Vivo Rat Model. Biopharm. Drug Dispos. 2013, 34, 396–401. [Google Scholar] [CrossRef]
- Kageyama, M.; Namiki, H.; Fukushima, H.; Terasaka, S.; Togawa, T.; Tanaka, A.; Ito, Y.; Shibata, N.; Takada, K. Effect of Chronic Administration of Ritonavir on Function of Cytochrome P450 3A and P-Glycoprotein in Rats. Biol. Pharm. Bull. 2005, 28, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Caillé, F.; Goutal, S.; Marie, S.; Auvity, S.; Cisternino, S.; Kuhnast, B.; Pottier, G.; Tournier, N. Positron Emission Tomography Imaging Reveals an Importance of Saturable Liver Uptake Transport for the Pharmacokinetics of Metoclopramide. Contrast Media Mol. Imaging 2018, 2018, 7310146. [Google Scholar] [CrossRef]
- Garcia-Varela, L.; Mossel, P.; Aguiar, P.; Vazquez-Matias, D.A.; van Waarde, A.; Willemsen, A.T.M.; Bartels, A.L.; Colabufo, N.A.; Dierckx, R.A.J.O.; Elsinga, P.H.; et al. Dose-Response Assessment of Cerebral P-Glycoprotein Inhibition in Vivo with [18F]MC225 and PET. J. Controlled Release 2022, 347, 500–507. [Google Scholar] [CrossRef]
- Gidal, B.E. P-Glycoprotein Expression and Pharmacoresistant Epilepsy: Cause or Consequence? Epilepsy Curr. 2014, 14, 136–138. [Google Scholar] [CrossRef]
- Bankstahl, J.P.; Bankstahl, M.; Kuntner, C.; Stanek, J.; Wanek, T.; Meier, M.; Ding, X.-Q.; Müller, M.; Langer, O.; Löscher, W. A Novel Positron Emission Tomography Imaging Protocol Identifies Seizure-Induced Regional Overactivity of P-Glycoprotein at the Blood-Brain Barrier. J. Neurosci. 2011, 31, 8803–8811. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Y.W.; Thom, M.; Catarino, C.B.; Martinian, L.; Figarella-Branger, D.; Bartolomei, F.; Koepp, M.; Sisodiya, S.M. Neuropathology of the Blood–Brain Barrier and Pharmaco-Resistance in Human Epilepsy. Brain 2012, 135, 3115–3133. [Google Scholar] [CrossRef]
- Brodie, M.J.; Mintzer, S.; Pack, A.M.; Gidal, B.E.; Vecht, C.J.; Schmidt, D. Enzyme Induction with Antiepileptic Drugs: Cause for Concern? Epilepsia 2013, 54, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Fuhr, L.M.; Marok, F.Z.; Hanke, N.; Selzer, D.; Lehr, T. Pharmacokinetics of the CYP3A4 and CYP2B6 Inducer Carbamazepine and Its Drug-Drug Interaction Potential: A Physiologically Based Pharmacokinetic Modeling Approach. Pharmaceutics 2021, 13, 270. [Google Scholar] [CrossRef] [PubMed]
- Kaukab, I.; Shah, S.N.H.; Abrar, M.A.; Anwer, N.; Murtaza, G. Influence of Rifampicin Pre-Treatment on the In Vivo Pharmacokinetics of Metoclopramide in Pakistani Healthy Volunteers Following Concurrent Oral Administration. Curr. Drug Metab. 2020, 21, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- FDA Clinical Drug Interaction Studies—Study Design, Data Analysis, and Clinical Implications—Guidance for Industry; FDA: Silver Spring, ML, USA, 2017.
