
SEDEX—Self-Emulsifying Delivery Via Hot Melt Extrusion: A Continuous Pilot-Scale Feasibility Study

 , and
, and
Abstract
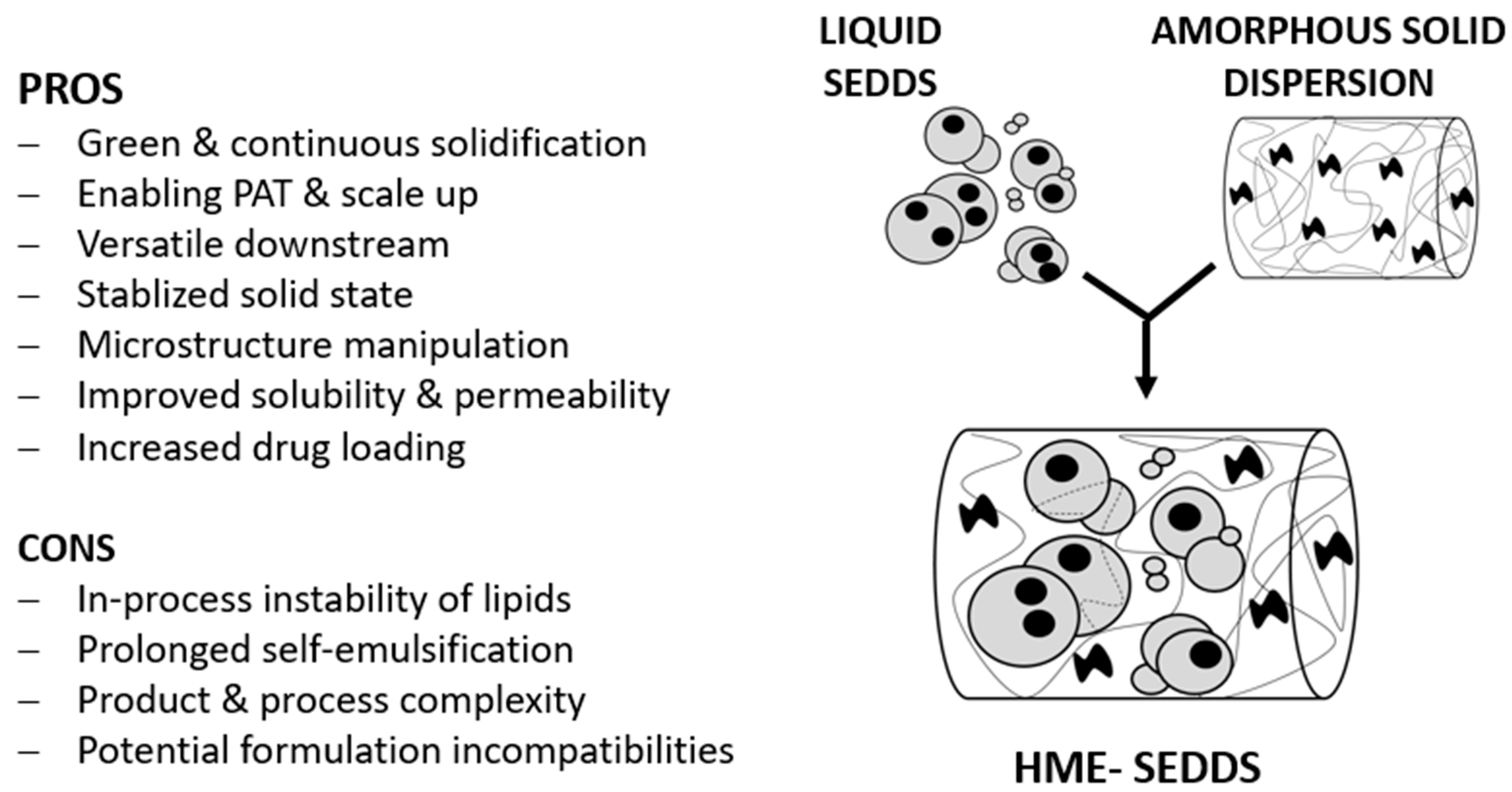
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Liquid SEDDS Preparation and Polymer Solubility
2.3. Drying of Polymers
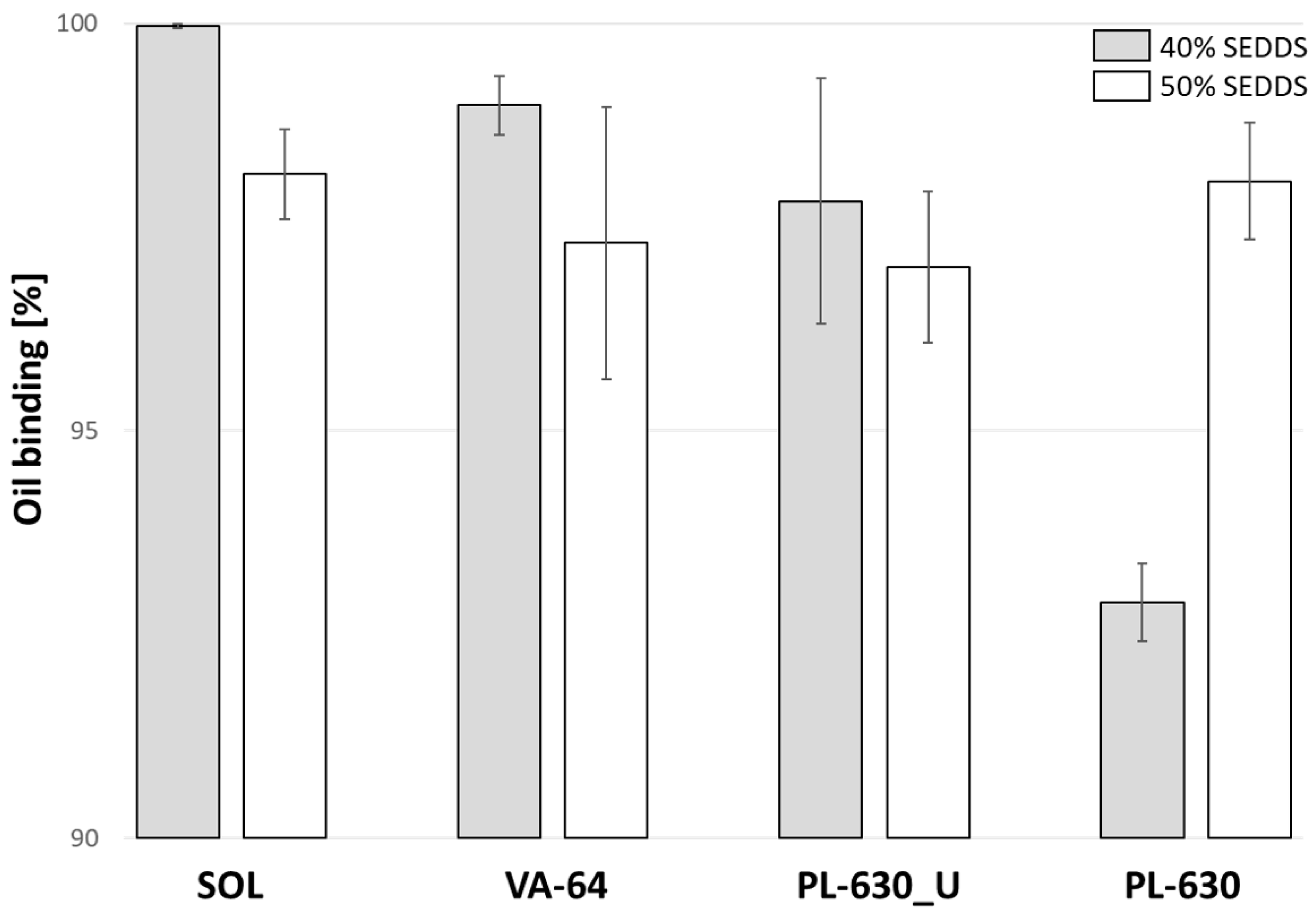
2.4. Oil-Binding Capacity
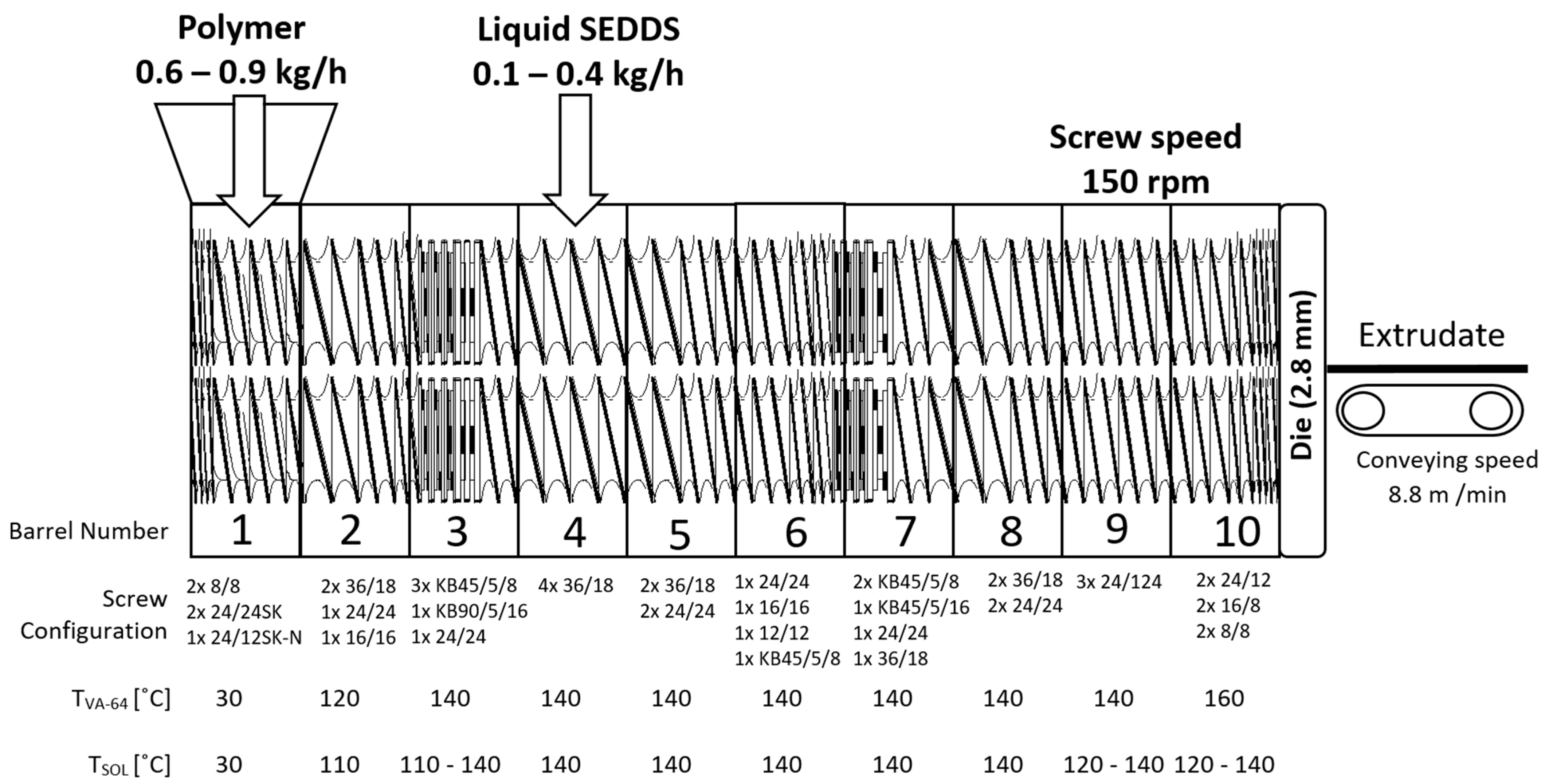
2.5. Hot Melt Extrusion
2.6. HME–SEDDSs Characterization
2.6.1. Polarized Optical Microscopy (POM)
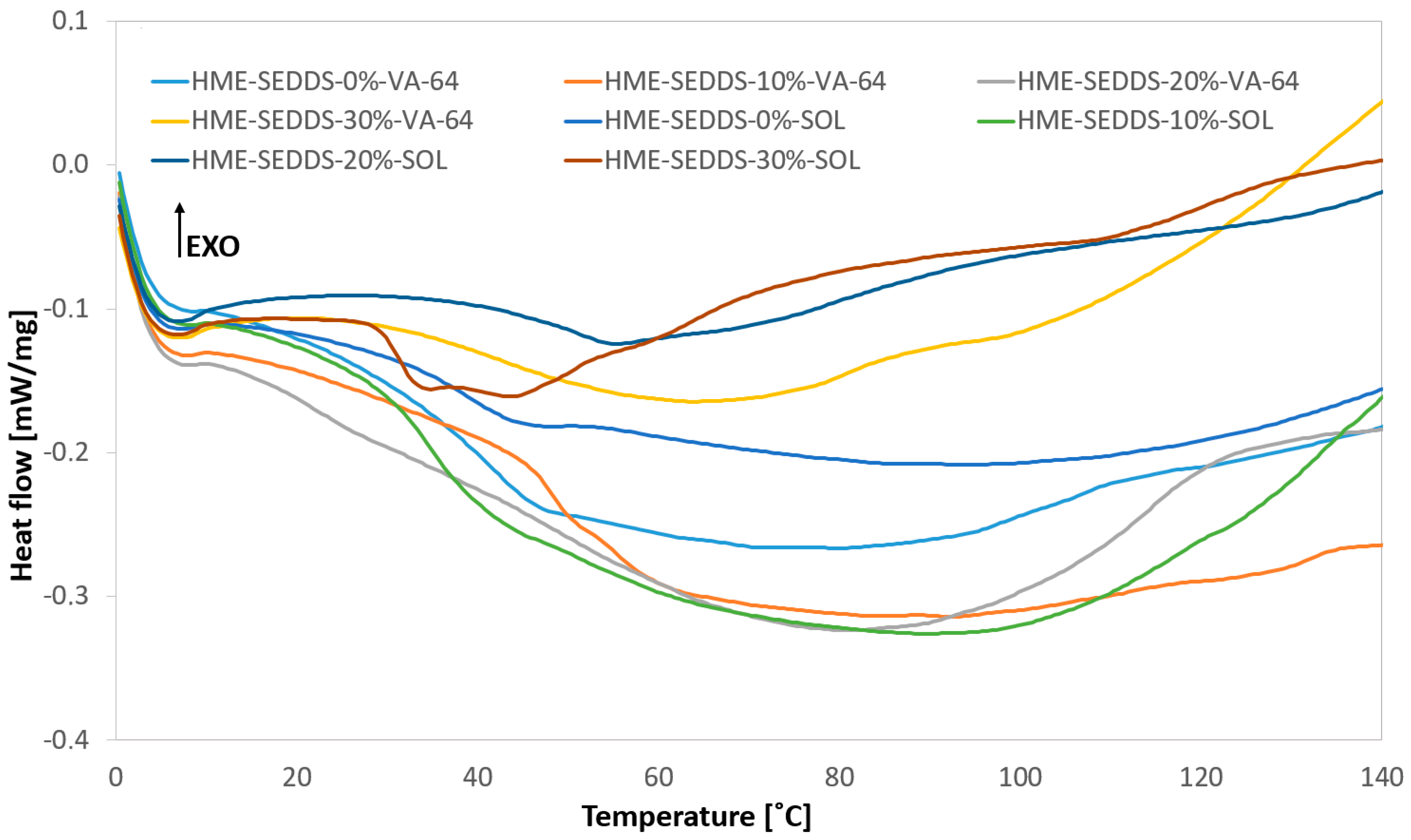
2.6.2. Differential Scanning Calorimetry (DSC)
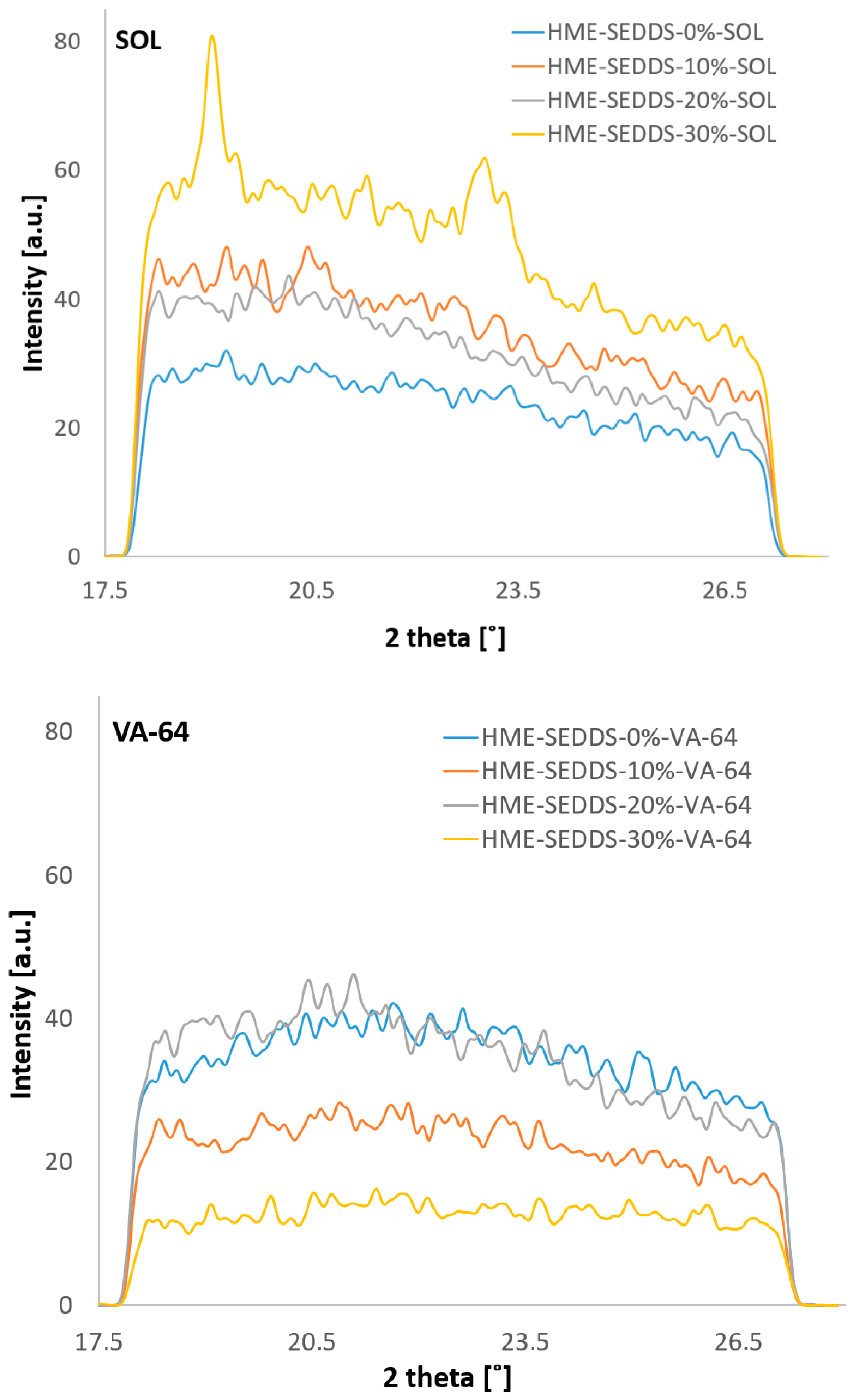
2.6.3. Wide-Angle X-ray Scattering (WAXS)
2.6.4. Emulsification Time
2.6.5. Droplet Size and Polydispersity Index (PDI)
2.6.6. Transmittance and Cloud Point
3. Results and Discussion
3.1. Oil-Binding Capacity
