
Prediction of Drug–Drug–Gene Interaction Scenarios of (E)-Clomiphene and Its Metabolites Using Physiologically Based Pharmacokinetic Modeling
 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
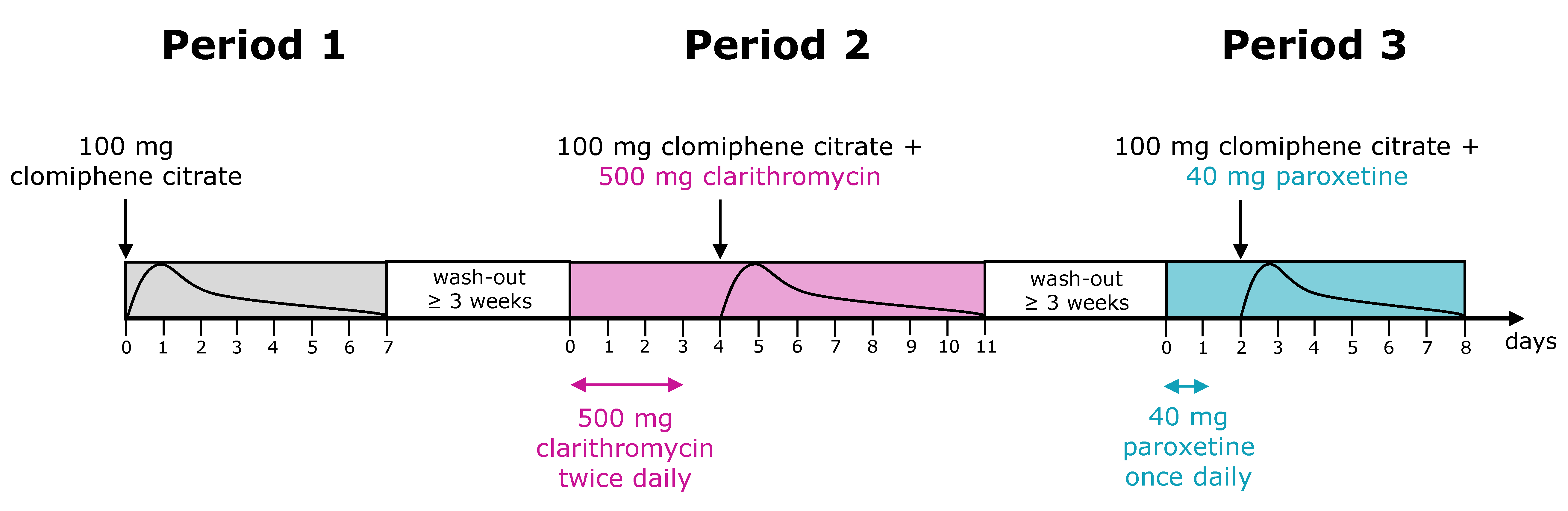
2.1. Clinical Study Data
2.2. Software
2.3. PBPK Model Development
2.4. DGI and DD(G)I Modeling
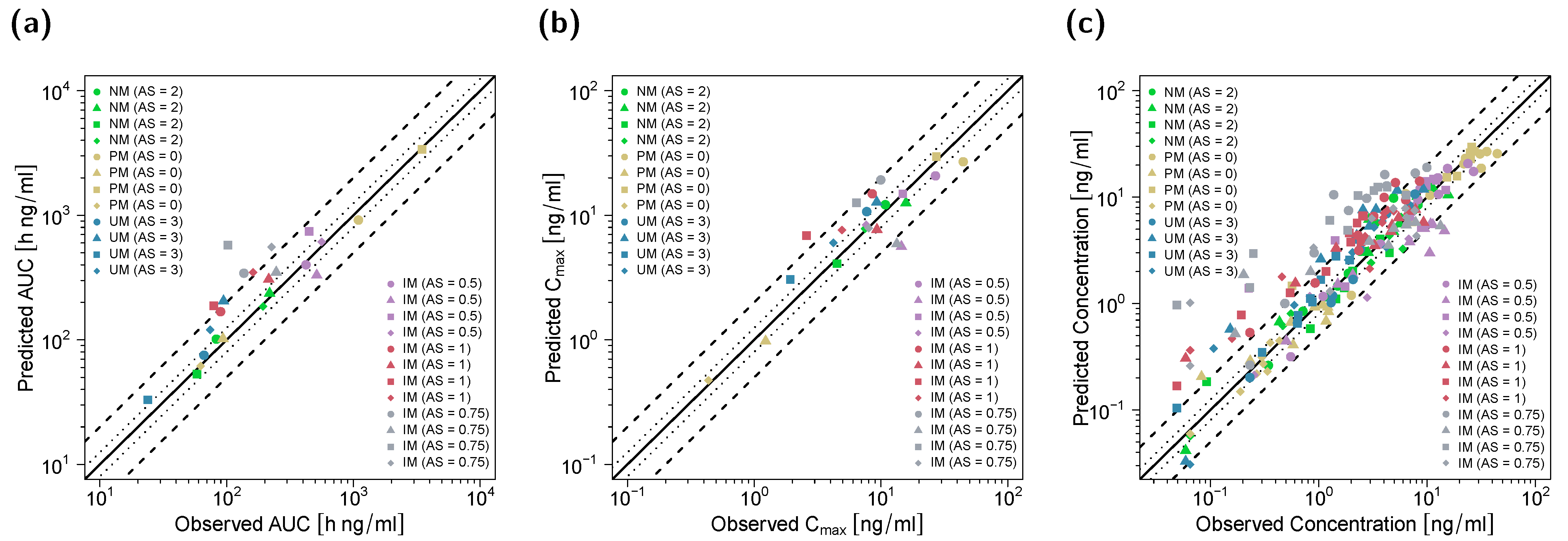
2.5. PBPK DGI and DD(G)I Model Evaluation
3. Results
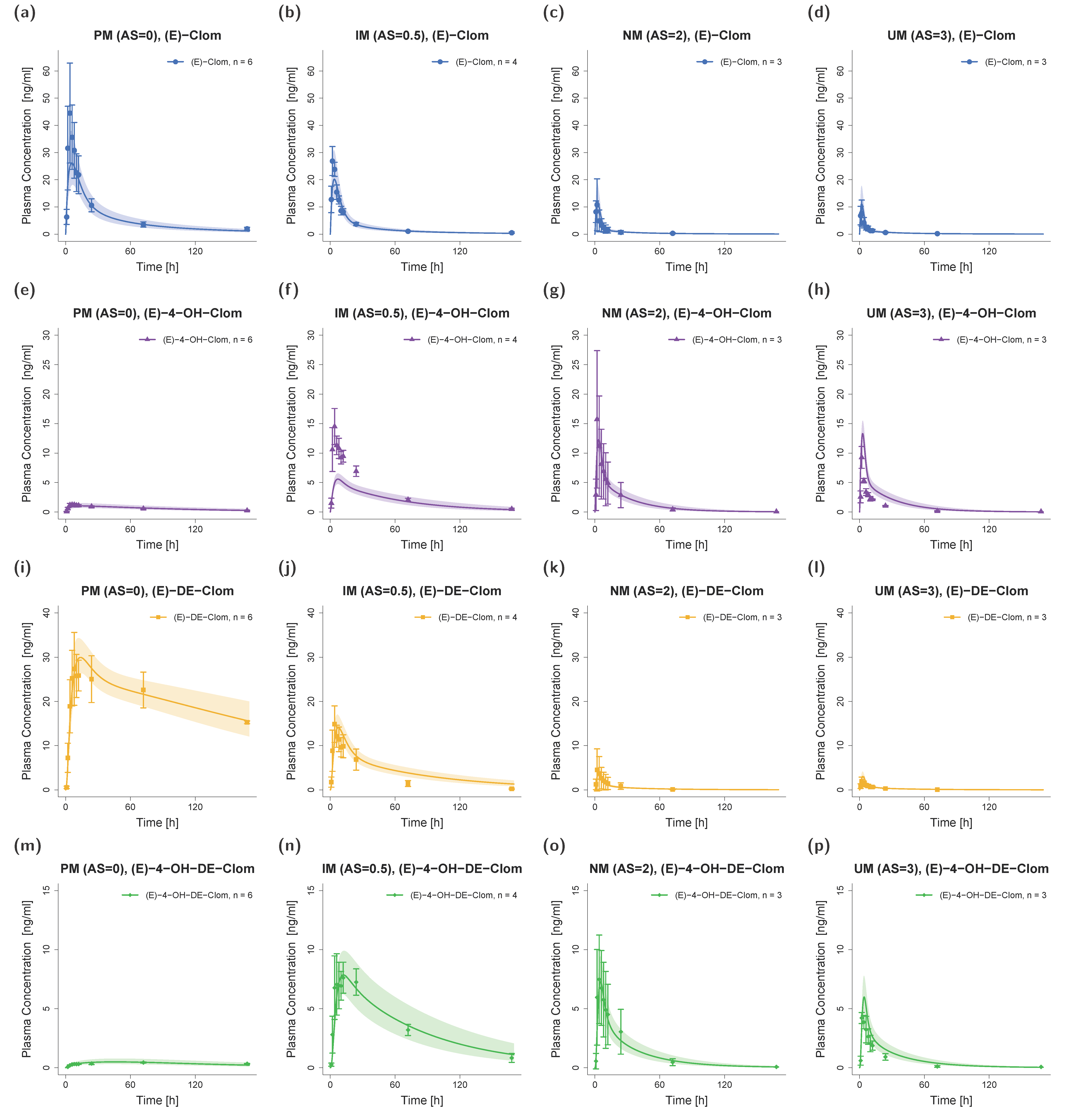