- Livezey, M.R.; Briggs, E.D.; Bolles, A.K.; Nagy, L.D.; Fujiwara, R.; Furge, L.L. Metoclopramide Is Metabolized by CYP2D6 and Is a Reversible Inhibitor, but Not Inactivator, of CYP2D6. Xenobiotica. 2014, 44, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luurtsema, G.; Molthoff, C.F.M.; Schuit, R.C.; Windhorst, A.D.; Lammertsma, A.A.; Franssen, E.J.F. Evaluation of (R)-[11C]Verapamil as PET Tracer of P-Glycoprotein Function in the Blood-Brain Barrier: Kinetics and Metabolism in the Rat. Nucl. Med. Biol. 2005, 32, 87–93. [Google Scholar] [CrossRef]
- Abrahim, A.; Luurtsema, G.; Bauer, M.; Karch, R.; Lubberink, M.; Pataraia, E.; Joukhadar, C.; Kletter, K.; Lammertsma, A.A.; Baumgartner, C.; et al. Peripheral Metabolism of (R)-[11C]Verapamil in Epilepsy Patients. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 116–123. [Google Scholar] [CrossRef]
- Lazarova, N.; Zoghbi, S.S.; Hong, J.; Seneca, N.; Tuan, E.; Gladding, R.L.; Liow, J.-S.; Taku, A.; Innis, R.B.; Pike, V.W. Synthesis and Evaluation of [N-Methyl-11C]N-Desmethyl-Loperamide as a New and Improved PET Radiotracer for Imaging P-Gp Function. J. Med. Chem. 2008, 51, 6034–6043. [Google Scholar] [CrossRef] [Green Version]
- Wanek, T.; Römermann, K.; Mairinger, S.; Stanek, J.; Sauberer, M.; Filip, T.; Traxl, A.; Kuntner, C.; Pahnke, J.; Bauer, F.; et al. Factors Governing P-Glycoprotein-Mediated Drug-Drug Interactions at the Blood-Brain Barrier Measured with Positron Emission Tomography. Mol. Pharm. 2015, 12, 3214–3225. [Google Scholar] [CrossRef]
- Savolainen, H.; Windhorst, A.D.; Elsinga, P.H.; Cantore, M.; Colabufo, N.A.; Willemsen, A.T.; Luurtsema, G. Evaluation of [18F]MC225 as a PET Radiotracer for Measuring P-Glycoprotein Function at the Blood–Brain Barrier in Rats: Kinetics, Metabolism, and Selectivity. J. Cereb. Blood Flow Metab. 2017, 37, 1286–1298. [Google Scholar] [CrossRef]
- Hernández-Lozano, I.; Mairinger, S.; Sauberer, M.; Stanek, J.; Filip, T.; Wanek, T.; Ciarimboli, G.; Tournier, N.; Langer, O. Influence of Cation Transporters (OCTs and MATEs) on the Renal and Hepatobiliary Disposition of [11C]Metoclopramide in Mice. Pharm. Res. 2021, 38, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Auvity, S.; Caillé, F.; Marie, S.; Wimberley, C.; Bauer, M.; Langer, O.; Buvat, I.; Goutal, S.; Tournier, N. P-Glycoprotein (ABCB1) Inhibits the Influx and Increases the Efflux of 11C-Metoclopramide Across the Blood-Brain Barrier: A PET Study on Nonhuman Primates. J. Nucl. Med. 2018, 59, 1609–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venhorst, J.; ter Laak, A.M.; Commandeur, J.N.M.; Funae, Y.; Hiroi, T.; Vermeulen, N.P.E. Homology Modeling of Rat and Human Cytochrome P450 2D (CYP2D) Isoforms and Computational Rationalization of Experimental Ligand-Binding Specificities. J. Med. Chem. 2003, 46, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.-W.; Oh, K.-Y.; Yoon, S.-J.; Shin, H.-B.; Jung, E.H.; Cho, C.-K.; Lim, C.W.; Kang, P.; Choi, C.-I.; Jang, C.-G.; et al. Effects of CYP2D6 Genetic Polymorphism on the Pharmacokinetics of Metoclopramide. Arch. Pharm. Res. 2020, 43, 1207–1213. [Google Scholar] [CrossRef]
- Wyen, C.; Fuhr, U.; Frank, D.; Aarnoutse, R.E.; Klaassen, T.; Lazar, A.; Seeringer, A.; Doroshyenko, O.; Kirchheiner, J.C.; Abdulrazik, F.; et al. Effect of an Antiretroviral Regimen Containing Ritonavir Boosted Lopinavir on Intestinal and Hepatic CYP3A, CYP2D6 and P-Glycoprotein in HIV-Infected Patients. Clin. Pharmacol. Ther. 2008, 84, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Drewe, J.; Gutmann, H.; Fricker, G.; Török, M.; Beglinger, C.; Huwyler, J. HIV Protease Inhibitor Ritonavir: A More Potent Inhibitor of P-Glycoprotein than the Cyclosporine Analog SDZ PSC 833. Biochem. Pharmacol. 1999, 57, 1147–1152. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breuil, L.; Ziani, N.; Leterrier, S.; Hugon, G.; Caillé, F.; Bouilleret, V.; Truillet, C.; Goislard, M.; El Biali, M.; Bauer, M.; et al. Impact of Cytochrome Induction or Inhibition on the Plasma and Brain Kinetics of [11C]metoclopramide, a PET Probe for P-Glycoprotein Function at the Blood-Brain Barrier. Pharmaceutics 2022, 14, 2650. https://doi.org/10.3390/pharmaceutics14122650
Breuil L, Ziani N, Leterrier S, Hugon G, Caillé F, Bouilleret V, Truillet C, Goislard M, El Biali M, Bauer M, et al. Impact of Cytochrome Induction or Inhibition on the Plasma and Brain Kinetics of [11C]metoclopramide, a PET Probe for P-Glycoprotein Function at the Blood-Brain Barrier. Pharmaceutics. 2022; 14(12):2650. https://doi.org/10.3390/pharmaceutics14122650
Chicago/Turabian StyleBreuil, Louise, Nora Ziani, Sarah Leterrier, Gaëlle Hugon, Fabien Caillé, Viviane Bouilleret, Charles Truillet, Maud Goislard, Myriam El Biali, Martin Bauer, and et al. 2022. "Impact of Cytochrome Induction or Inhibition on the Plasma and Brain Kinetics of [11C]metoclopramide, a PET Probe for P-Glycoprotein Function at the Blood-Brain Barrier" Pharmaceutics 14, no. 12: 2650. https://doi.org/10.3390/pharmaceutics14122650