3.2. Hot Melt Extrusion (HME)
3.3. HME–SEDDSs Characterization
3.3.1. Polarized Optical Microscopy (POM)
3.3.2. Thermal Phase Behavior of HME–SEDDSs Formulations
3.3.3. Crystallinity of HME–SEDDSs Observed Via WAXS
3.3.4. Trends in Emulsification Efficiency among HME–SEDDSs Formulations
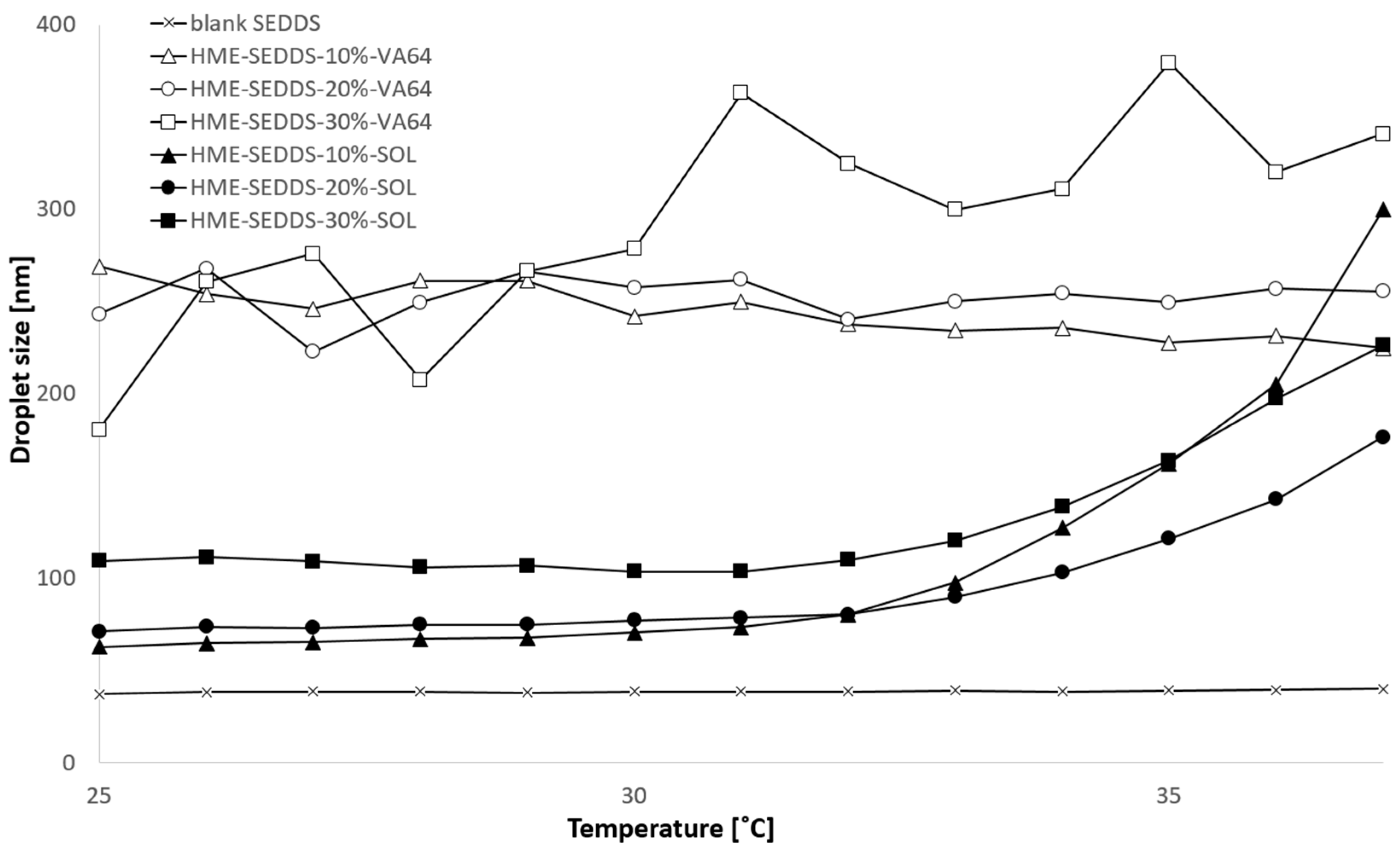
3.3.5. Droplet Size and Polydispersity Index (PDI) Obtained from HME–SEDDSs
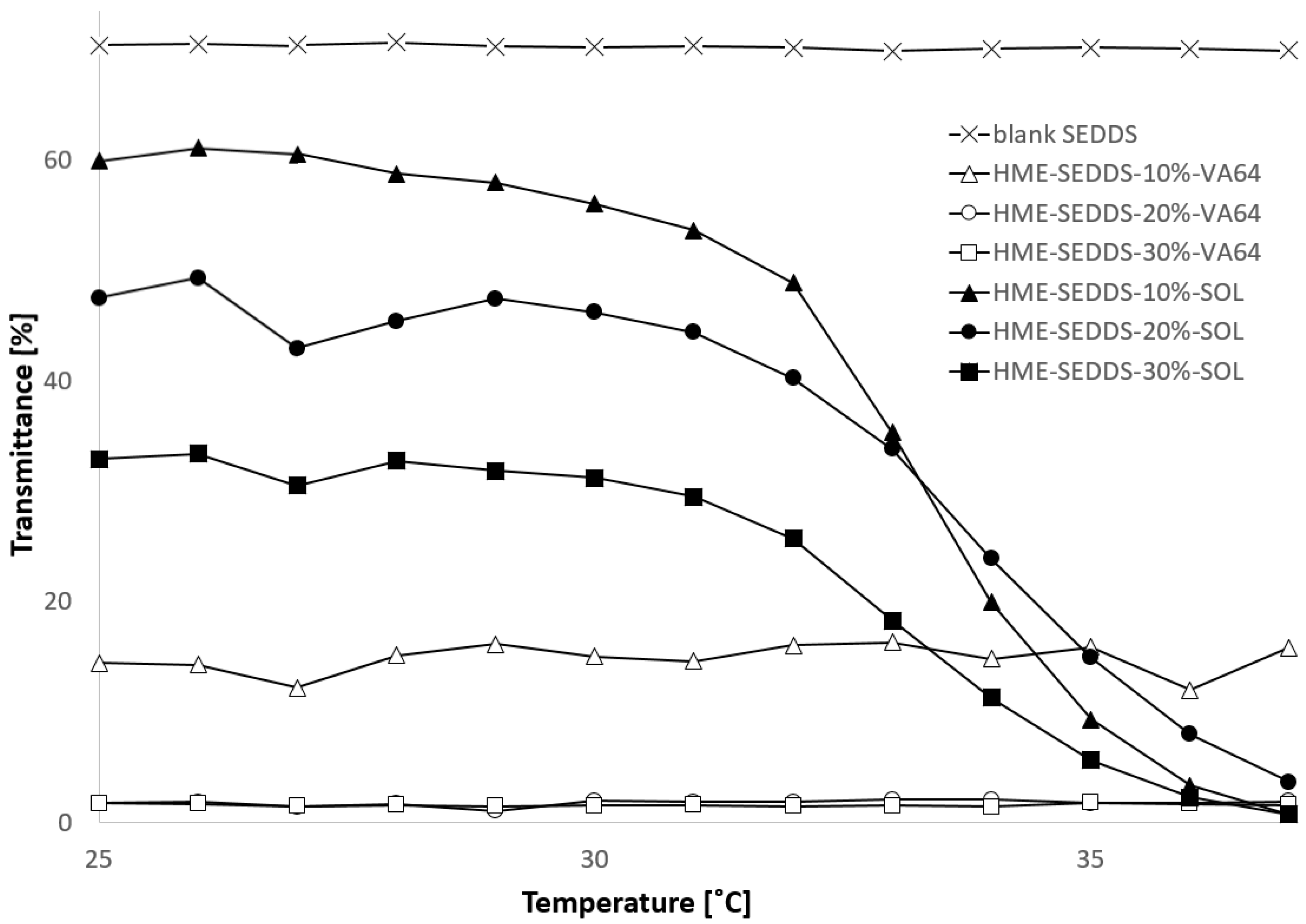
3.3.6. Transmittance and Cloud Point
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ASD | Amorphous solid dispersion |
| DSC | Differential scanning calorimetry |
| GMP | Good manufacturing practice |
| HME | Hot melt extrusion |
| HME–SEDDS | Solid SEDDSs prepared by hot melt extrusion |
| HPC | Hydroxy propyl cellulose |
| HPMCAS | Hydroxy propyl methylcellulose-acetate/succinate |