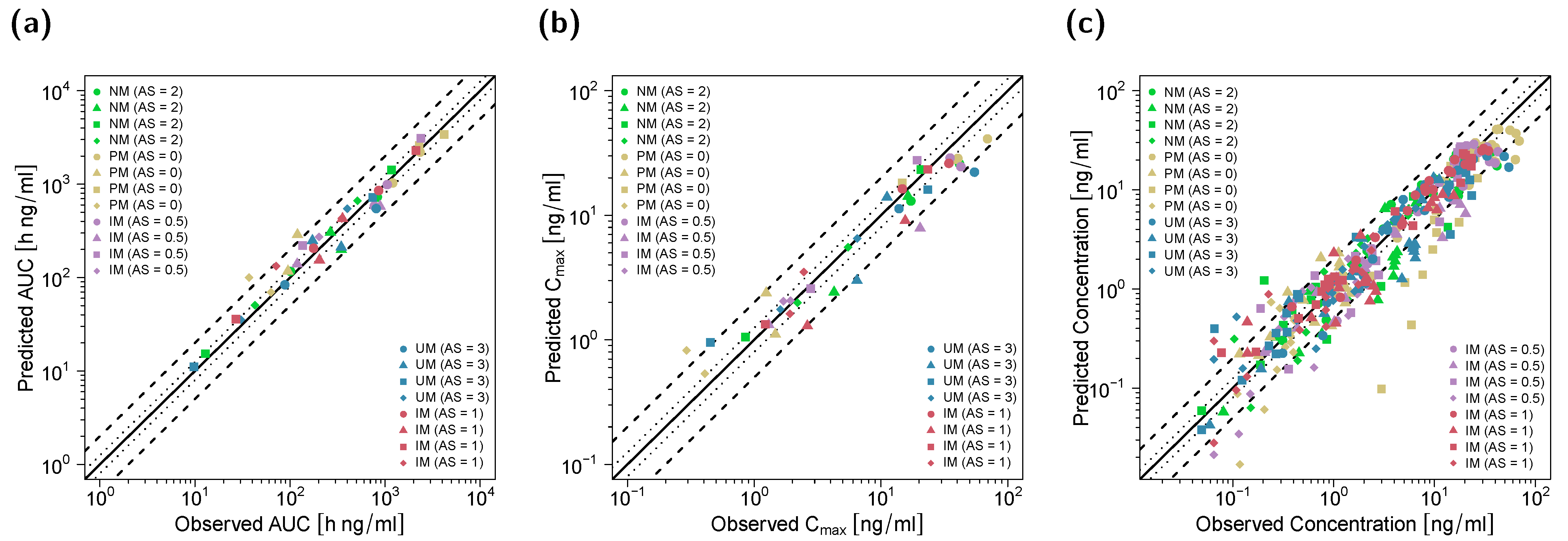
3.1. PBPK Model Building and Evaluation
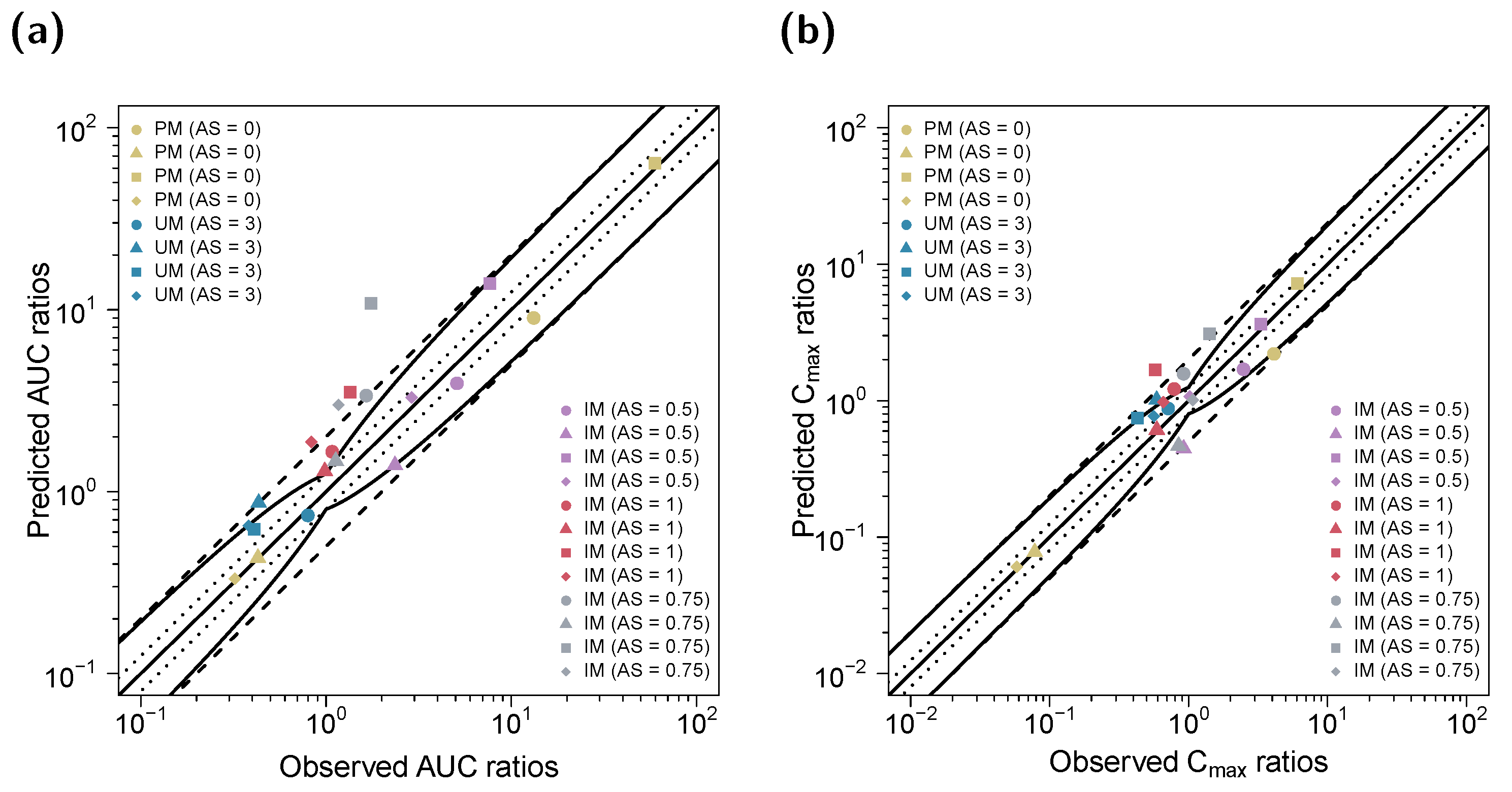
3.2. DGI Modeling and Evaluation
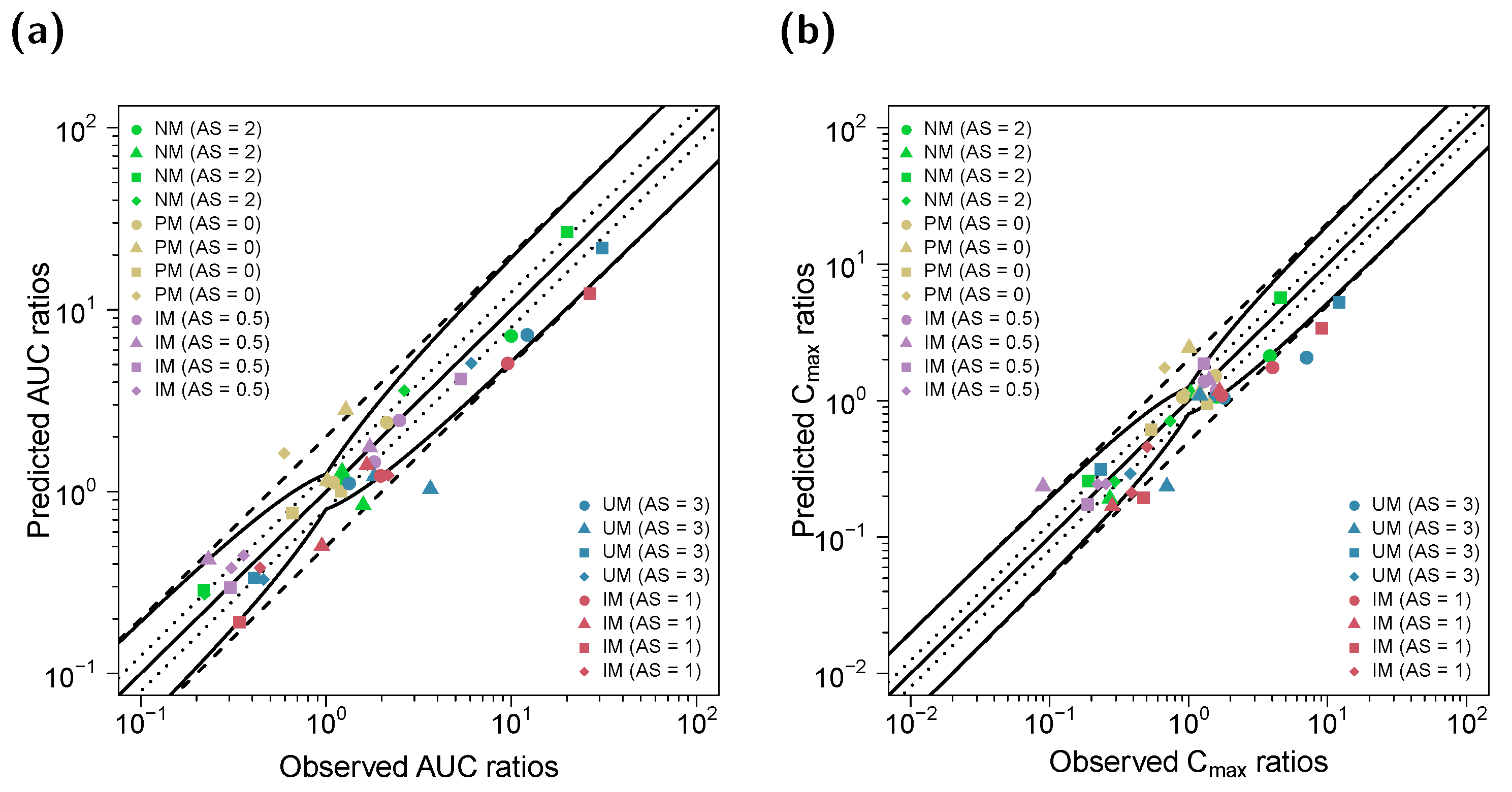
3.3. DD(G)I Modeling and Evaluation
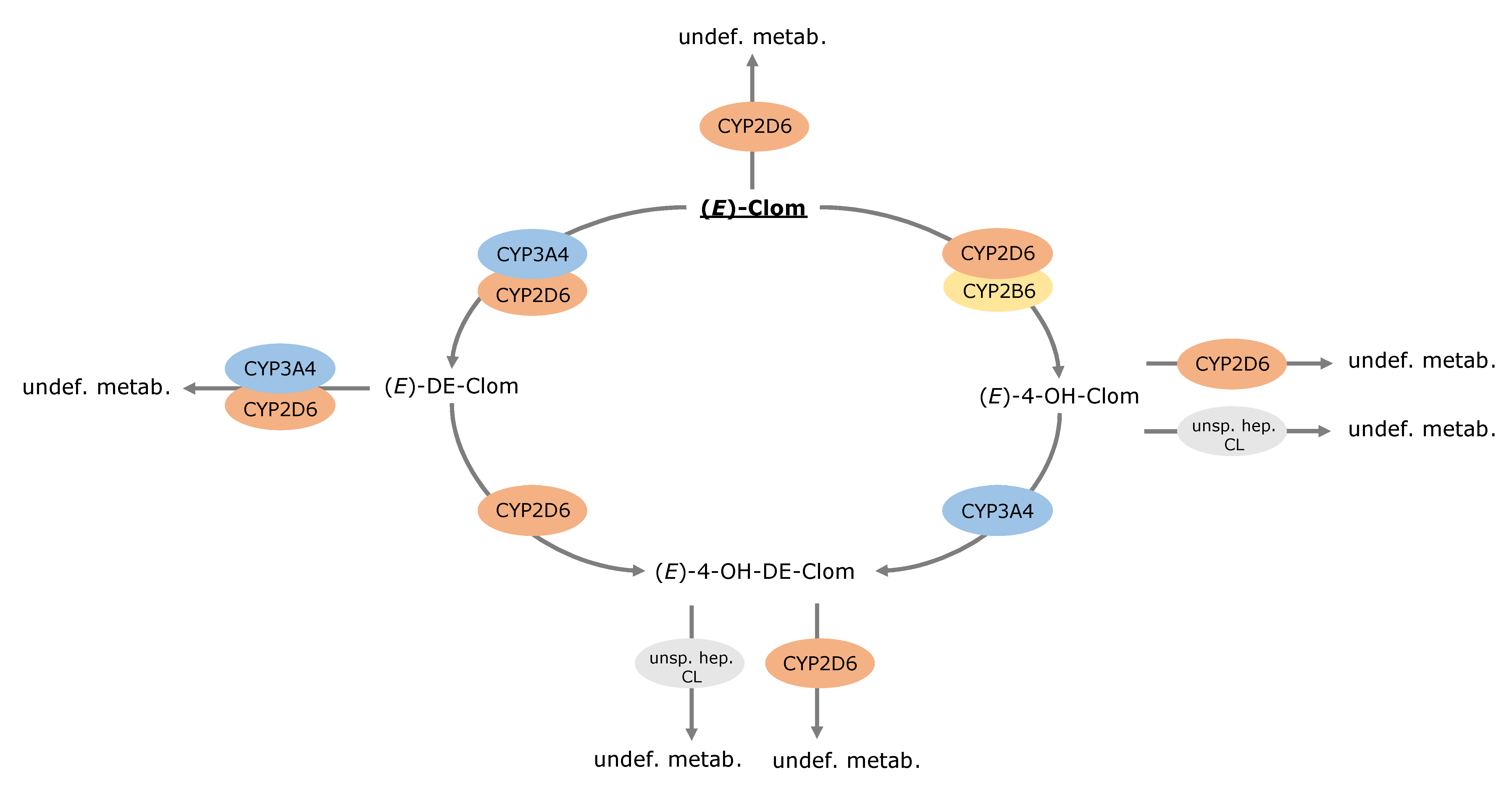
3.4. Contribution of Metabolic Pathways to (E)-Clom and Metabolite Disposition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Legro, R.S.; Barnhart, H.X.; Schlaff, W.D.; Carr, B.R.; Diamond, M.P.; Carson, S.A.; Steinkampf, M.P.; Coutifaris, C.; McGovern, P.G.; Cataldo, N.A.; et al. Clomiphene, metformin, or both for infertility in the polycystic ovary syndrome. N. Engl. J. Med. 2007, 356, 551–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deswal, R.; Narwal, V.; Dang, A.; Pundir, C.S. The prevalence of polycystic ovary syndrome: A brief systematic review. J. Hum. Reprod. Sci. 2022, 13, 261–271. [Google Scholar] [CrossRef]
- Jungheim, E.S.; Odibo, A.O. Fertility treatment in women with polycystic ovary syndrome: A decision analysis of different oral ovulation induction agents. Fertil. Steril. 2010, 94, 2659–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency (2018) Assessment Report EnCyzix. Available online: https://www.ema.europa.eu/en/documents/assessment-report/encyzix-epar-public-assessment-report_en.pdf (accessed on 12 August 2021).