| LOD | Loss on drying |
| MCC | Microcrystalline cellulose |
| PAT | Process analytical technology |
| PDI | Polydispersity index |
| PL_630 | Plasdone® 630 |
| PL_630_U | Plasdone® 630 Ultra |
| SEDDSs | Self-emulsifying drug delivery systems |
| SEDEXSOL | Self-emulsifying delivery via extrusionSoluplus® |
| SOL-HME–SEDDSs | Solid SEDDSs prepared by hot melt extrusion with Soluplus® |
| VA-64 | Kollidon® VA-64, copovidone |
| VA-64-HME–SEDDSs | Solid SEDDSs prepared by hot melt extrusion with Kollidon VA-64® |
| WAXS | Wide-angle X-ray scattering |
| ZSK18 | Co-rotating closely intermeshing twin screw extruder from Coperion with a nominal screw diameter of 18 mm |
References
- Singh, D.; Bedi, N.; Tiwary, A.K. Enhancing solubility of poorly aqueous soluble drugs: Critical appraisal of techniques. J. Pharm. Investig. 2017, 48, 509–526. [Google Scholar] [CrossRef]
- Hribar, M.; Trontelj, J.; Berglez, S.; Bevc, A.; Kuščer, L.; Diaci, J.; Legen, I. Design of an Innovative Advanced Gastric Simulator. Dissolution Technol. 2019, 26, 20–29. [Google Scholar] [CrossRef]
- Feeney, O.M.; Crum, M.F.; McEvoy, C.L.; Trevaskis, N.L.; Williams, H.D.; Pouton, C.W.; Charman, W.N.; Bergström, C.A.; Porter, C.J. 50 years of oral lipid-based formulations: Provenance, progress and future perspectives. Adv. Drug Deliv. Rev. 2016, 101, 167–194. [Google Scholar] [CrossRef] [PubMed]
- Maji, I.; Mahajan, S.; Sriram, A.; Medtiya, P.; Vasave, R.; Khatri, D.K.; Kumar, R.; Singh, S.B.; Madan, J.; Singh, P.K. Solid self emulsifying drug delivery system: Superior mode for oral delivery of hydrophobic cargos. J. Control. Release 2021, 337, 646–660. [Google Scholar] [CrossRef] [PubMed]
- Zupančič, O.; Spoerk, M.; Paudel, A. Lipid-based solubilization technology via hot melt extrusion: Promises and challenges. Expert Opin. Drug Deliv. 2022, 19, 1013–1032. [Google Scholar] [CrossRef]
- Zupančič, O.; Grieβinger, J.A.; Rohrer, J.; de Sousa, I.P.; Danninger, L.; Partenhauser, A.; Sündermann, N.E.; Laffleur, F.; Bernkop-Schnürch, A. Development, in vitro and in vivo evaluation of a self-emulsifying drug delivery system (SEDDS) for oral enoxaparin administration. Eur. J. Pharm. Biopharm. 2016, 109, 113–121. [Google Scholar] [CrossRef]
- Zupančič, O.; Partenhauser, A.; Lam, H.T.; Rohrer, J.; Bernkop-Schnürch, A. Development and in vitro characterisation of an oral self-emulsifying delivery system for daptomycin. Eur. J. Pharm. Sci. 2016, 81, 129–136. [Google Scholar] [CrossRef]
- Tan, A.; Rao, S.; Prestidge, C.A. Transforming Lipid-Based Oral Drug Delivery Systems into Solid Dosage Forms: An Overview of Solid Carriers, Physicochemical Properties, and Biopharmaceutical Performance. Pharm. Res. 2013, 30, 2993–3017. [Google Scholar] [CrossRef]
- Joyce, P.; Dening, T.J.; Meola, T.; Schultz, H.; Holm, R.; Thomas, N.; Prestidge, C.A. Solidification to improve the biopharmaceutical performance of SEDDS: Opportunities and challenges. Adv. Drug Deliv. Rev. 2019, 142, 102–117. [Google Scholar] [CrossRef]
- Wagh, M.P.; Singh, P.K.; Chaudhari, C.S.; Khairnar, D.A. Solid self-emulsifying drug delivery system: Preparation techniques and dosage forms. Int. J. Biopharm. 2014, 5, 101–108. Available online: https://scholar.google.de/scholar?cluster=14869124386309671947&hl=de&as_sdt=0,5 (accessed on 8 August 2022).