- Mürdter, T.E.; Kerb, R.; Turpeinen, M.; Schroth, W.; Ganchev, B.; Böhmer, G.M.; Igel, S.; Schaeffeler, E.; Zanger, U.; Brauch, H.; et al. Genetic polymorphism of cytochrome p450 2D6 determines oestrogen receptor activity of the major infertility drug clomiphene via its active metabolites. Hum. Mol. Genet. 2012, 21, 1145–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kröner, P. Hydroxylierte Metaboliten des Clomifens: In vitro und in vivo Untersuchungen zur Bildung, Aktivität und Konjugation. Ph.D. Thesis, Eberhard-Karls-University Tübingen, Tübingen, Germany, 2018. [Google Scholar]
- Dickey, R.P.; Holtkamp, D.E. Development, pharmacology and clinical experience with clomiphene citrate. Hum. Reprod. Update 1996, 2, 483–506. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Singh, B.; Sharma, R.; Singh, A.; Kumar, A. Selective Estrogen Receptor Modulators (SERMs): Mechanistic insights against microbial infections. Curr. Mol. Med. 2019, 20, 102–115. [Google Scholar] [CrossRef]
- Garg, A.; Singh, A.; Kumar, A. Selective estrogen receptor modulators against Gram-positive and Gram-negative bacteria: An experimental study. Future Microbiol. 2021, 16, 987–1001. [Google Scholar] [CrossRef]
- Homburg, R. Clomiphene citrate--end of an era? A mini-review. Hum. Reprod. 2005, 20, 2043–2051. [Google Scholar] [CrossRef]
- Rostami-Hodjegan, A.; Lennard, M.S.; Tucker, G.T.; Ledger, W.L. Monitoring plasma concentrations to individualize treatment with clomiphene citrate. Fertil. Steril. 2004, 81, 1187–1193. [Google Scholar] [CrossRef]
- Saha, L.; Kaur, S.; Saha, P.K. Pharmacotherapy of polycystic ovary syndrome—An update. Fundam. Clin. Pharmacol. 2012, 26, 54–62. [Google Scholar] [CrossRef]
- Kröner, P.; Heinkele, G.; Kerb, R.; Igel, S.; Schwab, M.; Mürdter, T.E. Stereoselective quantification of phase 1 and 2 metabolites of clomiphene in human plasma and urine. Talanta 2021, 221, 121658. [Google Scholar] [CrossRef]
- Ji, M.; Kim, K.-R.; Lee, W.; Choe, W.; Chun, S.; Min, W.-K. Genetic polymorphism of CYP2D6 and clomiphene concentrations in infertile patients with ovulatory dysfunction treated with clomiphene citrate. J. Korean Med. Sci. 2016, 31, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Fuhr, L.M.; Marok, F.Z.; Hanke, N.; Selzer, D.; Lehr, T. Pharmacokinetics of the CYP3A4 and CYP2B6 inducer carbamazepine and its drug-drug interaction potential: A physiologically based pharmacokinetic modeling approach. Pharmaceutics 2021, 13, 270. [Google Scholar] [CrossRef] [PubMed]
- Hanke, N.; Frechen, S.; Moj, D.; Britz, H.; Eissing, T.; Wendl, T.; Lehr, T. PBPK models for CYP3A4 and P-gp DDI prediction: A modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 647–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroth, W.; Winter, S.; Mürdter, T.; Schaeffeler, E.; Eccles, D.; Eccles, B.; Chowbay, B.; Khor, C.C.; Tfayli, A.; Zgheib, N.K.; et al. Improved prediction of endoxifen metabolism by CYP2D6 genotype in breast cancer patients treated with tamoxifen. Front. Pharmacol. 2017, 8, 582. [Google Scholar] [CrossRef] [Green Version]
- Caudle, K.E.; Sangkuhl, K.; Whirl-Carrillo, M.; Swen, J.J.; Haidar, C.E.; Klein, T.E.; Gammal, R.S.; Relling, M.V.; Scott, S.A.; Hertz, D.L.; et al. Standardizing CYP2D6 genotype to phenotype translation: Consensus recommendations from the clinical pharmacogenetics implementation consortium and dutch pharmacogenetics working group. Clin. Transl. Sci. 2020, 13, 116–124. [Google Scholar] [CrossRef] [Green Version]
- FDA Drug Development and Drug Interactions. Table of Substrates, Inhibitors and Inducers. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 13 January 2022).
- Ganchev, B.; Heinkele, G.; Kerb, R.; Schwab, M.; Mürdter, T.E. Quantification of clomiphene metabolite isomers in human plasma by rapid-resolution liquid chromatography-electrospray ionization-tandem mass spectrometry. Anal. Bioanal. Chem. 2011, 400, 3429–3441. [Google Scholar] [CrossRef] [PubMed]
- Mikkelson, T.J.; Kroboth, P.D.; Cameron, W.J.; Dittert, L.W.; Chungi, V.; Manberg, P.J. Single-dose pharmacokinetics of clomiphene citrate in normal volunteers **Supported by a grant from Serono Laboratories, Inc., Randolph, Massachusetts. Fertil. Steril. 1986, 46, 392–396. [Google Scholar] [CrossRef]
- Ratiopharm GmbH Clomifen-ratiopharm® 50 mg Tabletten (Study 1991). Available online: https://www.ratiopharm.de/produkte/details/praeparate/praeparatedaten/detail/pzn-3884844.html (accessed on 23 November 2021).