- Jannin, V.; Musakhanian, J.; Marchaud, D. Approaches for the development of solid and semi-solid lipid-based formulations. Adv. Drug Deliv. Rev. 2008, 60, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Nikolakakis, I.; Malamataris, S. Self-Emulsifying Pellets: Relations Between Kinetic Parameters of Drug Release and Emulsion Reconstitution—Influence of Formulation Variables. J. Pharm. Sci. 2014, 103, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- Diril, M.; Karasulu, Y.; Toskas, M.; Nikolakakis, I. Development and Permeability Testing of Self-Emulsifying Atorvastatin Calcium Pellets and Tablets of Compressed Pellets. Processes 2019, 7, 365. [Google Scholar] [CrossRef] [Green Version]
- Serratoni, M.; Newton, M.; Booth, S.; Clarke, A. Controlled drug release from pellets containing water-insoluble drugs dissolved in a self-emulsifying system. Eur. J. Pharm. Biopharm. 2007, 65, 94–98. [Google Scholar] [CrossRef]
- Sarabu, S.; Bandari, S.; Kallakunta, V.R.; Tiwari, R.; Patil, H.; Repka, M.A. An update on the contribution of hot-melt extrusion technology to novel drug delivery in the twenty-first century: Part II. Expert Opin. Drug Deliv. 2019, 16, 567–582. [Google Scholar] [CrossRef]
- Thakkar, R.; Thakkar, R.; Pillai, A.; Ashour, E.A.; Repka, M.A. Systematic screening of pharmaceutical polymers for hot melt extrusion processing: A comprehensive review. Int. J. Pharm. 2020, 576, 118989. [Google Scholar] [CrossRef]
- Khinast, J.; Rantanen, J.; Kleinebudde, P.; Khinast, J.; Rantanen, J. Chapter 1—Continuous Manufacturing: Definitions and Engineering Principles. In Continuous Manufacturing of Pharmaceuticals; Kleinebudde, P., Khinast, J., Rantanen, J., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 1–31. [Google Scholar] [CrossRef]
- Sacher, S.; Poms, J.; Rehrl, J.; Khinast, J.G. PAT implementation for advanced process control in solid dosage manufacturing—A practical guide. Int. J. Pharm. 2022, 613, 121408. [Google Scholar] [CrossRef]
- Rehrl, J.; Karttunen, A.-P.; Nicolaï, N.; Hörmann, T.; Horn, M.; Korhonen, O.; Nopens, I.; De Beer, T.; Khinast, J.G. Control of three different continuous pharmaceutical manufacturing processes: Use of soft sensors. Int. J. Pharm. 2018, 543, 60–72. [Google Scholar] [CrossRef]
- Rehrl, J.; Kirchengast, M.; Celikovic, S.; Sacher, S.; Kruisz, J.; Khinast, J.; Horn, M. Improving Pellet Quality in a Pharmaceutical Hot Melt Extrusion Process via PID Control and LOLIMOT-Based MPC. J. Pharm. Innov. 2020, 15, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Matić, J.; Paudel, A.; Bauer, H.; Garcia, R.A.L.; Biedrzycka, K.; Khinast, J.G. Developing HME-Based Drug Products Using Emerging Science: A Fast-Track Roadmap from Concept to Clinical Batch. AAPS PharmSciTech 2020, 21, 176. [Google Scholar] [CrossRef]
- Matić, J.; Alva, C.; Witschnigg, A.; Eder, S.; Reusch, K.; Paudel, A.; Khinast, J. Towards predicting the product quality in hot-melt extrusion: Small scale extrusion. Int. J. Pharm. X 2020, 2, 100062. [Google Scholar] [CrossRef] [PubMed]
- Matić, J.; Witschnigg, A.; Zagler, M.; Eder, S.; Khinast, J. A novel in silico scale-up approach for hot melt extrusion processes. Chem. Eng. Sci. 2019, 204, 257–269. [Google Scholar] [CrossRef]
- Matić, J.; Alva, C.; Eder, S.; Reusch, K.; Paudel, A.; Khinast, J. Towards predicting the product quality in hot-melt extrusion: Pilot plant scale extrusion. Int. J. Pharm. X 2021, 3, 100084. [Google Scholar] [CrossRef] [PubMed]
- Repka, M.A.; Bandari, S.; Kallakunta, V.R.; Vo, A.Q.; McFall, H.; Pimparade, M.B.; Bhagurkar, A.M. Melt extrusion with poorly soluble drugs—An integrated review. Int. J. Pharm. 2018, 535, 68–85. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, R.; Matić, J.; Schrank, S.; Laske, S.; Khinast, J.; Roblegg, E. NANEX: Process design and optimization. Int. J. Pharm. 2016, 506, 35–45. [Google Scholar] [CrossRef]
- Khinast, J.; Baumgartner, R.; Roblegg, E. Nano-extrusion: A One-Step Process for Manufacturing of Solid Nanoparticle Formulations Directly from the Liquid Phase. AAPS PharmSciTech 2013, 14, 601–604. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, R.; Eitzlmayr, A.; Matsko, N.; Tetyczka, C.; Khinast, J.; Roblegg, E. Nano-extrusion: A promising tool for continuous manufacturing of solid nano-formulations. Int. J. Pharm. 2014, 477, 1–11. [Google Scholar] [CrossRef]
- Silva, L.A.D.; Almeida, S.L.; Alonso, E.C.; Rocha, P.B.; Martins, F.T.; Freitas, L.A.; Taveira, S.F.; Cunha-Filho, M.S.; Marreto, R.N. Preparation of a solid self-microemulsifying drug delivery system by hot-melt extrusion. Int. J. Pharm. 2018, 541, 1–10. [Google Scholar] [CrossRef]
- Zupančič, O.; Leonaviciute, G.; Lam, H.T.; Partenhauser, A.; Podričnik, S.; Bernkop-Schnürch, A. Development andin vitroevaluation of an oral SEDDS for desmopressin. Drug Deliv. 2016, 23, 2074–2083. [Google Scholar] [CrossRef] [Green Version]