- Wiehle, R.; Cunningham, G.R.; Pitteloud, N.; Wike, J.; Hsu, K.; Fontenot, G.K.; Rosner, M.; Dwyer, A.; Podolski, J. Testosterone restoration by enclomiphene citrate in men with secondary hypogonadism: Pharmacodynamics and pharmacokinetics. BJU Int. 2013, 112, 1188–1200. [Google Scholar] [CrossRef] [Green Version]
- Miller, G.D.; Moore, C.; Nair, V.; Hill, B.; Willick, S.E.; Rogol, A.D.; Eichner, D. Hypothalamic-pituitary-testicular axis effects and urinary detection following clomiphene administration in males. J. Clin. Endocrinol. Metab. 2019, 104, 906–914. [Google Scholar] [CrossRef]
- Lippert, J.; Burghaus, R.; Edginton, A.; Frechen, S.; Karlsson, M.; Kovar, A.; Lehr, T.; Milligan, P.; Nock, V.; Ramusovic, S.; et al. Open systems pharmacology community-an open access, open source, open science approach to modeling and simulation in pharmaceutical sciences. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 878–882. [Google Scholar] [CrossRef] [Green Version]
- Wojtyniak, J.-G.; Britz, H.; Selzer, D.; Schwab, M.; Lehr, T. Data digitizing: Accurate and precise data extraction for quantitative systems pharmacology and physiologically-based pharmacokinetic modeling. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 322–331. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Ganchev, B. Charakterisierung der Metabolischen Bioaktivierung des Clomifens unter Besonderer Berücksichtigung Genetischer Polymorphismen. Ph.D. Thesis, Eberhard Karls University Tübingen, Tübingen, Germany, 2014. [Google Scholar]
- Obach, R.S.; Walsky, R.L.; Venkatakrishnan, K. Mechanism-based inactivation of human cytochrome p450 enzymes and the prediction of drug-drug interactions. Drug Metab. Dispos. 2007, 35, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rüdesheim, S.; Selzer, D.; Mürdter, T.; Igel, S.; Kerb, R.; Schwab, M.; Lehr, T. Physiologically based pharmacokinetic modeling to describe the CYP2D6 activity score-dependent metabolism of paroxetine, atomoxetine and risperidone. Pharmaceutics 2022, 14, 1734. [Google Scholar] [CrossRef] [PubMed]
- Open Systems Pharmacology Suite Community. Open Systems Pharmacology Suite Manual. Available online: https://raw.githubusercontent.com/Open-Systems-Pharmacology/OSPSuite.Documentation/master/OpenSystemsPharmacologySuite.pdf (accessed on 23 November 2021).
- Guest, E.J.; Aarons, L.; Houston, J.B.; Rostami-Hodjegan, A.; Galetin, A. Critique of the two-fold measure of prediction success for ratios: Application for the assessment of drug-drug interactions. Drug Metab. Dispos. 2011, 39, 170–173. [Google Scholar] [CrossRef] [Green Version]
- Les Laboratoires Servier Servier Medical Art. Available online: https://smart.servier.com/ (accessed on 12 August 2021).
- Kousta, E.; White, D.M.; Franks, S. Modern use of clomiphene citrate in induction of ovulation. Hum. Reprod. Update 1997, 3, 359–365. [Google Scholar] [CrossRef]
- Imani, B.; Eijkemans, M.J.C.; Te Velde, E.R.; Habbema, J.D.F.; Fauser, B.C.J.M. Predictors of patients remaining anovulatory during clomiphene citrate induction of ovulation in normogonadotropic oligoamenorrheic infertility. J. Clin. Endocrinol. Metab. 1998, 83, 2361–2365. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-J.; Byeon, J.-Y.; Kim, Y.-H.; Kim, S.-H.; Lee, C.-M.; Jung, E.H.; Chae, W.K.; Lee, Y.J.; Jang, C.-G.; Lee, S.-Y.; et al. Effect of the CYP2D6*10 allele on the pharmacokinetics of clomiphene and its active metabolites. Arch. Pharm. Res. 2018, 41, 347–353. [Google Scholar] [CrossRef]
- Ellakwa, H.E.; Sanad, Z.F.; Hamza, H.A.; Emara, M.A.; Elsayed, M.A. Predictors of patient responses to ovulation induction with clomiphene citrate in patients with polycystic ovary syndrome experiencing infertility. Int. J. Gynaecol. Obstet. 2016, 133, 59–63. [Google Scholar] [CrossRef]
- Mahran, A.; Abdelmeged, A.; El-Adawy, A.R.; Eissa, M.K.; Shaw, R.W.; Amer, S.A. The predictive value of circulating anti-Müllerian hormone in women with polycystic ovarian syndrome receiving clomiphene citrate: A prospective observational study. J. Clin. Endocrinol. Metab. 2013, 98, 4170–4175. [Google Scholar] [CrossRef]
- Amer, S.A.; Mahran, A.; Abdelmaged, A.; El-Adawy, A.R.; Eissa, M.K.; Shaw, R.W. The influence of circulating anti-Müllerian hormone on ovarian responsiveness to ovulation induction with gonadotrophins in women with polycystic ovarian syndrome: A pilot study. Reprod. Biol. Endocrinol. 2013, 11, 115. [Google Scholar] [CrossRef]
- Ghobadi, C.; Gregory, A.; Crewe, H.K.; Rostami-Hodjegan, A.; Lennard, M.S. CYP2D6 is primarily responsible for the metabolism of clomiphene. Drug Metab. Pharmacokinet. 2008, 23, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Rüdesheim, S.; Selzer, D.; Fuhr, U.; Schwab, M.; Lehr, T. Physiologically-based pharmacokinetic modeling of dextromethorphan to investigate interindividual variability within CYP2D6 activity score groups. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 494–511. [Google Scholar] [CrossRef]
- Gaedigk, A.; Dinh, J.C.; Jeong, H.; Prasad, B.; Leeder, J.S. Ten years’ experience with the CYP2D6 activity score: A perspective on future investigations to improve clinical predictions for precision therapeutics. J. Pers. Med. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzarino, M.; Biava, M.; de la Torre, X.; Fiacco, I.; Botrè, F. Characterization of the biotransformation pathways of clomiphene, tamoxifen and toremifene as assessed by LC-MS/(MS) following in vitro and excretion studies. Anal. Bioanal. Chem. 2013, 405, 5467–5487. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.; Arutchelvam, V.; Quinton, R. Enclomiphene, an estrogen receptor antagonist for the treatment of testosterone deficiency in men. IDrugs 2009, 12, 109–119. [Google Scholar]
- Szutu, M.; Morgan, D.J.; McLeish, M.; Phillipou, G.; Blackman, G.L.; Cox, L.W.; Dollman, W. Pharmacokinetics of intravenous clomiphene isomers. Br. J. Clin. Pharmacol. 1989, 27, 639–640. [Google Scholar] [CrossRef] [Green Version]
- Mazzarino, M.; Fiacco, I.; de la Torre, X.; Botrè, F. A mass spectrometric approach for the study of the metabolism of clomiphene, tamoxifen and toremifene by liquid chromatography time-of-flight spectroscopy. Eur. J. Mass Spectrom. 2008, 14, 171–180. [Google Scholar] [CrossRef]
- Lu, J.; He, G.; Wang, X.; Xu, Y.; Wu, Y.; Dong, Y.; He, Z.; Liu, X.; Bo, T.; Ouyang, G. Mass spectrometric identification and characterization of new clomiphene metabolites in human urine by liquid chromatography-quadrupole time-of-flight tandem mass spectrometry. J. Chromatogr. A 2012, 1243, 23–32. [Google Scholar] [CrossRef]
- Sheiner, L.B. Learning versus confirming in clinical drug development. Clin. Pharmacol. Ther. 1997, 61, 275–291. [Google Scholar] [CrossRef]
- Jones, H.; Rowland-Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, 63. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, M.; Allard, W.G.; Vossen, R.H.A.M.; Baak-Pablo, R.F.; Menafra, R.; Deiman, B.A.L.M.; Deenen, M.J.; Neven, P.; Johansson, I.; Gastaldello, S.; et al. Toward predicting CYP2D6-mediated variable drug response from CYP2D6 gene sequencing data. Sci. Transl. Med. 2021, 13, eabf3637. [Google Scholar] [CrossRef] [PubMed]
- Valentin, J. Basic anatomical and physiological data for use in radiological protection: Reference values. A report of age- and gender-related differences in the anatomical and physiological characteristics of reference individuals. ICRP Publication 89. Ann. ICRP. 2002, 32, 5–265. [Google Scholar]
- Open Systems Pharmacology Suite Community. PK-Sim® Ontogeny Database Documentation, Version 7.3. Available online: https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/master/PK-Sim%20Ontogeny%20Database%20Version%207.3.pdf (accessed on 25 March 2020).
- National Center for Health Statistics Hyattsville. 20782 Third National Health and Nutrition Examination Survey, NHANES III (1988–1994). Available online: https://wwwn.cdc.gov/nchs/nhanes/nhanes3/default.aspx (accessed on 25 March 2020).
- Willmann, S.; Höhn, K.; Edginton, A.; Sevestre, M.; Solodenko, J.; Weiss, W.; Lippert, J.; Schmitt, W. Development of a physiology-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J. Pharmacokinet. Pharmacodyn. 2007, 34, 401–431. [Google Scholar] [CrossRef]
- Rodrigues, A.D. Integrated cytochrome P450 reaction phenotyping. Attempting to bridge the gap between cDNA-expressed cytochromes P450 and native human liver microsomes. Biochem. Pharmacol. 1999, 57, 465–480. [Google Scholar] [CrossRef]
- Nishimura, M.; Naito, S. Tissue-specific mRNA Expression Profiles of Human ATP-binding Cassette and Solute Carrier Transporter Superfamilies. Drug Metab. Pharmacokinet. 2005, 20, 452–477. [Google Scholar] [CrossRef]
- Rowland Yeo, K.; Walsky, R.L.; Jamei, M.; Rostami-Hodjegan, A.; Tucker, G.T. Prediction of time-dependent CYP3A4 drug-drug interactions by physiologically based pharmacokinetic modelling: Impact of inactivation parameters and enzyme turnover. Eur. J. Pharm. Sci. 2011, 43, 160–173. [Google Scholar] [CrossRef]
- Greenblatt, D.J.; von Moltke, L.L.; Harmatz, J.S.; Chen, G.; Weemhoff, J.L.; Jen, C.; Kelley, C.J.; LeDuc, B.W.; Zinny, M.A. Time course of recovery of cytochrome p450 3A function after single doses of grapefruit juice. Clin. Pharmacol. Ther. 2003, 74, 121–129. [Google Scholar] [CrossRef]
- Tsamandouras, N.; Rostami-Hodjegan, A.; Aarons, L. Combining the ‘bottom up’ and ‘top down’ approaches in pharmacokinetic modelling: Fitting PBPK models to observed clinical data. Br. J. Clin. Pharmacol. 2015, 79, 48–55. [Google Scholar] [CrossRef]
- Austin, R.P.; Barton, P.; Cockroft, S.L.; Wenlock, M.C.; Riley, R.J. The influence of nonspecific microsomal binding on apparent intrinsic clearance, and its prediction from physicochemical properties. Drug Metab. Dispos. 2002, 30, 1497–1503. [Google Scholar] [CrossRef] [Green Version]
- Obach, R.S. Nonspecific binding to microsomes: Impact on scale-up of in vitro intrinsic clearance to hepatic clearance as assessed through examination of warfarin, imipramine, and propranolol. Drug Metab. Dispos. Biol. Fate Chem. 1997, 25, 1359–1369. [Google Scholar] [PubMed]
- Watanabe, R.; Esaki, T.; Kawashima, H.; Natsume-Kitatani, Y.; Nagao, C.; Ohashi, R.; Mizuguchi, K. Predicting Fraction Unbound in Human Plasma from Chemical Structure: Improved Accuracy in the Low Value Ranges. Mol. Pharm. 2018, 15, 5302–5311. [Google Scholar] [CrossRef] [PubMed]
- Siramshetty, V.B.; Grishagin, I.; Nguyễn, Ð.T.; Peryea, T.; Skovpen, Y.; Stroganov, O.; Katzel, D.; Sheils, T.; Jadhav, A.; Mathé, E.A.; et al. NCATS Inxight Drugs: A comprehensive and curated portal for translational research. Nucleic Acids Res. 2022, 50, D1307–D1316. [Google Scholar] [CrossRef] [PubMed]
- Developed by ChemAxon (2009), Chemicalize Was Used for Prediction of (E)-Clomiphene Properties. Available online: https://chemicalize.com/ (accessed on 9 August 2021).