- Kavimughil, M.; Leena, M.M.; Moses, J.; Anandharamakrishnan, C. Effect of material composition and 3D printing temperature on hot-melt extrusion of ethyl cellulose based medium chain triglyceride oleogel. J. Food Eng. 2022, 329, 111055. [Google Scholar] [CrossRef]
- Kushwah, V.; Lopes, D.G.; Saraf, I.; Koutsamanis, I.; Werner, B.; Zangger, K.; Roy, M.C.; Bartlett, J.A.; Schmidt, H.F.; Shamblin, S.L.; et al. Phase Behavior of Drug–Lipid–Surfactant Ternary Systems toward Understanding the Annealing-Induced Change. Mol. Pharm. 2022, 19, 532–546. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, V.; Saraf, I.; Yeoh, T.; Ardelean, I.; Weber, H.; Sarkar, A.; Chen, R.; Vogel, T.; Modhave, D.; Laggner, P.; et al. Interplay of Aging and Lot-to-Lot Variability on the Physical and Chemical Properties of Excipients: A Case Study of Mono- and Diglycerides. Mol. Pharm. 2021, 18, 862–877. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, V.; Lopes, D.G.; Koutsamanis, I.; Plank, H.; Ardelean, I.; Sarkar, A.; Prpich, A.; Ende, M.T.A.; Schmidt, H.F.; Doshi, P.; et al. Evolution of the microstructure and the drug release upon annealing the drug loaded lipid-surfactant microspheres. Eur. J. Pharm. Sci. 2020, 147, 105278. [Google Scholar] [CrossRef]
- Mandić, J.; Pirnat, V.; Luštrik, M.; Ilić, I.G.; Vrečer, F.; Gašperlin, M.; Pobirk, A.Z. Solidification of SMEDDS by fluid bed granulation and manufacturing of fast drug release tablets. Int. J. Pharm. 2020, 583, 119377. [Google Scholar] [CrossRef] [PubMed]
- Leonaviciute, G.; Zupančič, O.; Prüfert, F.; Rohrer, J.; Bernkop-Schnürch, A. Impact of lipases on the protective effect of SEDDS for incorporated peptide drugs towards intestinal peptidases. Int. J. Pharm. 2016, 508, 102–108. [Google Scholar] [CrossRef]
- Becker, K.; Salar-Behzadi, S.; Zimmer, A. Solvent-Free Melting Techniques for the Preparation of Lipid-Based Solid Oral Formulations. Pharm. Res. 2015, 32, 1519–1545. [Google Scholar] [CrossRef] [Green Version]
- Leichner, C.; Menzel, C.; Laffleur, F.; Bernkop-Schnürch, A. Development and in vitro characterization of a papain loaded mucolytic self-emulsifying drug delivery system (SEDDS). Int. J. Pharm. 2017, 530, 346–353. [Google Scholar] [CrossRef]
- Zupančič, O.; Grießinger, J.A.; Lam, H.T.; Bernkop-Schnürch, A. Storage Stability of Bivalirudin: Hydrophilic Versus Lipophilic Solutions. J. Pharm. Sci. 2017, 106, 1322–1330. [Google Scholar] [CrossRef]
- Gao, P.; Jiang, Z.; Luo, Q.; Mu, C.; Cui, M.; Yang, X. Preparation and Evaluation of Self-emulsifying Drug Delivery System (SEDDS) of Cepharanthine. AAPS PharmSciTech 2021, 22, 245. [Google Scholar] [CrossRef]
- Van Staden, D.; Du Plessis, J.; Viljoen, J. Development of a Self-Emulsifying Drug Delivery System for Optimized Topical Delivery of Clofazimine. Pharmaceutics 2020, 12, 523. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Composition |
|---|---|
| Liquid SEDDSs | 30% Labrafac® lipophile 30% Kolliphor® RH40 30% Capmul® MCM 10% Transcutol® |
| HME–SEDDSs 10% BLANK | 90% Polymer * 3% Labrafac® lipophile 3% Kolliphor® RH40 3% Capmul® MCM 1% Transcutol® |
| HME–SEDDSs 20% BLANK | 80% Polymer * 6% Labrafac® lipophile 6% Kolliphor® RH40 6% Capmul® MCM 2% Transcutol® |
| HME–SEDDSs 30% BLANK | 70% Polymer * 9% Labrafac® lipophile 9% Kolliphor® RH40 9% Capmul® MCM 1% Transcutol® |
| Concentration | 0.4% w/v | 2.0% w/v | ||||||
|---|---|---|---|---|---|---|---|---|
| Temperature | 25 °C | 37 °C | 25 °C | 37 °C | ||||
| Droplet Size (nm) | PDI | Droplet Size (nm) | PDI | Droplet Size (nm) | PDI | Droplet Size (nm) | PDI | |
| Blank SEDDSs | 31.8 | 0.051 | 35.8 | 0.035 | 32.5 | 0.048 | 36.0 | 0.041 |
| HME–SEDDSs–10%–VA-64 | 122.7 | 0.214 | 136.1 | 0.197 | 257.1 | 0.255 | 286.0 | 0.256 |
| HME–SEDDSs–20%–VA-64 | 152.9 | 0.193 | 170.6 | 0.132 | 259.1 | 0.246 | 234.4 | 0.269 |
| HME–SEDDSs–30%–VA-64 | 115.7 | 0.251 | 129.2 | 0.244 | 226.3 | 0.279 | 285.8 | 0.271 |
| HME–SEDDSs–10%–SOL | 61.8 | 0.061 | 90.2 | 0.089 | 63.9 | 0.047 | 139.1 | 0.152 |
| HME–SEDDSs–20%–SOL | 68.7 | 0.100 | 95.8 | 0.085 | 71.7 | 0.145 | 150.4 | 0.194 |
| HME–SEDDSs–30%–SOL | 73.5 | 0.128 | 95.1 | 0.119 | 120.1 | 0.264 | 145.8 | 0.182 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zupančič, O.; Doğan, A.; Matić, J.; Kushwah, V.; Alva, C.; Spoerk, M.; Paudel, A. SEDEX—Self-Emulsifying Delivery Via Hot Melt Extrusion: A Continuous Pilot-Scale Feasibility Study. Pharmaceutics 2022, 14, 2617. https://doi.org/10.3390/pharmaceutics14122617
Zupančič O, Doğan A, Matić J, Kushwah V, Alva C, Spoerk M, Paudel A. SEDEX—Self-Emulsifying Delivery Via Hot Melt Extrusion: A Continuous Pilot-Scale Feasibility Study. Pharmaceutics. 2022; 14(12):2617. https://doi.org/10.3390/pharmaceutics14122617
Chicago/Turabian StyleZupančič, Ožbej, Aygün Doğan, Josip Matić, Varun Kushwah, Carolina Alva, Martin Spoerk, and Amrit Paudel. 2022. "SEDEX—Self-Emulsifying Delivery Via Hot Melt Extrusion: A Continuous Pilot-Scale Feasibility Study" Pharmaceutics 14, no. 12: 2617. https://doi.org/10.3390/pharmaceutics14122617