- Das, P.; Prajapati, M.; Maity, A. Study of equilibrium solubility of Clomiphene Citrate as model compound by Saturation orbital shake flask method. J. Pharm. Adv. Res. 2020, 3, 843–847. [Google Scholar]
- Smith, D.A.; Dalvie, D. Why do metabolites circulate? Xenobiotica 2012, 42, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehe sive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Güngör, S.; Delgado-Charro, M.B.; Masini-Etévé, V.; Potts, R.O.; Guy, R.H. Transdermal flux predictions for selected selective oestrogen receptor modulators (SERMs): Comparison with experimental results. J. Control. Release Off. J. Control. Release Soc. 2013, 172, 601–606. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, W. General approach for the calculation of tissue to plasma partition coefficients. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2008, 22, 457–467. [Google Scholar] [CrossRef]
- Kawai, R.; Lemaire, M.; Steimer, J.L.; Bruelisauer, A.; Niederberger, W.; Rowland, M. Physiologically based pharmacokinetic study on a cyclosporin derivative, SDZ IMM 125. J. Pharmacokinet. Biopharm. 1994, 22, 327–365. [Google Scholar] [CrossRef] [PubMed]
- Developed by ChemAxon (2009), Chemicalize Was Used for Prediction of (E)-N-Desethylclomiphene Properties. Available online: https://chemicalize.com/ (accessed on 9 August 2021).
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically based pharmacokinetic modeling 1: Predicting the tissue distribution of moderate-to-strong bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Rowland, M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef] [PubMed]
- Developed by ChemAxon (2009), Chemicalize Was Used for Prediction of (E)-4-Hydroxyclomiphene Properties. Available online: https://chemicalize.com/ (accessed on 9 August 2021).
- Berezhkovskiy, L.M. Volume of distribution at steady state for a linear pharmacokinetic system with peripheral elimination. J. Pharm. Sci. 2004, 93, 1628–1640. [Google Scholar] [CrossRef]
- Developed by ChemAxon (2009), Chemicalize Was Used for Prediction of (E)-4-Hydroxy-N-desethylclomiphene Properties. Available online: https://chemicalize.com/ (accessed on 9 August 2021).
- T’jollyn, H.; Snoeys, J.; Vermeulen, A.; Michelet, R.; Cuyckens, F.; Mannens, G.; Van Peer, A.; Annaert, P.; Allegaert, K.; Van Bocxlaer, J.; et al. Physiologically Based Pharmacokinetic Predictions of Tramadol Exposure Throughout Pediatric Life: An Analysis of the Different Clearance Contributors with Emphasis on CYP2D6 Maturation. AAPS J. 2015, 17, 1376–1387. [Google Scholar] [CrossRef] [Green Version]
- United States Pharmacopeial Convention. United States Pharmacopeia and National Formulary (USP 29-NF 24). 2006, p. 553. Available online: http://www.pharmacopeia.cn/v29240/usp29nf24s0_m18490.html (accessed on 24 August 2021).
- Beal, S.L. Ways to fit a PK model with some data below the quantification limit. J. Pharmacokinet. Pharmacodyn. 2001, 28, 481–504. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AS = 0 | AS = 0.5 | AS = 0.75 | AS = 1 | AS = 2 | AS = 3 | |
|---|---|---|---|---|---|---|
| n | 6 # | 4 | 1 + | 2 | 3 | 3 |
| CYP2D6 phenotypes | PM | IM | IM | IM | NM | UM |
| CYP2D6 genotypes | *4/*4 *4/*5 *4/*6 | *4/*41 *4/*9 | *9/*10 | *1/*4 | *1/*1 | *1/*1 × 3 |
| Demographics | ||||||
| Age [years] | 25.2 (22–29) | 24.3 (21–30) | 22.0 (-) | 25.5 (23–28) | 32.3 (26–43) | 25.7 (22–28) |
| Weight [kg] | 62.3 (50.0–70.0) | 59.3 (55.5–64.0) | 63.0 (-) | 68.8 (63.5–74.0) | 56.5 (48.0–63.5) | 61.7 (54.0–73.0) |
| Height [cm] | 1.70 (1.53–1.75) | 1.68 (1.59–1.72) | 1.66 (-) | 1.71 (1.68–1.73) | 1.63 (1.60–1.67) | 1.65 (1.57–1.75) |
| BMI [kg/m2] | 21.6 (20.6–22.9) | 21.1 (20.3–22.0) | 22.9 (-) | 23.6 (22.5–24.7) | 21.3 (18.8–24.2) | 22.6 (20.3–23.8) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovar, C.; Kovar, L.; Rüdesheim, S.; Selzer, D.; Ganchev, B.; Kröner, P.; Igel, S.; Kerb, R.; Schaeffeler, E.; Mürdter, T.E.; et al. Prediction of Drug–Drug–Gene Interaction Scenarios of (E)-Clomiphene and Its Metabolites Using Physiologically Based Pharmacokinetic Modeling. Pharmaceutics 2022, 14, 2604. https://doi.org/10.3390/pharmaceutics14122604
Kovar C, Kovar L, Rüdesheim S, Selzer D, Ganchev B, Kröner P, Igel S, Kerb R, Schaeffeler E, Mürdter TE, et al. Prediction of Drug–Drug–Gene Interaction Scenarios of (E)-Clomiphene and Its Metabolites Using Physiologically Based Pharmacokinetic Modeling. Pharmaceutics. 2022; 14(12):2604. https://doi.org/10.3390/pharmaceutics14122604
Chicago/Turabian StyleKovar, Christina, Lukas Kovar, Simeon Rüdesheim, Dominik Selzer, Boian Ganchev, Patrick Kröner, Svitlana Igel, Reinhold Kerb, Elke Schaeffeler, Thomas E. Mürdter, and et al. 2022. "Prediction of Drug–Drug–Gene Interaction Scenarios of (E)-Clomiphene and Its Metabolites Using Physiologically Based Pharmacokinetic Modeling" Pharmaceutics 14, no. 12: 2604. https://doi.org/10.3390/pharmaceutics14122604